
Importance and Considerations of Antibody Engineering in Antibody-Drug Conjugates Development from a Clinical Pharmacologist’s Perspective


Abstract
:1. Introduction
2. Structural Considerations Affecting ADC Disposition
2.1. Antibody Selection
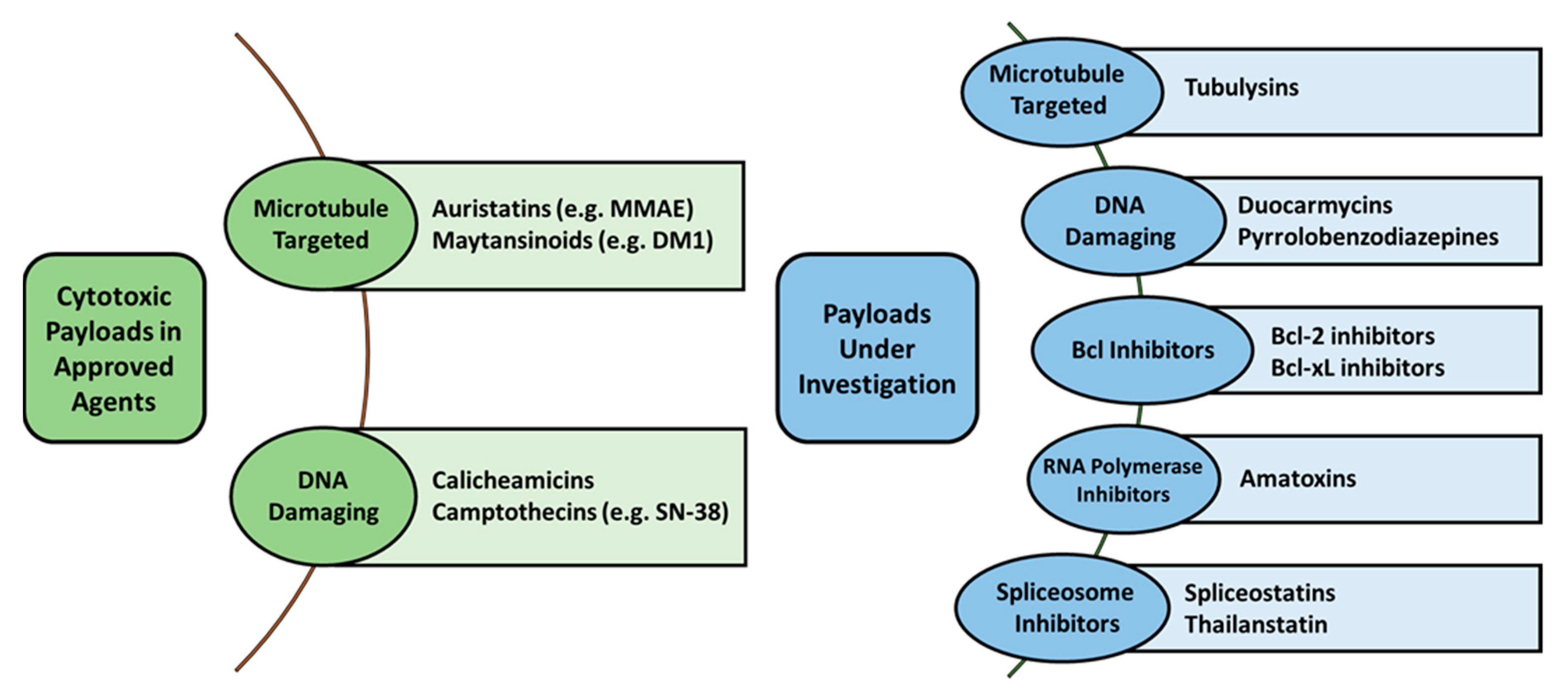
2.2. Payload Options
2.3. Linker-Drug Stability
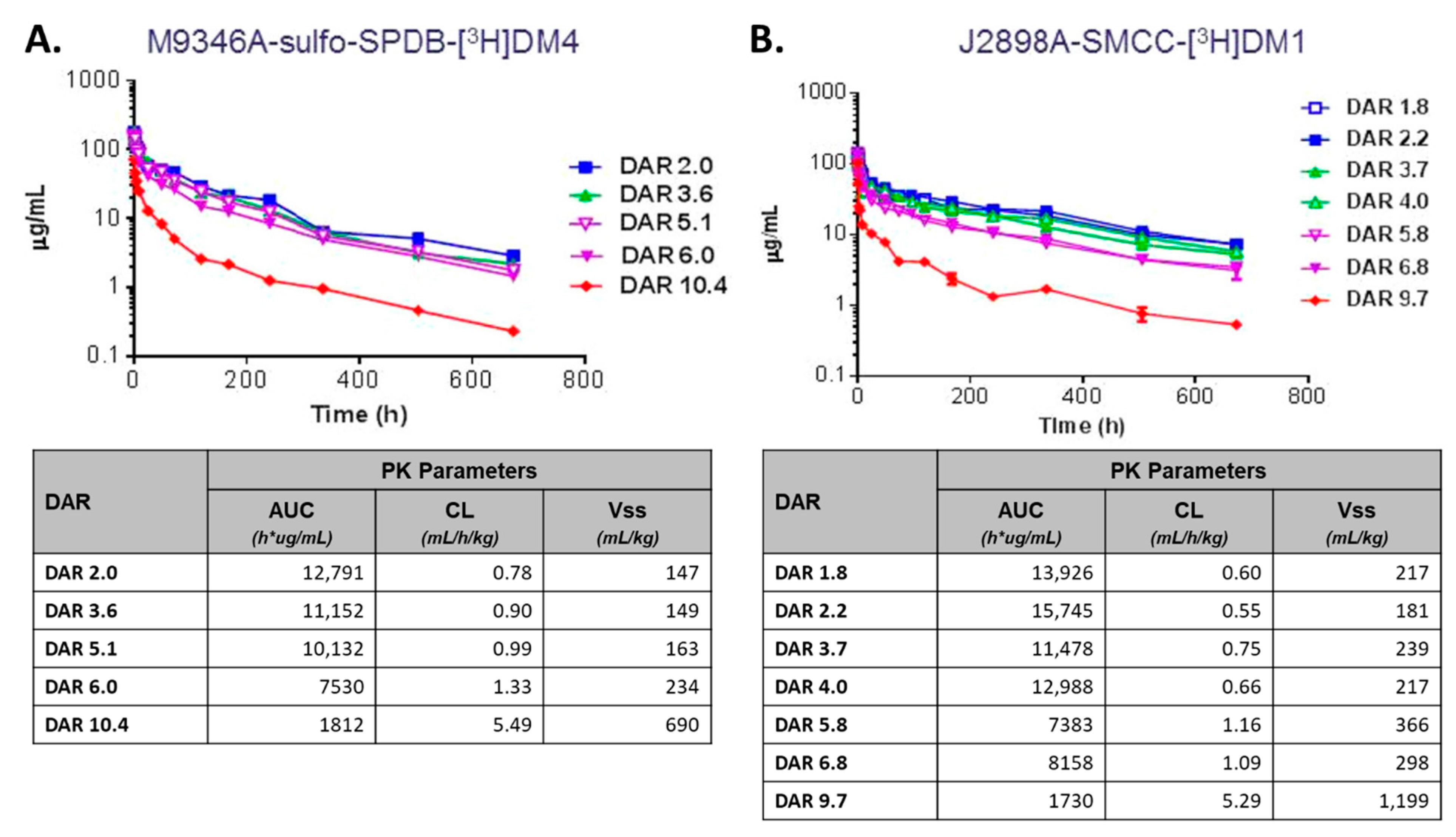
2.4. Drug-Antibody Ratio (DAR)
2.5. Surface Modification
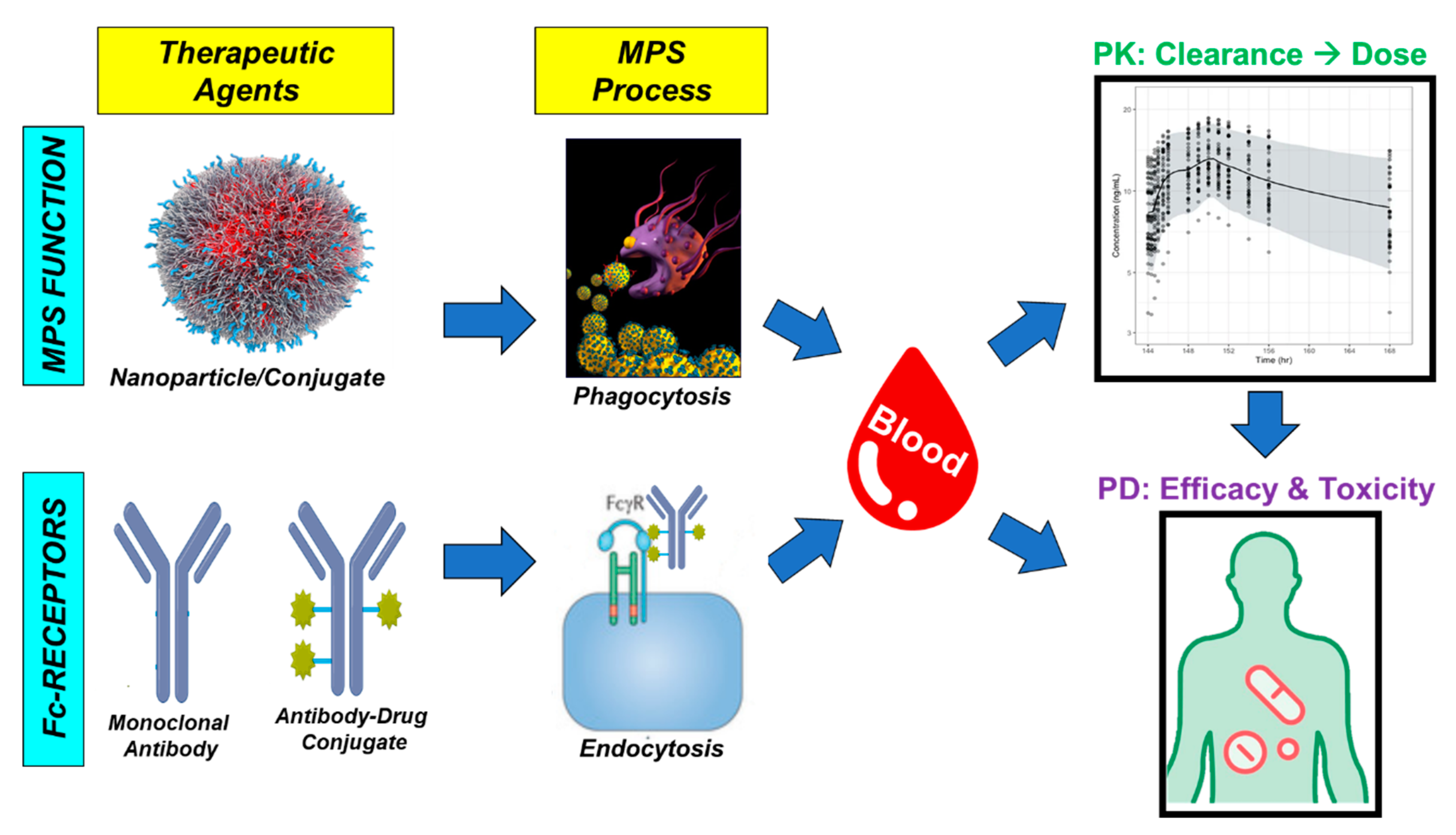
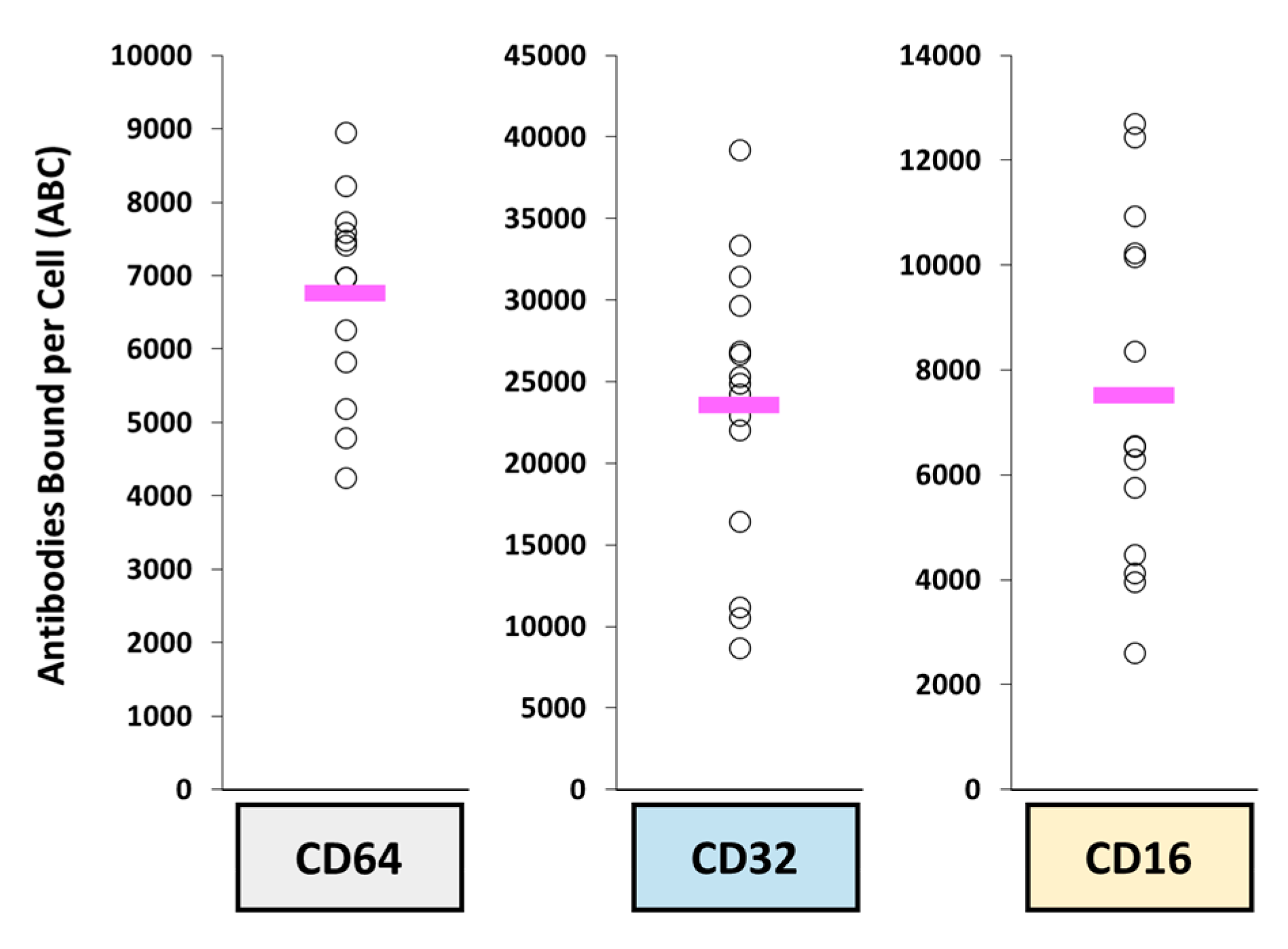
3. Biological Interactions Affecting ADC Pharmacologic Disposition
3.1. Target Expression and Affinity
3.2. Non-Specific Endocytosis
3.3. Antigen Targets Resistant to Internalization
3.4. Non-Antigen Receptor-Mediated Uptake
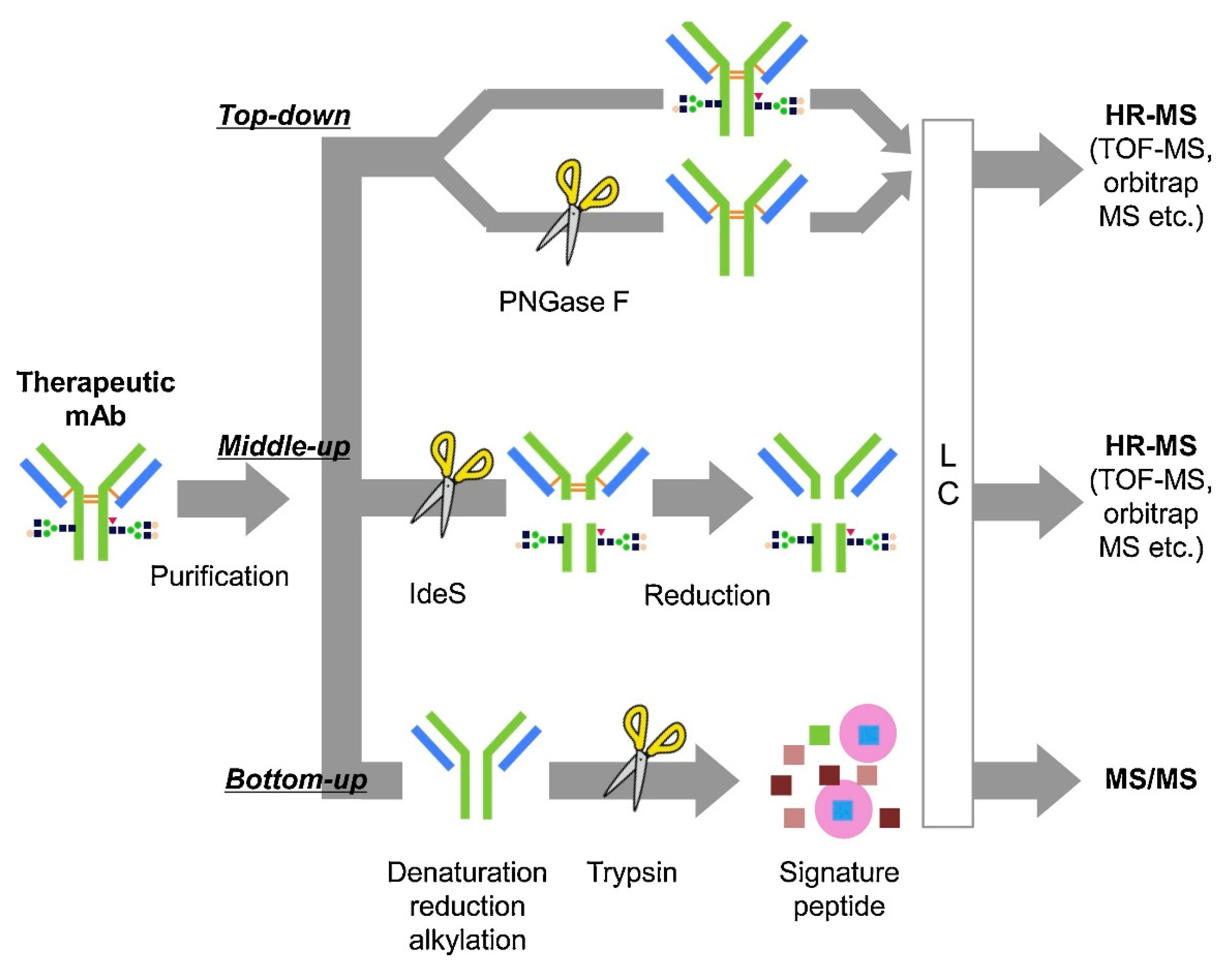
4. Bioanalytical Methods to Evaluate Novel Engineered ADC Disposition
4.1. Intact Mass Analysis
4.2. Middle-Down
4.3. Bottom-Up
5. Managing the Therapeutic Index (TI) of ADCs
5.1. Patient Selection Strategies
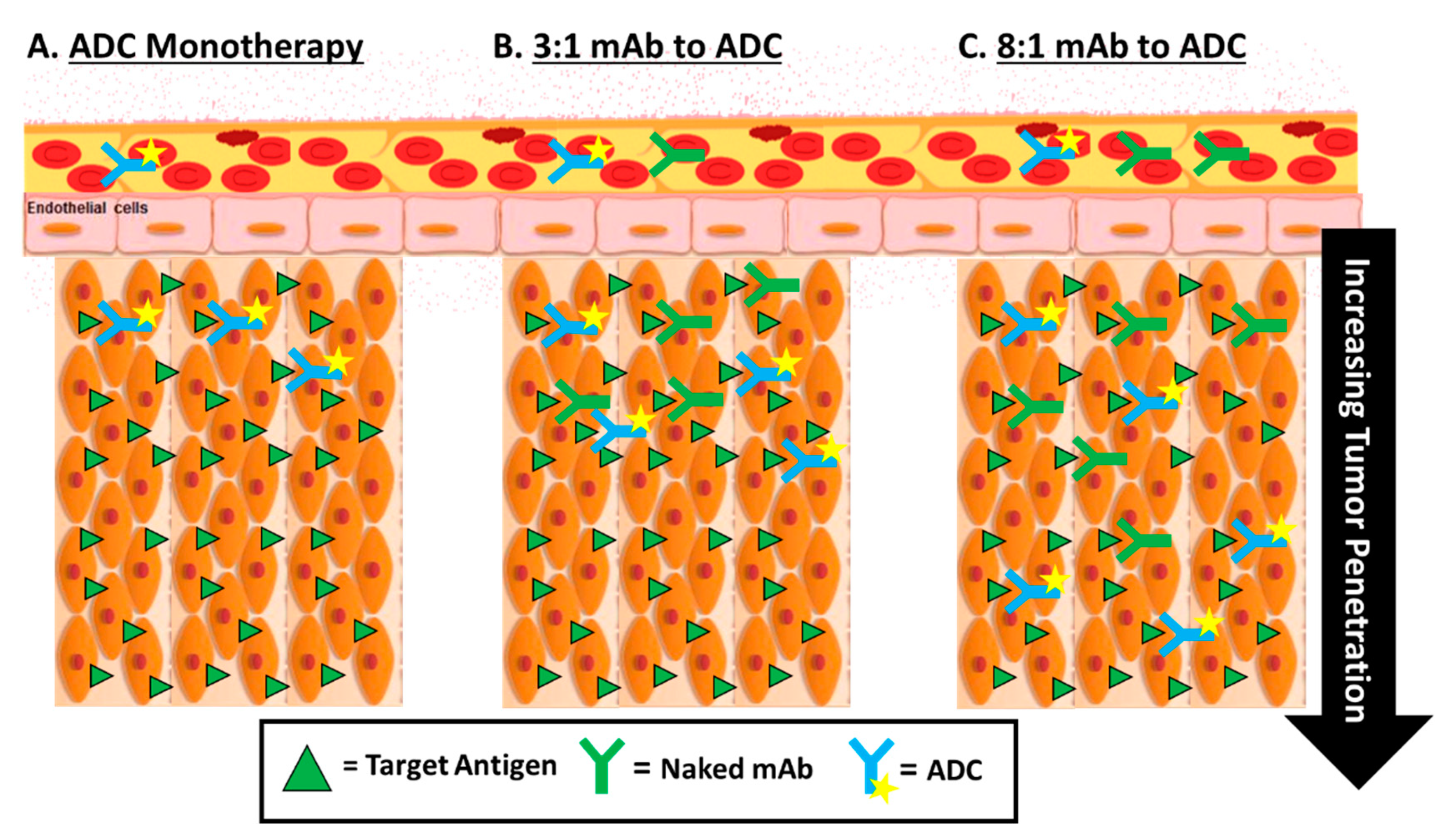
5.2. Optimizing the Delivery of ADCs—Tumor Penetration
6. The Next Generation
6.1. Formulation Strategies
6.1.1. Novel Target Antigens
6.1.2. Novel Antibodies
6.1.3. Novel Payloads
6.2. Therapeutic Strategies
6.2.1. Combination Therapy
6.2.2. Conditional Activation
6.3. Avoiding Resistance
6.4. ADCs as Immune Modulators
6.5. Therapeutic Drug Monitoring
6.6. Optimized Dosing Based on Biomarkers and Precision Medicine Methods for Individual Patients or Selected Patient Groups
7. The Next Generation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lucas, A.T.; Robinson, R.; Schorzman, A.N.; Piscitelli, J.A.; Razo, J.F.; Zamboni, W.C. Pharmacologic Considerations in the Disposition of Antibodies and Antibody-Drug Conjugates in Preclinical Models and in Patients. Antibodies 2019, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Maleki, L.A.; Baradaran, B.; Majidi, J.; Mohammadian, M.; Shahneh, F.Z. Future prospects of monoclonal antibodies as magic bullets in Immunotherapy. Hum. Antib. 2013, 22, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.R.; Canakci, M.; Li, L.; Zhuang, J.; Osborne, B.; Thayumanavan, S. Field Guide to Challenges and Opportunities in Antibody–Drug Conjugates for Chemists. Bioconjug. Chem. 2015, 26, 2198–2215. [Google Scholar] [CrossRef] [Green Version]
- Lucas, A.T.; Price, L.S.L.; Schorzman, A.N.; Storrie, M.; Piscitelli, J.A.; Razo, J.; Zamboni, W.C. Factors Affecting the Pharmacology of Antibody–Drug Conjugates. Antibodies 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hock, M.B.; Thudium, K.E.; Carrasco-Triguero, M.; Schwabe, N.F. Immunogenicity of antibody drug conjugates: Bioanalytical methods and monitoring strategy for a novel therapeutic modality. AAPS J. 2015, 17, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Tourdot, S.; Abdolzade-Bavil, A.; Bessa, J.; Broët, P.; Fogdell-Hahn, A.; Giorgi, M.; Jawa, V.; Kuranda, K.; Legrand, N.; Pattijn, S.; et al. European immunogenicity platform open symposium on immunogenicity of biopharmaceuticals. MAbs 2020, 12, 1725369. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.; Devanarayan, V.; Amaravadi, L.; Barrett, Y.C.; Bowsher, R.; Finco-Kent, D.; Fiscella, M.; Gorovits, B.; Kirschner, S.; Moxness, M.; et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J. Pharm. Biomed. Anal. 2008, 48, 1267–1281. [Google Scholar] [CrossRef]
- Vandivort, T.C.; Horton, D.B.; Johnson, S.B. Regulatory and strategic considerations for addressing immunogenicity and related responses in biopharmaceutical development programs. J. Clin. Transl. Sci. 2020, 4, 547–555. [Google Scholar] [CrossRef]
- Duhazé, J.; Caubet, M.; Hässler, S.; Bachelet, D.; Allez, M.; Deisenhammer, F.; Fogdell-Hahn, A.; Gleizes, A.; Hacein-Bey-Abina, S.; Mariette, X.; et al. Assessing the effect of genetic markers on drug immunogenicity from a mechanistic model-based approach. BMC Med. Res. Method. 2020, 20, 69. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G. Current challenges in assessing immunogenicity. Bioanalysis 2019, 11, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Iida, S.; Yamane-Ohnuki, N.; Kanda, Y.; Kuni-Kamochi, R.; Nakano, R.; Imai-Nishiya, H.; Okazaki, A.; Shinkawa, T.; Natsume, A.; et al. Non-fucosylated therapeutic antibodies: The next generation of therapeutic antibodies. Cytotechnology 2007, 55, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uppal, H.; Doudement, E.; Mahapatra, K.; Darbonne, W.C.; Bumbaca, D.; Shen, B.-Q.; Du, X.; Saad, O.; Bowles, K.; Olsen, S.; et al. Potential Mechanisms for Thrombocytopenia Development with Trastuzumab Emtansine (T-DM1). Clin. Cancer Res. 2015, 21, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiggins, B.; Liu-Shin, L.; Yamaguchi, H.; Ratnaswamy, G. Characterization of Cysteine-Linked Conjugation Profiles of Immunoglobulin G1 and Immunoglobulin G2 Antibody–Drug Conjugates. J. Pharm. Sci. 2015, 104, 1362–1372. [Google Scholar] [CrossRef]
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [Green Version]
- Yoo, E.M.; Wims, L.A.; Chan, L.A.; Morrison, S.L. Human IgG2 Can Form Covalent Dimers. J. Immunol. 2003, 170, 3134–3138. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Jefferis, R. Antibody therapeutics: Isotype and glycoform selection. Expert Opin. Biol. Ther. 2007, 7, 1401–1413. [Google Scholar] [CrossRef]
- Zhang, A.; Fang, J.; Chou, R.Y.-T.; Bondarenko, P.V.; Zhang, Z. Conformational Difference in Human IgG2 Disulfide Isoforms Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry. Biochemistry 2015, 54, 1956–1962. [Google Scholar] [CrossRef]
- McDonagh, C.F.; Kim, K.M.; Turcott, E.; Brown, L.L.; Westendorf, L.; Feist, T.; Sussman, D.; Stone, I.; Anderson, M.; Miyamoto, J.; et al. Engineered anti-CD70 antibody-drug conjugate with increased therapeutic index. Mol. Cancer Ther. 2008, 7, 2913–2923. [Google Scholar] [CrossRef] [Green Version]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar] [PubMed]
- Oflazoglu, E.; Stone, I.J.; Gordon, K.A.; Grewal, I.; Van Rooijen, N.; Law, C.-L.; Gerber, H.-P. Macrophages contribute to the antitumor activity of the anti-CD30 antibody SGN-30. Blood 2007, 110, 4370–4372. [Google Scholar] [CrossRef] [Green Version]
- Vafa, O.; Gilliland, G.L.; Brezski, R.; Strake, B.; Wilkinson, T.; Lacy, E.R.; Scallon, B.; Teplyakov, A.; Malia, T.J.; Strohl, W. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods 2014, 65, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Ling, W.L.; Lua, W.H.; Gan, S.K. Sagacity in antibody humanization for therapeutics, diagnostics and research purposes: Considerations of antibody elements and their roles. Antib. Ther. 2020, 3, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-Drug Conjugates: An Emerging Concept in Cancer Therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Trail, P.A.; Dubowchik, G.M.; Lowinger, T.B. Antibody drug conjugates for treatment of breast cancer: Novel targets and diverse approaches in ADC design. Pharmacol. Ther. 2018, 181, 126–142. [Google Scholar] [CrossRef]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.; Thurston, D.E.; Crescioli, S.; et al. Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs). OncoImmunology 2018, 7, e1395127. [Google Scholar] [CrossRef]
- Szot, C.; Saha, S.; Zhang, X.M.; Zhu, Z.; Hilton, M.B.; Morris, K.; Seaman, S.; Dunleavey, J.; Hsu, K.-S.; Yu, G.-J.; et al. Tumor stroma–targeted antibody-drug conjugate triggers localized anticancer drug release. J. Clin. Investig. 2018, 128, 2927–2943. [Google Scholar] [CrossRef] [Green Version]
- Samsudin, F.; Yeo, J.Y.; Gan, S.K.; Bond, P.J. Not all therapeutic antibody isotypes are equal: The case of IgM versus IgG in Pertuzumab and Trastuzumab. Chem. Sci. 2020, 11, 2843–2854. [Google Scholar] [CrossRef] [Green Version]
- Zolot, R.S.; Basu, S.; Million, R.P. Antibody–drug conjugates. Nat. Rev. Drug Discov. 2013, 12, 259–260. [Google Scholar] [CrossRef]
- Yaghoubi, S.; Karimi, M.H.; Lotfinia, M.; Gharibi, T.; Mahi-Birjand, M.; Kavi, E.; Hosseini, F.; Sepehr, K.S.; Khatami, M.; Bagheri, N.; et al. Potential drugs used in the antibody–drug conjugate (ADC) architecture for cancer therapy. J. Cell. Physiol. 2020, 235, 31–64. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.G.; Kim, K.M. Strategies and Advancement in Antibody-Drug Conjugate Optimization for Targeted Cancer Therapeutics. Biomol. Ther. 2015, 23, 493–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Nakada, T.; Masuda, T.; Naito, H.; Yoshida, M.; Ashida, S.; Morita, K.; Miyazaki, H.; Kasuya, Y.; Ogitani, Y.; Yamaguchi, J.; et al. Novel antibody drug conjugates containing exatecan derivative-based cytotoxic payloads. Bioorg. Med. Chem. Lett. 2016, 26, 1542–1545. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.L.; Fishkin, N.E.; Li, W.; Whiteman, K.R.; Kovtun, Y.; Reid, E.E.; Archer, K.E.; Maloney, E.K.; Audette, C.A.; Mayo, M.F.; et al. A New Class of Antibody–Drug Conjugates with Potent DNA Alkylating Activity. Mol. Cancer Ther. 2016, 15, 1870–1878. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.L.; Shizuka, M.; Wilhelm, A.; Salomon, P.; Reid, E.E.; Lanieri, L.; Sikka, S.; Maloney, E.K.; Harvey, L.; Qiu, Q.; et al. A DNA-Interacting Payload Designed to Eliminate Cross-Linking Improves the Therapeutic Index of Antibody–Drug Conjugates (ADCs). Mol. Cancer Ther. 2018, 17, 650–660. [Google Scholar] [CrossRef] [Green Version]
- Hennessy, E.J. Selective inhibitors of Bcl-2 and Bcl-xL: Balancing antitumor activity with on-target toxicity. Bioorg. Med. Chem. Lett. 2016, 26, 2105–2114. [Google Scholar] [CrossRef]
- Butler, M.S. Remediating Cancer via Splicing Modulation. J. Med. Chem. 2013, 56, 6573–6575. [Google Scholar] [CrossRef]
- Eustáquio, A.S.; Janso, J.E.; Ratnayake, A.S.; O’Donnell, C.J.; Koehn, F.E. Spliceostatin hemiketal biosynthesis in Burkholderia spp. is catalyzed by an iron/α-ketoglutarate-dependent dioxygenase. Proc. Natl. Acad. Sci. USA 2014, 111, E3376–E3385. [Google Scholar] [CrossRef] [Green Version]
- Kaida, D.; Schneider-Poetsch, T.; Yoshida, M. Splicing in oncogenesis and tumor suppression. Cancer Sci. 2012, 103, 1611–1616. [Google Scholar] [CrossRef] [Green Version]
- Dan, N.; Setua, S.; Kashyap, V.K.; Khan, S.; Jaggi, M.; Yallapu, M.; Chauhan, S.C. Antibody-Drug Conjugates for Cancer Therapy: Chemistry to Clinical Implications. Pharmaceuticals 2018, 11, 32. [Google Scholar] [CrossRef] [Green Version]
- Anderl, J.; Faulstich, H.; Hechler, T.; Kulke, M. Antibody–Drug Conjugate Payloads. In Methods in Molecular Biology; Clifton, N.J., Ed.; Springer Science and Business Media: Berlin, Germany, 2013; Volume 1045, pp. 51–70. [Google Scholar]
- Gerber, H.-P.; Koehn, F.E.; Abraham, R.T. The antibody-drug conjugate: An enabling modality for natural product-based cancer therapeutics. Nat. Prod. Rep. 2013, 30, 625. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody–drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCombs, J.R.; Owen, S.C. Antibody Drug Conjugates: Design and Selection of Linker, Payload and Conjugation Chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Capone, E.; Lamolinara, A.; Pastorino, F.; Gentile, R.; Ponziani, S.; Di Vittorio, G.; D’Agostino, D.; Bibbò, S.; Rossi, C.; Piccolo, E.; et al. Targeting Vesicular LGALS3BP by an Antibody-Drug Conjugate as Novel Therapeutic Strategy for Neuroblastoma. Cancers 2020, 12, 2989. [Google Scholar] [CrossRef] [PubMed]
- Casi, G.; Neri, D. Antibody–drug conjugates: Basic concepts, examples and future perspectives. J. Control. Release 2012, 161, 422–428. [Google Scholar] [CrossRef]
- Gébleux, R.; Wulhfard, S.; Casi, G.; Neri, D. Antibody Format and Drug Release Rate Determine the Therapeutic Activity of Noninternalizing Antibody–Drug Conjugates. Mol. Cancer Ther. 2015, 14, 2606–2612. [Google Scholar] [CrossRef] [Green Version]
- Catcott, K.C.; McShea, M.A.; Bialucha, C.U.; Miller, K.L.; Hicks, S.W.; Saxena, P.; Gesner, T.G.; Woldegiorgis, M.; Lewis, M.E.; Bai, C.; et al. Microscale screening of antibody libraries as maytansinoid antibody-drug conjugates. mAbs 2016, 8, 513–523. [Google Scholar] [CrossRef] [Green Version]
- King, H.D.; Dubowchik, G.M.; Mastalerz, H.; Willner, D.; Hofstead, S.J.; Firestone, R.A.; Lasch, S.J.; Trail, P.A. Monoclonal Antibody Conjugates of Doxorubicin Prepared with Branched Peptide Linkers: Inhibition of Aggregation by Methoxytriethyleneglycol Chains. J. Med. Chem. 2002, 45, 4336–4343. [Google Scholar] [CrossRef]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of Drug–Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody–Maytansinoid Conjugates. Bioconjug. Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—an emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef]
- Ponziani, S.; Di Vittorio, G.; Pitari, G.; Cimini, A.M.; Ardini, M.; Gentile, R.; Iacobelli, S.; Sala, G.; Capone, E.; Flavell, D.J.; et al. Antibody-Drug Conjugates: The New Frontier of Chemotherapy. Int. J. Mol. Sci. 2020, 21, 5510. [Google Scholar] [CrossRef] [PubMed]
- Yoder, N.C.; Bai, C.; Tavares, D.; Widdison, W.C.; Whiteman, K.R.; Wilhelm, A.; Wilhelm, S.D.; McShea, M.A.; Maloney, E.K.; Ab, O.; et al. A Case Study Comparing Heterogeneous Lysine- and Site-Specific Cysteine-Conjugated Maytansinoid Antibody-Drug Conjugates (ADCs) Illustrates the Benefits of Lysine Conjugation. Mol. Pharm. 2019, 16, 3926–3937. [Google Scholar] [CrossRef]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-selective modification strategies in antibody-drug conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef] [PubMed]
- Sadiki, A.; Vaidya, S.R.; Abdollahi, M.; Bhardwaj, G.; Dolan, M.E.; Turna, H.; Arora, V.; Sanjeev, A.; Robinson, T.D.; Koid, A.; et al. Site-specific conjugation of native antibody. Antib. Ther. 2020, 3, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Ito, Y. Recent Chemical Approaches for Site-Specific Conjugation of Native Antibodies: Technologies toward Next-Generation Antibody–Drug Conjugates. ChemBioChem 2019, 20, 2729–2737. [Google Scholar] [CrossRef]
- Burke, P.J.; Hamilton, J.Z.; Jeffrey, S.C.; Hunter, J.H.; Doronina, S.O.; Okeley, N.M.; Miyamoto, J.B.; Anderson, M.E.; Stone, I.J.; Ulrich, M.L.; et al. Optimization of a PEGylated Glucuronide-Monomethylauristatin E Linker for Antibody–Drug Conjugates. Mol. Cancer Ther. 2016, 16, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caliceti, P.; Veronese, F.M. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)-protein conjugates. Adv. Drug Deliv. Rev. 2003, 55, 1261–1277. [Google Scholar] [CrossRef]
- Gefen, T.; Vaya, J.; Khatib, S.; Harkevich, N.; Artoul, F.; Heller, E.D.; Pitcovski, J.; Aizenshtein, E. The impact of PEGylation on protein immunogenicity. Int. Immunopharmacol. 2013, 15, 254–259. [Google Scholar] [CrossRef]
- Veronese, F.M.; Mero, A. The Impact of PEGylation on Biological Therapies. BioDrugs 2008, 22, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Goetze, A.M.; Liu, Y.D.; Zhang, Z.; Shah, B.; Lee, E.; Bondarenko, P.V.; Flynn, G.C. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiol. 2011, 21, 949–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata-Koyama, M.; Iida, S.; Misaka, H.; Mori, K.; Yano, K.; Shitara, K.; Satoh, M. Nonfucosylated rituximab potentiates human neutrophil phagocytosis through its high binding for FcgammaRIIIb and MHC class II expression on the phagocytotic neutrophils. Exp. Hematol. 2009, 37, 309–321. [Google Scholar] [CrossRef]
- Tibbitts, J.; Canter, D.; Graff, R.; Smith, A.M.; Khawli, L.A. Key factors influencing ADME properties of therapeutic proteins: A need for ADME characterization in drug discovery and development. mAbs 2016, 8, 229–245. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Stefano, J.E.; Manning, C.; Kyazike, J.; Chen, B.; Gianolio, D.A.; Park, A.; Busch, M.; Bird, J.; Zheng, X.; et al. Site-Specific Antibody–Drug Conjugation through Glycoengineering. Bioconjug. Chem. 2014, 25, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Stork, R.; Zettlitz, K.A.; Müller, D.; Rether, M.; Hanisch, F.-G.; Kontermann, R.E. N-Glycosylation as Novel Strategy to Improve Pharmacokinetic Properties of Bispecific Single-chain Diabodies. J. Biol. Chem. 2008, 283, 7804–7812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.; Morrison, S.L. Effect of altered CH2-associated carbohydrate structure on the functional properties and in vivo fate of chimeric mouse-human immunoglobulin G1. J. Exp. Med. 1994, 180, 1087–1096. [Google Scholar] [CrossRef]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and Anticancer Drug Conjugate Assemblies: The Role of the Linkage between Components. Toxins 2011, 3, 848–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.M.; Morris, C.Q. Antibody–Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Adv. Ther. 2017, 34, 1015–1035. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W. The Evolution of Antibody-Drug Conjugates: A Positive Inflexion Point. In American Society of Clinical Oncology Educational Book, Proceedings of the American Society of Clinical Oncology Annual Meeting, Chicago, IL, USA, 29 May–2 June 2020; American Society of Clinical Oncology: Alexandria, VA, USA, 2020; Volume 40, pp. 127–134. [Google Scholar] [CrossRef]
- Tolcher, A.W. Antibody drug conjugates: Lessons from 20 years of clinical experience. Ann. Oncol. 2016, 27, 2168–2172. [Google Scholar] [CrossRef]
- Burris, H.A., III; Rugo, H.S.; Vukelja, S.J.; Vogel, C.L.; Borson, R.A.; Limentani, S.; Tan-Chiu, E.; Krop, I.E.; Michaelson, R.A.; Girish, S.; et al. Phase II Study of the Antibody Drug Conjugate Trastuzumab-DM1 for the Treatment of Human Epidermal Growth Factor Receptor 2 (HER2) –Positive Breast Cancer After Prior HER2-Directed Therapy. J. Clin. Oncol. 2011, 29, 398–405. [Google Scholar] [CrossRef]
- Sharma, S.; Li, Z.; Bussing, D.; Shah, D.K. Evaluation of Quantitative Relationship Between Target Expression and Antibody-Drug Conjugate Exposure Inside Cancer Cells. Drug Metab. Dispos. 2020, 48, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Phillips, G.D.L.; Verma, S.; Ro, J.; Huober, J.; Guardino, A.E.; Samant, M.K.; Olsen, S.; De Haas, S.L.; Pegram, M.D. Relationship between Tumor Biomarkers and Efficacy in EMILIA, a Phase III Study of Trastuzumab Emtansine in HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2016, 22, 3755–3763. [Google Scholar] [CrossRef] [Green Version]
- Lapusan, S.; Vidriales, M.-B.; Thomas, X.; DE Botton, S.; Vekhoff, A.; Tang, R.; Dumontet, C.; Morariu-Zamfir, R.; Lambert, J.M.; Ozoux, M.-L.; et al. Phase I studies of AVE9633, an anti-CD33 antibody-maytansinoid conjugate, in adult patients with relapsed/refractory acute myeloid leukemia. Investig. New Drugs 2011, 30, 1121–1131. [Google Scholar] [CrossRef]
- Polson, A.G.; Williams, M.; Gray, A.M.; Fuji, R.N.; A Poon, K.; McBride, J.; Raab, H.; Januario, T.; Go, M.; Lau, J.; et al. Anti-CD22-MCC-DM1: An antibody-drug conjugate with a stable linker for the treatment of non-Hodgkin’s lymphoma. Leukemia 2010, 24, 1566–1573. [Google Scholar] [CrossRef] [Green Version]
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A.; Robert, F., III; et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017, 18, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Morgensztern, D.; Besse, B.; Greillier, L.; Santana-Davila, R.; Ready, N.; Hann, C.L.; Glisson, B.S.; Farago, A.F.; Dowlati, A.; Rudin, C.M.; et al. Efficacy and Safety of Rovalpituzumab Tesirine in Third-Line and Beyond Patients with DLL3-Expressing, Relapsed/Refractory Small-Cell Lung Cancer: Results from the Phase II TRINITY Study. Clin. Cancer Res. 2019, 25, 6958–6966. [Google Scholar] [CrossRef] [Green Version]
- Puca, L.; Sailor, V.; Gavyert, K.; Dardenne, E.; Isse, K.; Sigouros, M.; Nanus, D.M.; Tagawa, S.T.; Mosquera, J.M.; Saunders, L.; et al. Abstract 1947: Rovalpituzumab tesirine as a therapeutic agent for neuroendocrine prostate cancer. Exp. Mol. Ther. 2018, 78 (Suppl. 13), 1947. [Google Scholar] [CrossRef] [Green Version]
- Columbus, G. Phase III Trial Evaluating Rova-T in Small Cell Lung Cancer Put on Hold. Targeted Oncology. 2018. Available online: https://www.targetedonc.com/news/phase-iii-trialevaluating-rovat-in-small-cell-lung-cancer-put-on-hold (accessed on 10 January 2021).
- Martin, L.P.; Konner, J.A.; Moore, K.N.; Seward, S.M.; Matulonis, U.A.; Perez, R.P.; Su, Y.; Berkenblit, A.; Ruiz-Soto, R.; Birrer, M.J. Characterization of folate receptor alpha (FRα) expression in archival tumor and biopsy samples from relapsed epithelial ovarian cancer patients: A phase I expansion study of the FRα-targeting antibody-drug conjugate mirvetuximab soravtansine. Gynecol. Oncol. 2017, 147, 402–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.N.; Borghaei, H.; O’Malley, D.M.; Jeong, W.; Seward, S.M.; Bauer, T.M.; Perez, R.P.; Matulonis, U.A.; Running, K.L.; Zhang, X.; et al. Phase 1 dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody-drug conjugate, in patients with solid tumors. Cancer 2017, 123, 3080–3087. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Martin, L.P.; O’Malley, D.M.; Matulonis, U.A.; Konner, J.A.; Perez, R.; Bauer, T.M.; Ruiz-Soto, R.; Birrer, M.J. Safety and Activity of Mirvetuximab Soravtansine (IMGN853), a Folate Receptor Alpha–Targeting Antibody–Drug Conjugate, in Platinum-Resistant Ovarian, Fallopian Tube, or Primary Peritoneal Cancer: A Phase I Expansion Study. J. Clin. Oncol. 2017, 35, 1112–1118. [Google Scholar] [CrossRef]
- Tijink, B.M.; Buter, J.; De Bree, R.; Giaccone, G.; Lang, M.S.; Staab, A.; Leemans, C.R.; Van Dongen, G.A. A Phase I Dose Escalation Study with Anti-CD44v6 Bivatuzumab Mertansine in Patients with Incurable Squamous Cell Carcinoma of the Head and Neck or Esophagus. Clin. Cancer Res. 2006, 12, 6064–6072. [Google Scholar] [CrossRef] [Green Version]
- Solal-Céligny, P. Safety of rituximab maintenance therapy in follicular lymphomas. Leuk. Res. 2006, 30 (Suppl. 1), S16–S21. [Google Scholar] [CrossRef]
- Xiao, G.; Gan, L.-S. Receptor-Mediated Endocytosis and Brain Delivery of Therapeutic Biologics. Int. J. Cell Biol. 2013, 2013, 703545. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S.; Mg, S.; Mayor, S. Endocytosis unplugged: Multiple ways to enter the cell. Cell Res. 2010, 20, 256–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulcahy, L.; Pink, R.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Vercauteren, D.; Vandenbroucke, R.E.; Jones, A.T.; Rejman, J.; Demeester, J.; De Smedt, S.C.; Sanders, N.N.; Braeckmans, K. The Use of Inhibitors to Study Endocytic Pathways of Gene Carriers: Optimization and Pitfalls. Mol. Ther. 2010, 18, 561–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtun, Y.V.; Goldmacher, V.S. Cell killing by antibody–drug conjugates. Cancer Lett. 2007, 255, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Schrama, D.; Reisfeld, R.A.; Becker, J.C. Antibody targeted drugs as cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 147–159. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [Green Version]
- Popova, N.V.; Deyev, I.E.; Petrenko, A.G. Clathrin-Mediated Endocytosis and Adaptor Proteins. Acta Nat. 2013, 5, 62–73. [Google Scholar] [CrossRef]
- Adler, M.; Mayo, A.; Zhou, X.; Franklin, R.A.; Jacox, J.B.; Medzhitov, R.; Alon, U. Endocytosis as a stabilizing mechanism for tissue homeostasis. Proc. Natl. Acad. Sci. USA 2018, 115, E1926–E1935. [Google Scholar] [CrossRef] [Green Version]
- Kraynov, E.; Kamath, A.V.; Walles, M.; Tarcsa, E.; Deslandes, A.; A Iyer, R.; Datta-Mannan, A.; Sriraman, P.; Bairlein, M.; Yang, J.J.; et al. Current Approaches for Absorption, Distribution, Metabolism, and Excretion Characterization of Antibody-Drug Conjugates: An Industry White Paper. Drug Metab. Dispos. 2015, 44, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Iznaga-Escobar, N.; Mishra, A.; Pérez-Rodriguez, R. Factors affecting pharmacokinetics of monoclonal antibodies: A review article. Methods Find. Exp. Clin. Pharmacol. 2004, 26, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Sialic acids in human health and disease. Trends Mol. Med. 2008, 14, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Wiig, H.; Gyenge, C.; Iversen, P.O.; Gullberg, D.; Tenstad, O. The Role of the Extracellular Matrix in Tissue Distribution of Macromolecules in Normal and Pathological Tissues: Potential Therapeutic Consequences. Microcirculation 2008, 15, 283–296. [Google Scholar] [CrossRef]
- Boswell, C.; Tesar, D.B.; Mukhyala, K.; Theil, F.-P.; Fielder, P.J.; Khawli, L.A. Effects of Charge on Antibody Tissue Distribution and Pharmacokinetics. Bioconjug. Chem. 2010, 21, 2153–2163. [Google Scholar] [CrossRef]
- Drake, P.M.; Rabuka, D. Recent Developments in ADC Technology: Preclinical Studies Signal Future Clinical Trends. BioDrugs 2017, 31, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Lyon, R.P.; Bovee, T.D.; O Doronina, S.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; E Anderson, M.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Loganzo, F.; Tan, X.; Sung, M.; Jin, G.; Myers, J.S.; Melamud, E.; Wang, F.; Diesl, V.; Follettie, M.T.; Musto, S.; et al. Tumor Cells Chronically Treated with a Trastuzumab–Maytansinoid Antibody–Drug Conjugate Develop Varied Resistance Mechanisms but Respond to Alternate Treatments. Mol. Cancer Ther. 2015, 14, 952–963. [Google Scholar] [CrossRef] [Green Version]
- Al-Rohil, R.N.; A Torres-Cabala, C.; Patel, A.; Tetzlaff, M.T.; Ivan, D.; Nagarajan, P.; Curry, J.L.; Miranda, R.N.; Duvic, M.; Prieto, V.G.; et al. Loss of CD30 expression after treatment with brentuximab vedotin in a patient with anaplastic large cell lymphoma: A novel finding. J. Cutan. Pathol. 2016, 43, 1161–1166. [Google Scholar] [CrossRef]
- Van Der Velden, V.H.J.; Boeckx, N.; Jedema, I.; Marvelde, J.G.T.; Hoogeveen, P.G.; Boogaerts, M.; Van Dongen, J. High CD33-antigen loads in peripheral blood limit the efficacy of gemtuzumab ozogamicin (Mylotarg®) treatment in acute myeloid leukemia patients. Leukemia 2004, 18, 983–988. [Google Scholar] [CrossRef] [Green Version]
- Phillips, G.D.L.; Fields, C.T.; Li, G.; Dowbenko, D.; Schaefer, G.; Miller, K.; Andre, F.; Burris, H.A., III; Albain, K.S.; Harbeck, N.; et al. Dual Targeting of HER2-Positive Cancer with Trastuzumab Emtansine and Pertuzumab: Critical Role for Neuregulin Blockade in Antitumor Response to Combination Therapy. Clin. Cancer Res. 2014, 20, 456–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Guo, J.; Shen, B.-Q.; Yadav, D.B.; Sliwkowski, M.X.; Crocker, L.M.; Lacap, J.A.; Phillips, G.D.L. Mechanisms of Acquired Resistance to Trastuzumab Emtansine in Breast Cancer Cells. Mol. Cancer Ther. 2018, 17, 1441–1453. [Google Scholar] [CrossRef] [Green Version]
- Luci, C.R.; García-Alonso, S.; Díaz-Rodriguez, E.; Nadal-Serrano, M.; Arribas, J.; Ocana, A.; Pandiella, A. Resistance to the Antibody–Drug Conjugate T-DM1 Is Based in a Reduction in Lysosomal Proteolytic Activity. Cancer Res. 2017, 77, 4639–4651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamblett, K.J.; Jacob, A.P.; Gurgel, J.L.; Tometsko, M.E.; Rock, B.M.; Patel, S.K.; Milburn, R.R.; Siu, S.; Ragan, S.P.; Rock, D.A.; et al. SLC46A3 Is Required to Transport Catabolites of Noncleavable Antibody Maytansine Conjugates from the Lysosome to the Cytoplasm. Cancer Res. 2015, 75, 5329–5340. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Ocana, A.; Tannock, I.F. Reversal of ATP-binding cassette drug transporter activity to modulate chemoresistance: Why has it failed to provide clinical benefit? Cancer Metastasis Rev. 2013, 32, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.; Stover, D. Using the Lessons Learned From the Clinic to Improve the Preclinical Development of Antibody Drug Conjugates. Pharm. Res. 2015, 32, 3458–3469. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Jimi, S.; Hara, S.; Takamatsu, Y.; Suzumiya, J.; Tamura, K. Importance of inducible multidrug resistance 1 expression in HL-60 cells resistant to gemtuzumab ozogamicin. Leuk. Lymphoma 2012, 53, 1399–1405. [Google Scholar] [CrossRef]
- Walter, R.B.; Gooley, T.A.; van der Velden, V.H.; Loken, M.R.; van Dongen, J.J.; Flowers, D.A.; Bernstein, I.D.; Appelbaum, F.R. CD33 expression and P-glycoprotein-mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood 2007, 109, 4168–4170. [Google Scholar] [CrossRef]
- Takeshita, A.; Shinjo, K.; Yamakage, N.; Ono, T.; Hirano, I.; Matsui, H.; Shigeno, K.; Nakamura, S.; Tobita, T.; Maekawa, M.; et al. CMC-544 (inotuzumab ozogamicin) shows less effect on multidrug resistant cells: Analyses in cell lines and cells from patients with B-cell chronic lymphocytic leukaemia and lymphoma. Br. J. Haematol. 2009, 146, 34–43. [Google Scholar] [CrossRef]
- Chan, K.R.; Ong, E.Z.; Mok, D.Z.; Ooi, E.E. Fc receptors and their influence on efficacy of therapeutic antibodies for treatment of viral diseases. Expert Rev. Anti-Infect. Ther. 2015, 13, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.W.; Wang, Y.; Wei, Y.H.; Zhao, P.P.; Wang, X.B.; Rong, J.J.; Zhong, W.Y.; Zhang, X.W.; Wang, L.; Zheng, H.F. Comprehensive Assessment of the Association between FCGRs polymorphisms and the risk of systemic lupus erythematosus: Evidence from a Meta-Analysis. Sci. Rep. 2016, 6, 31617. [Google Scholar] [CrossRef] [Green Version]
- Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Bruhns, P.; Saeys, Y.; Hammad, H.; Lambrecht, B.N. The function of Fcγ receptors in dendritic cells and macrophages. Nat. Rev. Immunol. 2014, 14, 94–108. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Sockolosky, J.T.; Szoka, F.C. The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Adv. Drug Deliv. Rev. 2015, 91, 109–124. [Google Scholar] [CrossRef] [Green Version]
- Gurbaxani, B.; Dela Cruz, L.L.; Chintalacharuvu, K.; Morrison, S.L. Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Mol. Immunol. 2006, 43, 1462–1473. [Google Scholar] [CrossRef]
- Dall’Acqua, W.F.; Woods, R.M.; Ward, E.S.; Palaszynski, S.R.; Patel, N.K.; Brewah, Y.A.; Wu, H.; Kiener, P.A.; Langermann, S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: Biological consequences. J. Immunol. 2002, 169, 5171–5180. [Google Scholar] [CrossRef] [Green Version]
- Datta-Mannan, A.; Witcher, D.R.; Tang, Y.; Watkins, J.; Jiang, W.; Wroblewski, V.J. Humanized IgG1 variants with differential binding properties to the neonatal Fc receptor: Relationship to pharmacokinetics in mice and primates. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Ward, E.S.; Devanaboyina, S.C.; Ober, R.J. Targeting FcRn for the modulation of antibody dynamics. Mol. Immunol. 2015, 67, 131–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, T.T.; Aveson, V.G. Neonatal Fc receptor and IgG-based therapeutics. MAbs 2011, 3, 422–430. [Google Scholar] [CrossRef]
- Su, C.T.; Lua, W.H.; Ling, W.L.; Gan, S.K. Allosteric Effects between the Antibody Constant and Variable Regions: A Study of IgA Fc Mutations on Antigen Binding. Antibodies 2018, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmeister, K.M.; Falet, H. Platelet clearance by the hepatic Ashwell-Morrell receptor: Mechanisms and biological significance. Thromb. Res. 2016, 141 (Suppl. 2), S68–S72. [Google Scholar] [CrossRef] [Green Version]
- Rowley, J.W.; Schwertz, H.; Weyrich, A.S. Platelet mRNA: The meaning behind the message. Curr. Opin. Hematol. 2012, 19, 385–391. [Google Scholar] [CrossRef]
- Godwin, C.D.; McDonald, G.B.; Walter, R.B. Sinusoidal obstruction syndrome following CD33-targeted therapy in acute myeloid leukemia. Blood 2017, 129, 2330–2332. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.M. Incorporation of FcRn-mediated disposition model to describe the population pharmacokinetics of therapeutic monoclonal IgG antibody in clinical patients. Biopharm. Drug Dispos. 2016, 37, 107–119. [Google Scholar] [CrossRef]
- Ternant, D.; Arnoult, C.; Pugnière, M.; Dhommée, C.; Drocourt, D.; Perouzel, E.; Passot, C.; Baroukh, N.; Mulleman, D.; Tiraby, G.; et al. IgG1 Allotypes Influence the Pharmacokinetics of Therapeutic Monoclonal Antibodies through FcRn Binding. J. Immunol. 2016, 196, 607–613. [Google Scholar] [CrossRef]
- Bruno, R.; Washington, C.B.; Lu, J.F.; Lieberman, G.; Banken, L.; Klein, P. Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother. Pharmacol. 2005, 56, 361–369. [Google Scholar] [CrossRef]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 2008, 3, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Sadauskas, E.; Wallin, H.; Stoltenberg, M.; Vogel, U.; Doering, P.; Larsen, A.; Danscher, G. Kupffer cells are central in the removal of nanoparticles from the organism. Part. Fibre Toxicol. 2007, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, K.M.; MacParland, S.A.; Ma, X.Z.; Spetzler, V.N.; Echeverri, J.; Ouyang, B.; Fadel, S.M.; Sykes, E.A.; Goldaracena, N.; Kaths, J.M.; et al. Mechanism of hard-nanomaterial clearance by the liver. Nat. Mater. 2016, 15, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.X.; Cao, H.; Xing, C.G.; Wei, S.H.; Jiang, G.Q.; Liu, Z.L. Visualization and body distribution of [¹³¹I]-herceptin in nude mice with BT-474 breast carcinoma. Genet. Mol. Res. 2014, 13, 6804–6812. [Google Scholar] [CrossRef] [PubMed]
- Abuqayyas, L.; Zhang, X.; Balthasar, J.P. Application of knockout mouse models to investigate the influence of FcγR on the pharmacokinetics and anti-platelet effects of MWReg30, a monoclonal anti-GPIIb antibody. Int. J. Pharm. 2013, 444, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Colbern, G.T.; Hiller, A.J.; Musterer, R.S.; Working, P.K.; Henderson, I.C. Antitumor activity of Herceptin in combination with STEALTH liposomal cisplatin or nonliposomal cisplatin in a HER2 positive human breast cancer model. J. Inorg. Biochem. 1999, 77, 117–120. [Google Scholar] [CrossRef]
- Yamashita-Kashima, Y.; Iijima, S.; Yorozu, K.; Furugaki, K.; Kurasawa, M.; Ohta, M.; Fujimoto-Ouchi, K. Pertuzumab in combination with trastuzumab shows significantly enhanced antitumor activity in HER2-positive human gastric cancer xenograft models. Clin. Cancer Res. 2011, 17, 5060–5070. [Google Scholar] [CrossRef] [Green Version]
- Caron, W.P.; Lay, J.C.; Fong, A.M.; La-Beck, N.M.; Kumar, P.; Newman, S.E.; Zhou, H.; Monaco, J.H.; Clarke-Pearson, D.L.; Brewster, W.R.; et al. Translational studies of phenotypic probes for the mononuclear phagocyte system and liposomal pharmacology. J. Pharmacol. Exp. Ther. 2013, 347, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Starling, B.R.; Kumar, P.; Lucas, A.T.; Barrow, D.; Farnan, L.; Hendrix, L.; Giovinazzo, H.; Song, G.; Gehrig, P.; Bensen, J.T.; et al. Mononuclear phagocyte system function and nanoparticle pharmacology in obese and normal weight ovarian and endometrial cancer patients. Cancer Chemother. Pharmacol. 2019, 83, 61–70. [Google Scholar] [CrossRef]
- Kirschbrown, W.P.; Lucas, A.T.; Li, C.; Girish, S.; Zamboni, W.C.; Garg, A. (Eds.) Biomarkers of Fc-gamma receptors (FcɣRs) on mononuclear phagocyte system (MPS) cells in blood of patients with advanced gastric cancer are upregulated as compared to patients with metastatic breast cancer. In Proceedings of the 30th EORTC-NCI-AACR Symposium, Dublin, Ireland, 13–16 December 2018. [Google Scholar]
- Rozman, S.; Grabnar, I.; Novaković, S.; Mrhar, A.; Jezeršek Novaković, B. Population pharmacokinetics of rituximab in patients with diffuse large B-cell lymphoma and association with clinical outcome. Br. J. Clin. Pharmacol. 2017, 83, 1782–1790. [Google Scholar] [CrossRef] [Green Version]
- Salar, A.; Avivi, I.; Bittner, B.; Bouabdallah, R.; Brewster, M.; Catalani, O.; Follows, G.; Haynes, A.; Hourcade-Potelleret, F.; Janikova, A.; et al. Comparison of subcutaneous versus intravenous administration of rituximab as maintenance treatment for follicular lymphoma: Results from a two-stage, phase IB study. J. Clin. Oncol. 2014, 32, 1782–1791. [Google Scholar] [CrossRef]
- Berinstein, N.L.; Grillo-López, A.J.; White, C.A.; Bence-Bruckler, I.; Maloney, D.; Czuczman, M.; Green, D.; Rosenberg, J.; McLaughlin, P.; Shen, D. Association of serum Rituximab (IDEC-C2B8) concentration and anti-tumor response in the treatment of recurrent low-grade or follicular non-Hodgkin’s lymphoma. Ann. Oncol. 1998, 9, 995–1001. [Google Scholar] [CrossRef]
- Krop, I.E.; Beeram, M.; Modi, S.; Jones, S.F.; Holden, S.N.; Yu, W.; Girish, S.; Tibbitts, J.; Yi, J.H.; Sliwkowski, M.X.; et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J. Clin. Oncol. 2010, 28, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M.X. Trastuzumab emtansine: A unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin. Cancer Res. 2011, 17, 6437–6447. [Google Scholar] [CrossRef] [Green Version]
- Advani, A.; Coiffier, B.; Czuczman, M.S.; Dreyling, M.; Foran, J.; Gine, E.; Gisselbrecht, C.; Ketterer, N.; Nasta, S.; Rohatiner, A.; et al. Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-Hodgkin’s lymphoma: Results of a phase I study. J. Clin. Oncol. 2010, 28, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Fayad, L.; Offner, F.; Smith, M.R.; Verhoef, G.; Johnson, P.; Kaufman, J.L.; Rohatiner, A.; Advani, A.; Foran, J.; Hess, G.; et al. Safety and clinical activity of a combination therapy comprising two antibody-based targeting agents for the treatment of non-Hodgkin lymphoma: Results of a phase I/II study evaluating the immunoconjugate inotuzumab ozogamicin with rituximab. J. Clin. Oncol. 2013, 31, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Thomas, D.; Jorgensen, J.; Kebriaei, P.; Jabbour, E.; Rytting, M.; York, S.; Ravandi, F.; Garris, R.; Kwari, M.; et al. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer 2013, 119, 2728–2736. [Google Scholar] [CrossRef]
- Boesch, A.W.; Brown, E.P.; Cheng, H.D.; Ofori, M.O.; Normandin, E.; Nigrovic, P.A.; Alter, G.; Ackerman, M.E. Highly parallel characterization of IgG Fc binding interactions. MAbs 2014, 6, 915–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorovits, B.; Krinos-Fiorotti, C. Proposed mechanism of off-target toxicity for antibody-drug conjugates driven by mannose receptor uptake. Cancer Immunol. Immunother. 2013, 62, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Chiffoleau, E. C-Type Lectin-Like Receptors as Emerging Orchestrators of Sterile Inflammation Represent Potential Therapeutic Targets. Front. Immunol. 2018, 9, 227. [Google Scholar] [CrossRef] [Green Version]
- Masters, J.C.; Nickens, D.J.; Xuan, D.; Shazer, R.L.; Amantea, M. Clinical toxicity of antibody drug conjugates: A meta-analysis of payloads. Investig. New Drugs 2018, 36, 121–135. [Google Scholar] [CrossRef]
- Guffroy, M.; Falahatpisheh, H.; Biddle, K.; Kreeger, J.; Obert, L.; Walters, K.; Goldstein, R.; Boucher, G.; Coskran, T.; Reagan, W.; et al. Liver Microvascular Injury and Thrombocytopenia of Antibody-Calicheamicin Conjugates in Cynomolgus Monkeys-Mechanism and Monitoring. Clin. Cancer Res. 2017, 23, 1760–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolcher, A.W.; Ochoa, L.; Hammond, L.A.; Patnaik, A.; Edwards, T.; Takimoto, C.; Smith, L.; de Bono, J.; Schwartz, G.; Mays, T.; et al. Cantuzumab mertansine, a maytansinoid immunoconjugate directed to the CanAg antigen: A phase I, pharmacokinetic, and biologic correlative study. J. Clin. Oncol. 2003, 21, 211–222. [Google Scholar] [CrossRef]
- Stahl, P.; Schlesinger, P.H.; Sigardson, E.; Rodman, J.S.; Lee, Y.C. Receptor-mediated pinocytosis of mannose glycoconjugates by macrophages: Characterization and evidence for receptor recycling. Cell 1980, 19, 207–215. [Google Scholar] [CrossRef]
- Wang, Q.; Zhao, G.; Lin, J.; Li, C.; Jiang, N.; Xu, Q.; Wang, Q.; Zhang, J. Role of the Mannose Receptor During Aspergillus fumigatus Infection and Interaction With Dectin-1 in Corneal Epithelial Cells. Cornea 2016, 35, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Todoroki, K.; Mizuno, H.; Sugiyama, E.; Toyo’oka, T. Bioanalytical methods for therapeutic monoclonal antibodies and antibody-drug conjugates: A review of recent advances and future perspectives. J. Pharm. Biomed. Anal. 2020, 179, 112991. [Google Scholar] [CrossRef]
- Xu, K.; Liu, L.; Saad, O.M.; Baudys, J.; Williams, L.; Leipold, D.; Shen, B.; Raab, H.; Junutula, J.R.; Kim, A.; et al. Characterization of intact antibody-drug conjugates from plasma/serum in vivo by affinity capture capillary liquid chromatography-mass spectrometry. Anal. Biochem. 2011, 412, 56–66. [Google Scholar] [CrossRef]
- He, J.; Yu, S.F.; Yee, S.; Kaur, S.; Xu, K. Characterization of in vivo biotransformations for trastuzumab emtansine by high-resolution accurate-mass mass spectrometry. MAbs 2018, 10, 960–967. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Alba, O.; Houel, S.; Hessmann, S.; Erb, S.; Rabuka, D.; Huguet, R.; Josephs, J.; Beck, A.; Drake, P.M.; Cianférani, S. A Case Study to Identify the Drug Conjugation Site of a Site-Specific Antibody-Drug-Conjugate Using Middle-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 2419–2429. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Huo, S.; Xue, C.; An, B.; Qu, J. Current LC-MS-based strategies for characterization and quantification of antibody-drug conjugates. J. Pharm. Anal. 2020, 10, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Hyung, S.J.; Li, D.; Koppada, N.; Kaur, S.; Saad, O.M. Method development of a novel PK assay for antibody-conjugated drug measurement of ADCs using peptide-linker drug analyte. Anal. Bioanal. Chem. 2019, 411, 2587–2596. [Google Scholar] [CrossRef] [PubMed]
- Ileana Dumbrava, E.; Meric-Bernstam, F.; Yap, T.A. Challenges with biomarkers in cancer drug discovery and development. Expert Opin. Drug Discov. 2018, 13, 685–690. [Google Scholar] [CrossRef]
- Dornan, D.; Bennett, F.; Chen, Y.; Dennis, M.; Eaton, D.; Elkins, K.; French, D.; Go, M.A.; Jack, A.; Junutula, J.R.; et al. Therapeutic potential of an anti-CD79b antibody-drug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma. Blood 2009, 114, 2721–2729. [Google Scholar] [CrossRef]
- Morschhauser, F.; Flinn, I.W.; Advani, R.; Sehn, L.H.; Diefenbach, C.; Kolibaba, K.; Press, O.W.; Salles, G.; Tilly, H.; Chen, A.I.; et al. Polatuzumab vedotin or pinatuzumab vedotin plus rituximab in patients with relapsed or refractory non-Hodgkin lymphoma: Final results from a phase 2 randomised study (ROMULUS). Lancet Haematol. 2019, 6, e254–e265. [Google Scholar] [CrossRef]
- Modi, S.; Park, H.; Murthy, R.K.; Iwata, H.; Tamura, K.; Tsurutani, J.; Moreno-Aspitia, A.; Doi, T.; Sagara, Y.; Redfern, C.; et al. Antitumor Activity and Safety of Trastuzumab Deruxtecan in Patients with HER2-Low-Expressing Advanced Breast Cancer: Results from a Phase Ib Study. J. Clin. Oncol. 2020, 38, 1887–1896. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Epenetos, A.A.; Snook, D.; Durbin, H.; Johnson, P.M.; Taylor-Papadimitriou, J. Limitations of radiolabeled monoclonal antibodies for localization of human neoplasms. Cancer Res. 1986, 46, 3183–3191. [Google Scholar]
- Merten, H.; Brandl, F.; Plückthun, A.; Zangemeister-Wittke, U. Antibody-Drug Conjugates for Tumor Targeting-Novel Conjugation Chemistries and the Promise of non-IgG Binding Proteins. Bioconjug. Chem. 2015, 26, 2176–2185. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 1990, 50 (Suppl. 3), 814s–819s. [Google Scholar]
- Pruszynski, M.; Koumarianou, E.; Vaidyanathan, G.; Revets, H.; Devoogdt, N.; Lahoutte, T.; Lyerly, H.K.; Zalutsky, M.R. Improved tumor targeting of anti-HER2 nanobody through N-succinimidyl 4-guanidinomethyl-3-iodobenzoate radiolabeling. J. Nucl. Med. 2014, 55, 650–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altunay, B.; Morgenroth, A.; Beheshti, M.; Vogg, A.; Wong, N.C.L.; Ting, H.H.; Biersack, H.J.; Stickeler, E.; Mottaghy, F.M. HER2-directed antibodies, affibodies and nanobodies as drug-delivery vehicles in breast cancer with a specific focus on radioimmunotherapy and radioimmunoimaging. Eur. J. Nucl. Med. Mol. Imaging 2020, 48, 1371–1389. [Google Scholar] [CrossRef]
- Singh, A.P.; Guo, L.; Verma, A.; Wong, G.G.; Thurber, G.M.; Shah, D.K. Antibody Coadministration as a Strategy to Overcome Binding-Site Barrier for ADCs: A Quantitative Investigation. AAPS J. 2020, 22, 28. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef]
- Boucher, Y.; Baxter, L.T.; Jain, R.K. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: Implications for therapy. Cancer Res. 1990, 50, 4478–4484. [Google Scholar]
- Jain, R.K.; Baxter, L.T. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: Significance of elevated interstitial pressure. Cancer Res. 1988, 48 Pt 1, 7022–7032. [Google Scholar]
- Jain, R.K. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev. 1990, 9, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Coats, S.; Williams, M.; Kebble, B.; Dixit, R.; Tseng, L.; Yao, N.S.; Tice, D.A.; Soria, J.C. Antibody-Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin. Cancer 2019, 25, 5441–5448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodack, D.P.; Askoxylakis, V.; Ferraro, G.B.; Sheng, Q.; Badeaux, M.; Goel, S.; Qi, X.; Shankaraiah, R.; Cao, Z.A.; Ramjiawan, R.R.; et al. The brain microenvironment mediates resistance in luminal breast cancer to PI3K inhibition through HER3 activation. Sci. Transl. Med. 2017, 9, eaal4682. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Liu, S.; Lyu, H.; Riker, A.I.; Zhang, Y.; Liu, B. Development of Effective Therapeutics Targeting HER3 for Cancer Treatment. Biol. Proced. Online 2019, 21, 5. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Koyama, K.; Kamai, Y.; Hirotani, K.; Ogitani, Y.; Zembutsu, A.; Abe, M.; Kaneda, Y.; Maeda, N.; Shiose, Y.; et al. A Novel HER3-Targeting Antibody-Drug Conjugate, U3-1402, Exhibits Potent Therapeutic Efficacy through the Delivery of Cytotoxic Payload by Efficient Internalization. Clin. Cancer Res. 2019, 25, 7151–7161. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Chang, Y.; Rios, A.; An, Z. HER3/ErbB3, an emerging cancer therapeutic target. Acta Biochim. Biophys. Sin. 2016, 48, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daiichi Sankyo Co., Ltd.; Daiichi Sankyo, Inc. Phase I/II Study of U3-1402 in Subjects with Human Epidermal Growth Factor Receptor 3 (HER3) Positive Metastatic Breast Cancer; Daiichi Sankyo Co, Ltd.: Tokyo, Japan, 2021. [Google Scholar]
- Vlachostergios, P.J.; Jakubowski, C.D.; Niaz, M.J.; Lee, A.; Thomas, C.; Hackett, A.L.; Patel, P.; Rashid, N.; Tagawa, S.T. Antibody-Drug Conjugates in Bladder Cancer. Bladder Cancer 2018, 4, 247–259. [Google Scholar] [CrossRef] [Green Version]
- Tati, S.; Fisk, J.C.; Abdullah, J.; Karacosta, L.; Chrisikos, T.; Philbin, P.; Morey, S.; Ghazal, D.; Zazala, F.; Jessee, J.; et al. Humanization of JAA-F11, a Highly Specific Anti-Thomsen-Friedenreich Pancarcinoma Antibody and InVitro Efficacy Analysis. Neoplasia 2017, 19, 716–733. [Google Scholar] [CrossRef] [PubMed]
- Elzoghby, A.O.; Abdelmoneem, M.A.; Hassanin, I.A.; Abd Elwakil, M.M.; Elnaggar, M.A.; Mokhtar, S.; Fang, J.Y.; Elkhodairy, K.A. Lactoferrin, a multi-functional glycoprotein: Active therapeutic, drug nanocarrier & targeting ligand. Biomaterials 2020, 263, 120355. [Google Scholar] [CrossRef] [PubMed]
- Vaghasiya, K.; Ray, E.; Singh, R.; Jadhav, K.; Sharma, A.; Khan, R.; Katare, O.P.; Verma, R.K. Efficient, enzyme responsive and tumor receptor targeting gelatin nanoparticles decorated with concanavalin-A for site-specific and controlled drug delivery for cancer therapy. Mater. Sci. Eng. Mater. Biol. Appl. 2021, 123, 112027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, J.; Song, J.; Liu, Y.; Ren, X.; Zhao, Y. Protein-Based Nanomedicine for Therapeutic Benefits of Cancer. ACS Nano 2021, 15, 8001–8038. [Google Scholar] [CrossRef] [PubMed]
- Asrorov, A.M.; Gu, Z.; Li, F.; Liu, L.; Huang, Y. Biomimetic camouflage delivery strategies for cancer therapy. Nanoscale 2021, 13, 8693–8706. [Google Scholar] [CrossRef]
- Wong, K.H.; Lu, A.; Chen, X.; Yang, Z. Natural Ingredient-Based Polymeric Nanoparticles for Cancer Treatment. Molecules 2020, 25, 3620. [Google Scholar] [CrossRef] [PubMed]
- Deonarain, M.P. Miniaturised ‘antibody’-drug conjugates for solid tumours? Drug Discov. Today Technol. 2018, 30, 47–53. [Google Scholar] [CrossRef]
- Deonarain, M.P.; Yahioglu, G.; Stamati, I.; Pomowski, A.; Clarke, J.; Edwards, B.M.; Diez-Posada, S.; Stewart, A.C. Small-Format Drug Conjugates: A Viable Alternative to ADCs for Solid Tumours? Antibodies 2018, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Richards, D.A. Exploring alternative antibody scaffolds: Antibody fragments and antibody mimics for targeted drug delivery. Drug Discov. Today Technol. 2018, 30, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Lipovšek, D.; Carvajal, I.; Allentoff, A.J.; Barros, A.; Brailsford, J., Jr.; Cong, Q.; Cotter, P.; Gangwar, S.; Hollander, C.; Lafont, V.; et al. Adnectin-drug conjugates for Glypican-3-specific delivery of a cytotoxic payload to tumors. Protein Eng. Des. Sel. 2018, 31, 159–171. [Google Scholar] [CrossRef]
- Li, Z.; Krippendorff, B.F.; Sharma, S.; Walz, A.C.; Lavé, T.; Shah, D.K. Influence of molecular size on tissue distribution of antibody fragments. MAbs 2016, 8, 113–119. [Google Scholar] [CrossRef] [Green Version]
- White, B.H.; Whalen, K.; Kriksciukaite, K.; Alargova, R.; Au Yeung, T.; Bazinet, P.; Brockman, A.; DuPont, M.; Oller, H.; Lemelin, C.A.; et al. Discovery of an SSTR2-Targeting Maytansinoid Conjugate (PEN-221) with Potent Activity in vitro and in vivo. J. Med. Chem. 2019, 62, 2708–2719. [Google Scholar] [CrossRef]
- Bennett, G.; Lutz, R.; Park, P.; Harrison, H.; Lee, K. Abstract 1167: Development of BT1718, a novel Bicycle Drug Conjugate for the treatment of lung cancer. Cancer Res. 2017, 77 (Suppl. 13), 1167. [Google Scholar] [CrossRef]
- Bennett, G.; Rigby, M.; Lutz, B.; Park, P.; Keen, N. Abstract B135: The mechanism of action of BT1718, a novel small-molecule drug conjugate for the treatment of solid tumors expressing MT1-MMP. Mol. Cancer Ther. 2018, 17 (Suppl. 1), B135. [Google Scholar] [CrossRef]
- Sau, S.; Alsaab, H.O.; Kashaw, S.K.; Tatiparti, K.; Iyer, A.K. Advances in antibody-drug conjugates: A new era of targeted cancer therapy. Drug Discov. Today 2017, 22, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Dev. Ther. 2018, 12, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Shim, H. Bispecific Antibodies and Antibody-Drug Conjugates for Cancer Therapy: Technological Considerations. Biomolecules 2020, 10, 360. [Google Scholar] [CrossRef] [Green Version]
- Andreev, J.; Thambi, N.; Perez Bay, A.E.; Delfino, F.; Martin, J.; Kelly, M.P.; Kirshner, J.R.; Rafique, A.; Kunz, A.; Nittoli, T.; et al. Bispecific Antibodies and Antibody-Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol. Cancer Ther. 2017, 16, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Comer, F.; Gao, C.; Coats, S. Bispecific and Biparatopic Antibody Drug Conjugates. In Innovations for Next-Generation Antibody-Drug Conjugates; Damelin, M., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 267–280. [Google Scholar]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.; Bezabeh, B.Z.; Fleming, R.L.; Dimasi, N.; et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2019, 35, 948–949. [Google Scholar] [CrossRef] [Green Version]
- Oganesyan, V.; Peng, L.; Bee, J.S.; Li, J.; Perry, S.R.; Comer, F.; Xu, L.; Cook, K.; Senthil, K.; Clarke, L.; et al. Structural insights into the mechanism of action of a biparatopic anti-HER2 antibody. J. Biol. Chem. 2018, 293, 8439–8448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarian, F.; Alibakhshi, A.; Eyvazi, S.; Arezumand, R.; Ahangarzadeh, S. Antibody-drug therapeutic conjugates: Potential of antibody-siRNAs in cancer therapy. J. Cell. Physiol. 2019, 234, 16724–16738. [Google Scholar] [CrossRef]
- Zavrtanik, U.; Lukan, J.; Loris, R.; Lah, J.; Hadži, S. Structural Basis of Epitope Recognition by Heavy-Chain Camelid Antibodies. J. Mol. Biol. 2018, 430, 4369–4386. [Google Scholar] [CrossRef] [PubMed]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef]
- Beghein, E.; Gettemans, J. Nanobody Technology: A Versatile Toolkit for Microscopic Imaging, Protein-Protein Interaction Analysis, and Protein Function Exploration. Front. Immunol. 2017, 8, 771. [Google Scholar] [CrossRef]
- Könning, D.; Zielonka, S.; Grzeschik, J.; Empting, M.; Valldorf, B.; Krah, S.; Schröter, C.; Sellmann, C.; Hock, B.; Kolmar, H. Camelid and shark single domain antibodies: Structural features and therapeutic potential. Curr. Opin. Struct. Biol. 2017, 45, 10–16. [Google Scholar] [CrossRef]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knöbl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Wang, Y.; Luo, X.; Ma, Z.; Xu, Y.; Zhang, X.; Lv, T.; Zhang, Y.; Wang, M.; Huang, Z.; et al. Anti-CD24 Antibody-Nitric Oxide Conjugate Selectively and Potently Suppresses Hepatic Carcinoma. Cancer Res. 2019, 79, 3395–3405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickok, J.R.; Thomas, D.D. Nitric oxide and cancer therapy: The emperor has NO clothes. Curr. Pharm. Des. 2010, 16, 381–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lares, M.R.; Rossi, J.J.; Ouellet, D.L. RNAi and small interfering RNAs in human disease therapeutic applications. Trends Biotechnol. 2010, 28, 570–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannon, G.J.; Rossi, J.J. Unlocking the potential of the human genome with RNA interference. Nature 2004, 431, 371–378. [Google Scholar] [CrossRef]
- McManus, M.T.; Sharp, P.A. Gene silencing in mammals by small interfering RNAs. Nat. Rev. Genet. 2002, 3, 737–747. [Google Scholar] [CrossRef]
- Schroeder, A.; Levins, C.G.; Cortez, C.; Langer, R.; Anderson, D.G. Lipid-based nanotherapeutics for siRNA delivery. J. Intern. Med. 2010, 267, 9–21. [Google Scholar] [CrossRef]
- Foster, D.J.; Brown, C.R.; Shaikh, S.; Trapp, C.; Schlegel, M.K.; Qian, K.; Sehgal, A.; Rajeev, K.G.; Jadhav, V.; Manoharan, M.; et al. Advanced siRNA Designs Further Improve In vivo Performance of GalNAc-siRNA Conjugates. Mol. Ther. 2018, 26, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.D.; Rhodes, D.G.; Burgess, D.J. DNA-based therapeutics and DNA delivery systems: A comprehensive review. AAPS J. 2005, 7, E61–E77. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Flynn, M.J.; Hartley, J.A. The emerging role of anti-CD25 directed therapies as both immune modulators and targeted agents in cancer. Br. J. Haematol. 2017, 179, 20–35. [Google Scholar] [CrossRef] [Green Version]
- Vargas, F.A.; Furness, A.J.S.; Solomon, I.; Joshi, K.; Mekkaoui, L.; Lesko, M.H.; Rota, E.M.; Dahan, R.; Georgiou, A.; Sledzinska, A.; et al. Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity 2017, 46, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- Horwitz, S.M.; Fanale, M.A.; Spira, A.I.; Havenith, K.; He, S.; Feingold, J.M.; Hamadani, M. Interim data from the first clinical study of adct-301, a novel pyrrolobenzodiazapine-based antibody drug conjugate, in relapsed/refractory hodgkin/non-hodgkin lymphoma. Hematol. Oncol. 2017, 35 (Suppl. 2), 270–271. [Google Scholar] [CrossRef] [Green Version]
- Sau, S.; Petrovici, A.; Alsaab, H.O.; Bhise, K.; Iyer, A.K. PDL-1 Antibody Drug Conjugate for Selective Chemo-Guided Immune Modulation of Cancer. Cancers 2019, 11, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, R.; Weiner, G.J. Picking the optimal target for antibody-drug conjugates. In American Society of Clinical Oncology Educational Book, Proceedings of the American Society of Clinical Oncology Annual Meeting, Chicago, IL, USA, 31 May–4 June 2013; American Society of Clinical Oncology: Alexandria, VA, USA, 2013. [Google Scholar] [CrossRef]
- Gébleux, R.; Stringhini, M.; Casanova, R.; Soltermann, A.; Neri, D. Non-internalizing antibody-drug conjugates display potent anti-cancer activity upon proteolytic release of monomethyl auristatin E in the subendothelial extracellular matrix. Int. J. Cancer. 2017, 140, 1670–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giansanti, F.; Capone, E.; Ponziani, S.; Piccolo, E.; Gentile, R.; Lamolinara, A.; Di Campli, A.; Sallese, M.; Iacobelli, V.; Cimini, A.; et al. Secreted Gal-3BP is a novel promising target for non-internalizing Antibody-Drug Conjugates. J. Control. Release 2019, 294, 176–184. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [Green Version]
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; van den Heuvel, E.G.; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.Y.; et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat. Med. 2018, 24, 203–212. [Google Scholar] [CrossRef]
- Daiichi Sankyo Inc.; Bristol-Myers Squibb; AstraZeneca. Trastuzumab Deruxtecan with Nivolumab in Advanced Breast and Urothelial Cancer; Daiichi Sankyo, Inc.: Tokyo, Japan, 2021. [Google Scholar]
- Daiichi Sankyo Inc.; AstraZeneca UK Limited; Merck Sharp & Dohme Corp. DS8201a and Pembrolizumab in Participants with Locally Advanced/Metastatic Breast or Non-Small Cell Lung Cancer; Daiichi Sankyo, Inc.: Tokyo, Japan, 2021. [Google Scholar]
- National Cancer Institute. Brentuximab Vedotin and Nivolumab with or without Ipilimumab in Treating Patients with Relapsed or Refractory Hodgkin Lymphoma; National Cancer Institute: Tokyo, Japan, 2021.
- Dana-Farber Cancer Institute; Merck Sharp & Dohme Corp. A Study of Pembrolizumab in Combination with Trastuzumab-DM1; National Cancer Institute: Boston, MA, USA, 2021.
- Hoffmann-La Roche. A Study to Evaluate the Efficacy and Safety of Trastuzumab Emtansine in Combination with Atezolizumab or Atezolizumab-Placebo in Participants with Human Epidermal Growth Factor-2 (HER2) Positive Locally Advanced or Metastatic Breast Cancer (BC) Who Received Prior Trastuzumab and Taxane Based Therapy; Hoffmann-La Roche: Basel, Switzerland, 2017. [Google Scholar]
- Keenan, B.P.; Fong, L. Conditional Cancer Immunotherapy as a Safer Way to Step on the Gas. Cancer Discov. 2021, 11, 20–22. [Google Scholar] [CrossRef]
- Kamata-Sakurai, M.; Narita, Y.; Hori, Y.; Nemoto, T.; Uchikawa, R.; Honda, M.; Hironiwa, N.; Taniguchi, K.; Shida-Kawazoe, M.; Metsugi, S.; et al. Antibody to CD137 Activated by Extracellular Adenosine Triphosphate Is Tumor Selective and Broadly Effective In vivo without Systemic Immune Activation. Cancer Discov. 2021, 11, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Sharp, L.L.; Chang, C.; Frey, G.; Wang, J.; Liu, H.; Xing, C.; Yalcin, S.; Walls, M.; Ben, Y.; Boyle, W.J.; et al. Abstract 827: Anti-tumor efficacy of BA3011, a novel Conditionally Active Biologic (CAB) anti-AXL-ADC. Cancer Res. 2018, 78 (Suppl. 13), 827. [Google Scholar] [CrossRef]
- Sharp, L.L.; Chang, C.; Frey, G.; Wang, J.; Liu, H.; Xing, C.; Yalcin, S.; Walls, M.; Ben, Y.; Boyle, W.J.; et al. Abstract 833: Anti-tumor efficacy of BA3021, a novel Conditionally Active Biologic (CAB) anti-ROR2 ADC. Cancer Res. 2018, 78 (Suppl. 13), 833. [Google Scholar] [CrossRef]
- BioAtla LLC. CAB-AXL-ADC Safety and Efficacy Study in Adult and Adolescent Patients with Solid Tumors; BioAtla LLC: San Diego, CA, USA, 2018. [Google Scholar]
- BioAtla LLC. CAB-ROR2-ADC Safety and Efficacy Study in Patients with Solid Tumors; BioAtla LLC: San Diego, CA, USA, 2018. [Google Scholar]
- Serwer, L.; Singh, S.; Krebber, C.; Liu, S.; Chauhan, N.; Leanna, R.; Lu, H.; Badagnani, I.; Henriques, T.; Morgan-Lappe, S.; et al. Abstract B103: A multi-analyte HPLC-MS/MS approach to assessing exposure of a Probody drug conjugate in preclinical studies. Mol. Cancer Ther. 2018, 17 (Suppl. 1), B103. [Google Scholar] [CrossRef]
- Weaver, A.Y.; Singh, S.; DuPage, A.; Sagert, J.; Flandez, J.; Menendez, E.; Ford, J.; Krimm, M.; Moore, S.; Nguyen, M.; et al. Abstract C165: Development of a probody drug conjugate (PDC) against CD166 for the treatment of multiple cancers. Mol. Cancer Ther. 2015, 14 (Suppl. 2), C165. [Google Scholar] [CrossRef]
- Lin, J.; Sagert, J. Targeting Drug Conjugates to the Tumor Microenvironment: Probody Drug Conjugates. In Innovations for Next-Generation Antibody-Drug Conjugates; Damelin, M., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 281–298. [Google Scholar]
- CytomX Therapeutics. PROCLAIM-CX-2029: A Trial to Find Safe and Active Doses of an Investigational Drug CX-2029 for Patients with Solid Tumors or DLBCL; CytomX Therapeutics: South San Francisco, CA, USA, 2018. [Google Scholar]
- CytomX Therapeutics. PROCLAIM-CX-2009: A Trial to Find Safe and Active Doses of an Investigational Drug CX-2009 for Patients with Selected Solid Tumors; CytomX Therapeutics: South San Francisco, CA, USA, 2017. [Google Scholar]
- Chen, X.; Soma, L.A.; Fromm, J.R. Targeted therapy for Hodgkin lymphoma and systemic anaplastic large cell lymphoma: Focus on brentuximab vedotin. OncoTargets Ther. 2013, 7, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Cianfriglia, M. The biology of MDR1-P-glycoprotein (MDR1-Pgp) in designing functional antibody drug conjugates (ADCs): The experience of gemtuzumab ozogamicin. Ann. Dell’istituto Super. Sanita 2013, 49, 150–168. [Google Scholar] [CrossRef]
- Loganzo, F.; Sung, M.; Gerber, H.P. Mechanisms of Resistance to Antibody-Drug Conjugates. Mol. Cancer Ther. 2016, 15, 2825–2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyes, K.; Brender, T.; Smith, S.W.; Xu, H.; Setter, B.; Fan, L.-Q.; Brunette, R.; Killebrew, J.; Tan, P.; Coburn, C.; et al. Abstract 3271: A systemically administered, conditionally active TLR8 agonist for the treatment of HER2-expressing tumors. Cancer Res. 2019, 79 (Suppl. 13), 3271. [Google Scholar] [CrossRef]
- Sharma, M.; Dumbrava, E.I.; Carvajal, R.; Catenacci, D.; Emens, L.; Hanna, G.; Juric, D.; Kang, Y.-K.; Lee, J.; Lee, K.-W.; et al. 401 Phase 1/2 study of novel HER2-targeting, TLR7/8 immune-stimulating antibody conjugate (ISAC) BDC-1001 with or without immune checkpoint inhibitor in patients with advanced HER2-expressing solid tumors. J. Immunother. Cancer 2020, 8 (Suppl. 3), A244. [Google Scholar] [CrossRef]
- Bolt Biotherapeutics, Inc. A First-in-Human Study Using BDC-1001 in Advanced HER2-Expressing Solid Tumors; Bolt Biotherapeutics, Inc.: Redwood City, CA, USA, 2021. [Google Scholar]
- Regazzi, M.; Golay, J.; Molinaro, M. Monoclonal Antibody Monitoring: Clinically Relevant Aspects, A Systematic Critical Review. Ther. Drug Monit. 2020, 42, 45–56. [Google Scholar] [CrossRef]
- Mould, D.R. The Pharmacokinetics of Biologics: A Primer. Dig. Dis. 2015, 33 (Suppl. 1), 61–69. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, P.L.C.; Shackelton, L.M.; Vande Casteele, N. Factors Influencing Drug Disposition of Monoclonal Antibodies in Inflammatory Bowel Disease: Implications for Personalized Medicine. BioDrugs 2019, 33, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Cheifetz, A. Overview of Therapeutic Drug Monitoring of Biologic Agents in Patients with Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2017, 13, 556–559. [Google Scholar]
- Fleisher, B.; Ait-Oudhia, S. A retrospective examination of the US Food and Drug Administration’s clinical pharmacology reviews of oncology biologics for potential use of therapeutic drug monitoring. OncoTargets Ther. 2018, 11, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Mould, D.R.; Sweeney, K.R. The pharmacokinetics and pharmacodynamics of monoclonal antibodies--mechanistic modeling applied to drug development. Curr. Opin. Drug Discov. Dev. 2007, 10, 84–96. [Google Scholar]
- Azzopardi, N.; Lecomte, T.; Ternant, D.; Boisdron-Celle, M.; Piller, F.; Morel, A.; Gouilleux-Gruart, V.; Vignault-Desvignes, C.; Watier, H.; Gamelin, E.; et al. Cetuximab pharmacokinetics influences progression-free survival of metastatic colorectal cancer patients. Clin. Cancer Res. 2011, 17, 6329–6337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fracasso, P.M.; Burris, H., III; Arquette, M.A.; Govindan, R.; Gao, F.; Wright, L.P.; Goodner, S.A.; Greco, F.A.; Jones, S.F.; Willcut, N.; et al. A phase 1 escalating single-dose and weekly fixed-dose study of cetuximab: Pharmacokinetic and pharmacodynamic rationale for dosing. Clin. Cancer Res. 2007, 13, 986–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glassman, P.M.; Balthasar, J.P. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol. Med. 2014, 11, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Mulleman, D.; Chu Miow Lin, D.; Ducourau, E.; Emond, P.; Ternant, D.; Magdelaine-Beuzelin, C.; Valat, J.P.; Paintaud, G.; Goupille, P. Trough infliximab concentrations predict efficacy and sustained control of disease activity in rheumatoid arthritis. Ther. Drug Monit. 2010, 32, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Dotan, I.; Ron, Y.; Yanai, H.; Becker, S.; Fishman, S.; Yahav, L.; Ben Yehoyada, M.; Mould, D.R. Patient factors that increase infliximab clearance and shorten half-life in inflammatory bowel disease: A population pharmacokinetic study. Inflamm. Bowel Dis. 2014, 20, 2247–2259. [Google Scholar] [CrossRef]
- Ordás, I.; Feagan, B.G.; Sandborn, W.J. Therapeutic drug monitoring of tumor necrosis factor antagonists in inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2012, 10, 1079–1087. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generic Name | Brand Name (Manufacturer) | Indication | Approval Year | ADC Characteristics | |||
|---|---|---|---|---|---|---|---|
| Antibody Isotype | Target Antigen | Linker | Payload | ||||
| Brentuximab vedotin | Adcetris (Seattle Genetics) | HL, ALCL | 2011 | IgG1 | CD30 | Cleavable (protease) | MMAE |
| Ado-trastuzumab emtasine | Kadcyla (Genentech) | HER2+ mBC HER2+ eBC | 2013 | IgG1 | HER2 | Non-cleavable | DM1 |
| Inotuzumab ozogamicin | Besponsa (Pfizer) | ALL | 2017 | IgG4 | CD22 | Cleavable (acid labile) | Calicheamicin |
| Gemtuzumab ozogamicin | Mylotarg (Pfizer) | CD33+ AML | 2017 | IgG4 | CD33 | Cleavable (disulfide) | Calicheamicin |
| Moxetumomab pasudotox-tdfk | Lumoxiti (AstraZeneca) | r/r HCL | 2018 | IgG1 | CD22 | Cleavable (disulfide) | Pseudomonas exotoxin |
| Polatuzumab vedotin | Polivy (Genentech) | r/r DLBCL | 2019 | IgG1 | CD79b | Cleavable (protease) | MMAE |
| Enfortumab vedotin | Padcev (Seattle Genetics) | mUC | 2019 | IgG1 | Nectin-4 | Cleavable (protease) | MMAE |
| Trastuzumab deruxtecan | Enhertu (AstraZeneca) (Daiichi-Sankyo) | HER2+ BC HER2+ GC/GEJ | 2019 | IgG1 | HER2 | Cleavable (peptides) | DXd |
| Sacituzumab govitecan | Trodelvy (Imunomedics) | mTNBC | 2020 | IgG1 | Trop-2 | Cleavable (acid labile) | SN-38 |
| Belantamab mafodotin-blmf | Blenrep (GlaxoSmithKline) | Multiple myeloma | 2020 | IgG1 | BCMA | Cleavable (protease) | MMAF |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucas, A.T.; Moody, A.; Schorzman, A.N.; Zamboni, W.C. Importance and Considerations of Antibody Engineering in Antibody-Drug Conjugates Development from a Clinical Pharmacologist’s Perspective. Antibodies 2021, 10, 30. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10030030
Lucas AT, Moody A, Schorzman AN, Zamboni WC. Importance and Considerations of Antibody Engineering in Antibody-Drug Conjugates Development from a Clinical Pharmacologist’s Perspective. Antibodies. 2021; 10(3):30. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10030030
Chicago/Turabian StyleLucas, Andrew T., Amber Moody, Allison N. Schorzman, and William C. Zamboni. 2021. "Importance and Considerations of Antibody Engineering in Antibody-Drug Conjugates Development from a Clinical Pharmacologist’s Perspective" Antibodies 10, no. 3: 30. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10030030