
Fc-Independent Protection from SARS-CoV-2 Infection by Recombinant Human Monoclonal Antibodies

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Expression of Proteins and Antibodies
2.2. ELISA
2.3. Binding to Recombinant FcγRs
2.4. Biolayer Interferometry Assays
2.4.1. Complement Binding
2.4.2. Affinity to Human FcRn
2.5. CD107a Degranulation Assay
2.6. FCγ Receptor Potency Assay
2.7. Plaque Reduction Neutralization Test (PRNT)
2.8. Animal Experiments
2.9. Measurement of Viral RNA by qRT-PCR
2.10. Lung Histology
3. Results
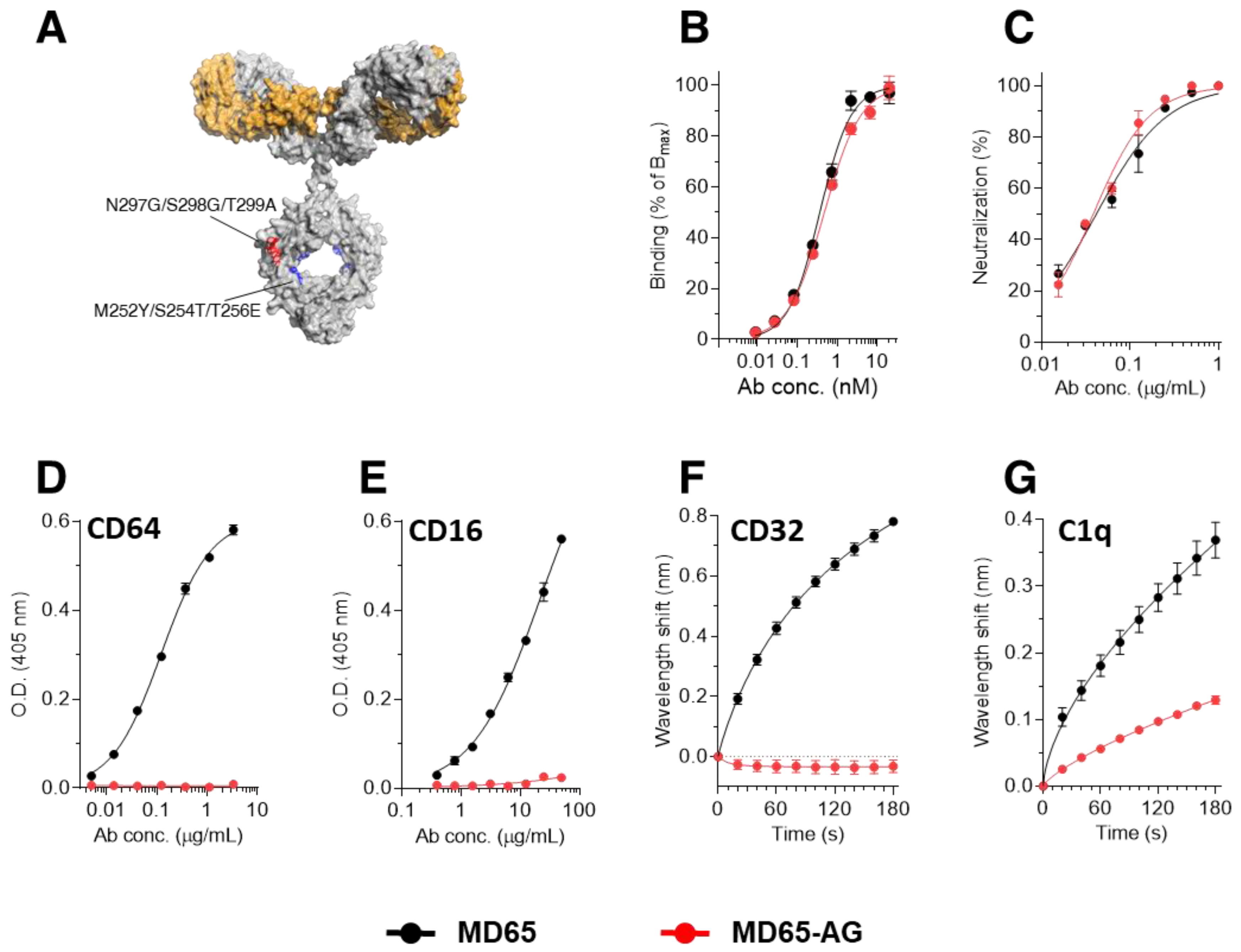
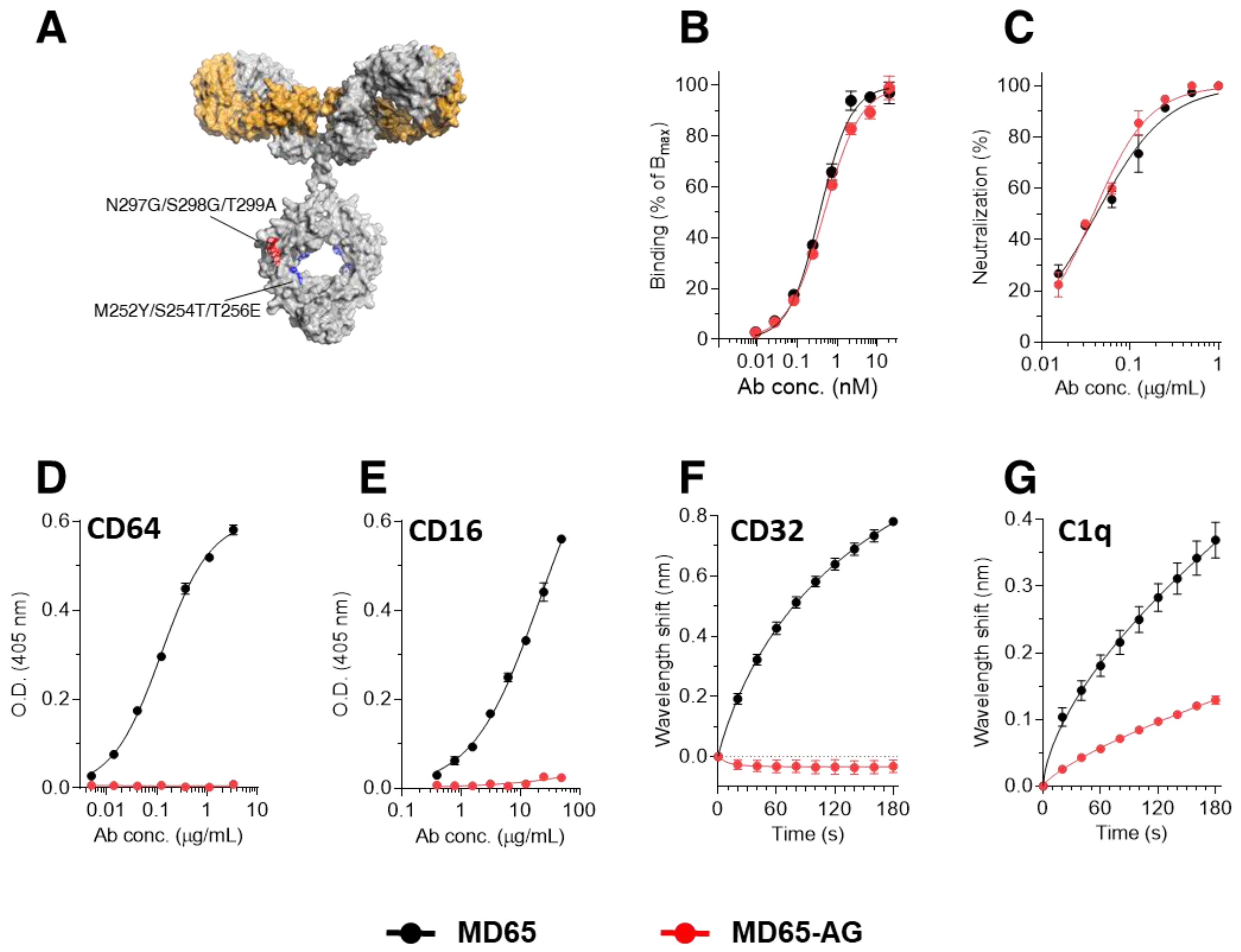
3.1. Construction of Fc-Engineered Antibodies
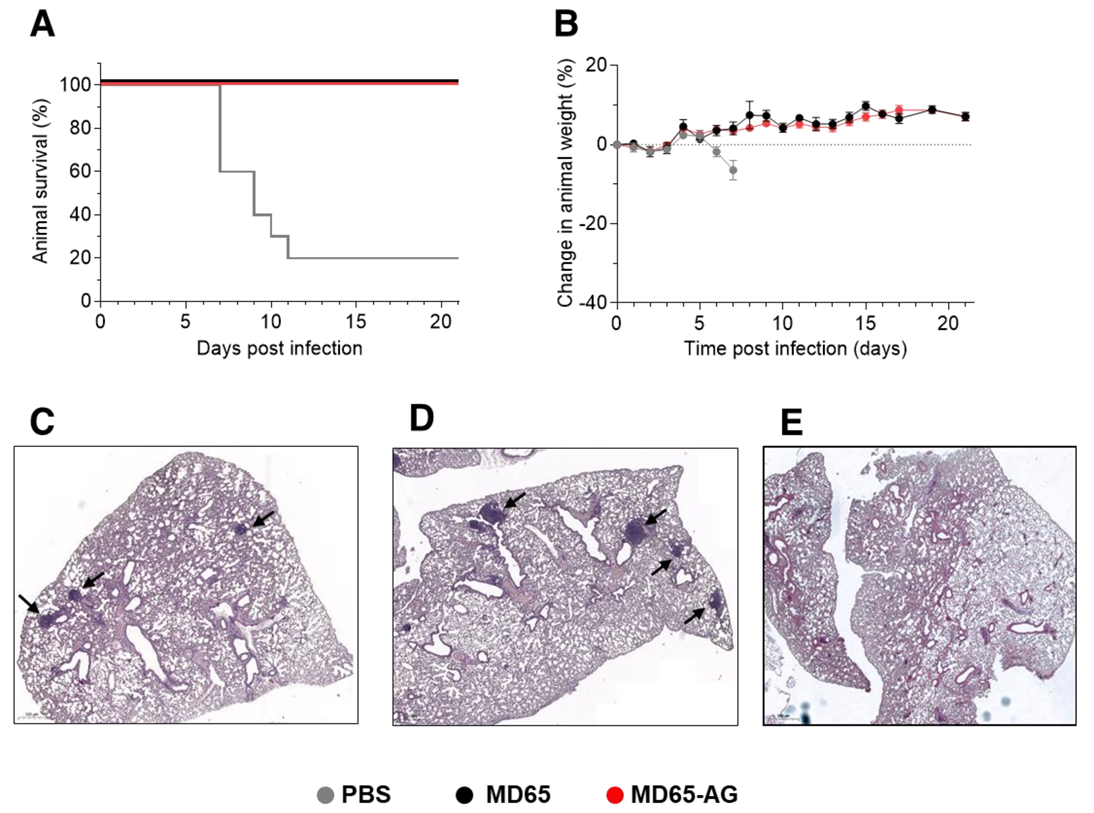
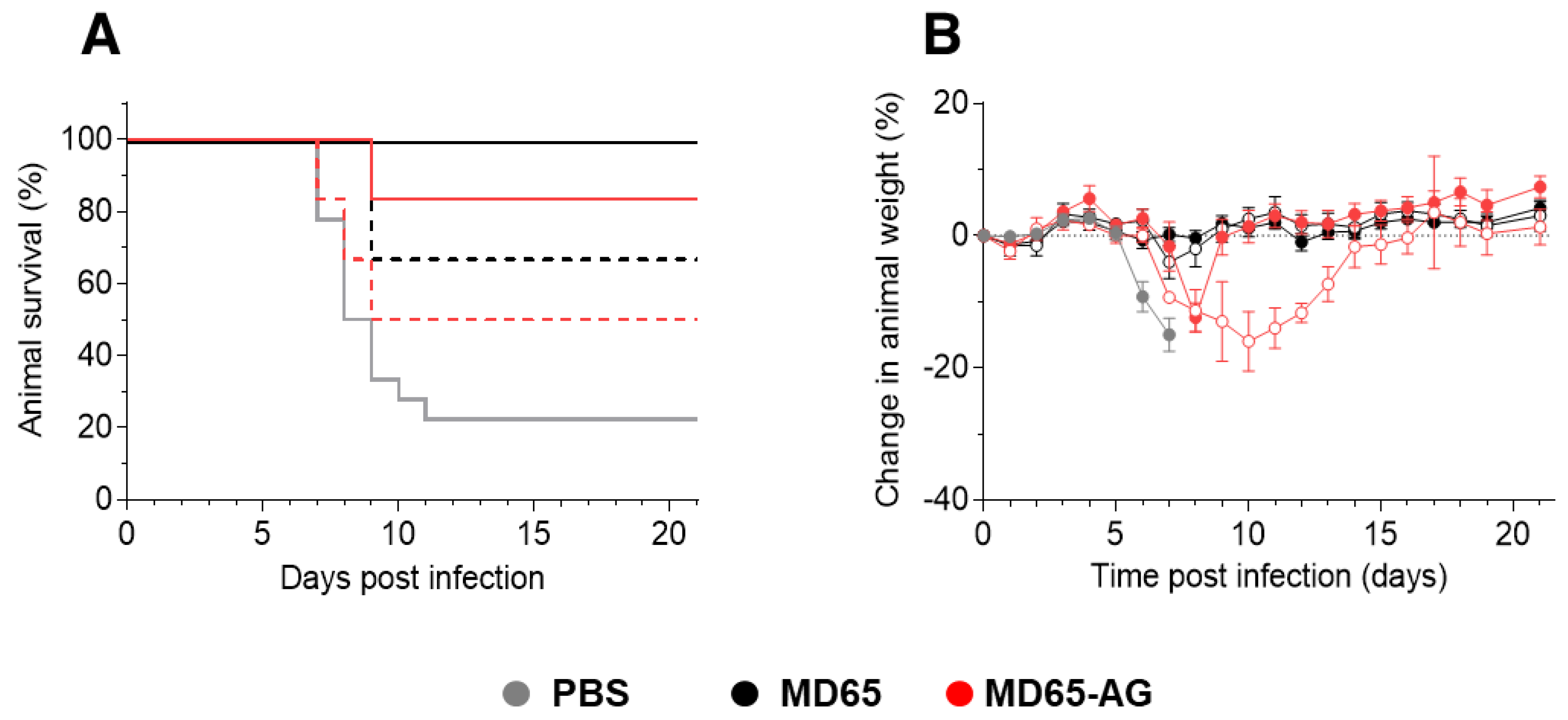
3.2. Prophylactic Activity of MD65-AG In-Vivo
3.3. Fc-Independent Post-Exposure Protection of SARS-CoV-2 Infected Mice
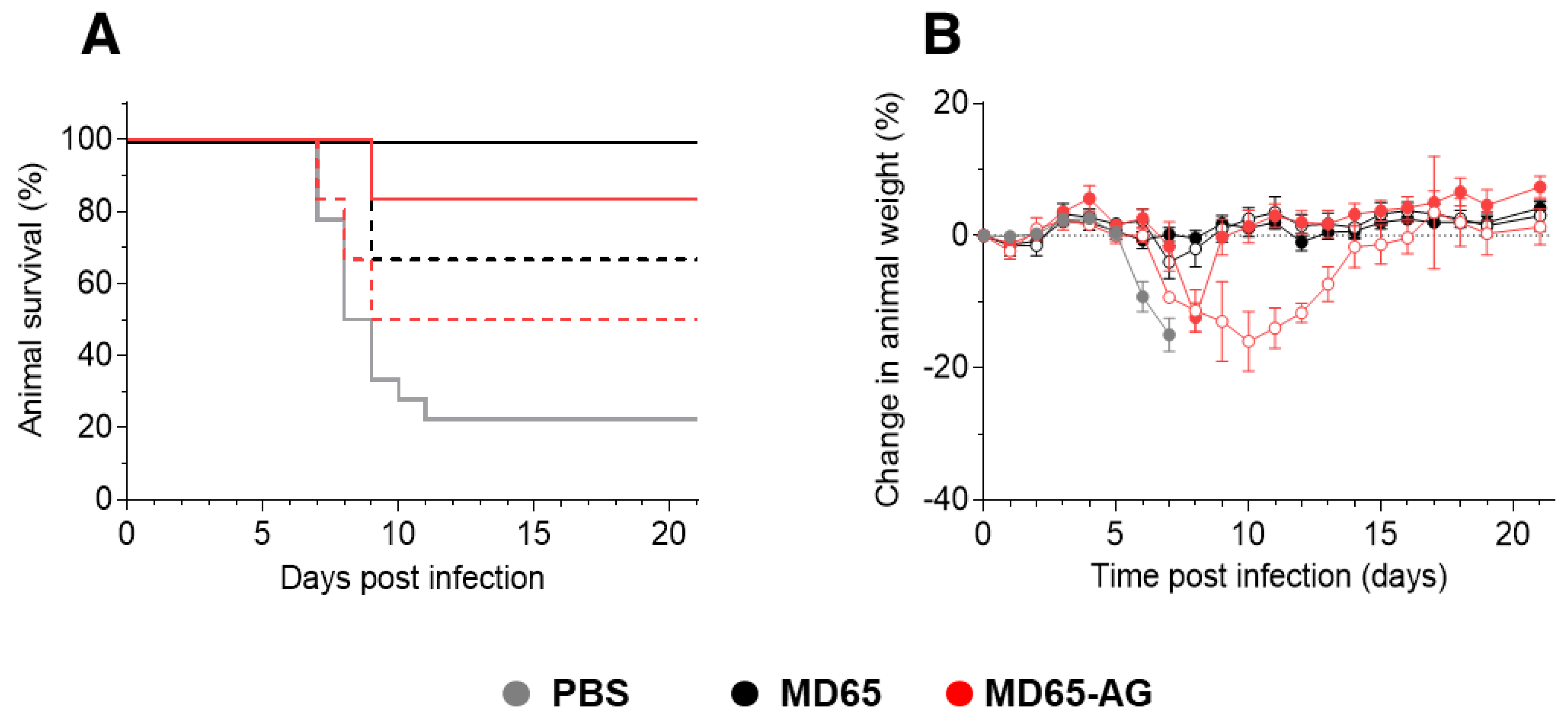
3.4. Dose-Dependent Therapeutic Efficacy of MD65-AG
3.5. Fc-Independent Post-Exposure Protection by Anti-NTD Antibody
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Z.; Bao, L.; Chen, C.; Zou, T.; Xue, Y.; Li, F.; Lv, Q.; Gu, S.; Gao, X.; Cui, S.; et al. Human Neutralizing Monoclonal Antibody Inhibition of Middle East Respiratory Syndrome Coronavirus Replication in the Common Marmoset. J. Infect. Dis. 2017, 215, 1807–1815. [Google Scholar] [CrossRef] [Green Version]
- Corti, D.; Zhao, J.; Pedotti, M.; Simonelli, L.; Agnihothram, S.; Fett, C.; Fernandez-Rodriguez, B.; Foglierini, M.; Agatic, G.; Vanzetta, F.; et al. Prophylactic and postexposure efficacy of a potent human monoclonal antibody against MERS coronavirus. Proc. Natl. Acad. Sci. USA 2015, 112, 10473–10478. [Google Scholar] [CrossRef] [Green Version]
- Rockx, B.; Corti, D.; Donaldson, E.; Sheahan, T.; Stadler, K.; Lanzavecchia, A.; Baric, R. Structural basis for potent cross-neutralizing human monoclonal antibody protection against lethal human and zoonotic severe acute respiratory syndrome coronavirus challenge. J. Virol. 2008, 82, 3220–3235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, R.; Noy-Porat, T.; Mechaly, A.; Makdasi, E.; Levy, Y.; Alcalay, R.; Falach, R.; Aftalion, M.; Epstein, E.; Gur, D.; et al. Post-exposure protection of SARS-CoV-2 lethal infected K18-hACE2 transgenic mice by neutralizing human monoclonal antibody. Nat. Commun. 2021, 12, 944. [Google Scholar] [CrossRef]
- Scheid, J.F.; Mouquet, H.; Feldhahn, N.; Seaman, M.S.; Velinzon, K.; Pietzsch, J.; Ott, R.G.; Anthony, R.M.; Zebroski, H.; Hurley, A.; et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 2009, 458, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chakraborti, S.; He, Y.; Roberts, A.; Sheahan, T.; Xiao, X.; Hensley, L.E.; Prabakaran, P.; Rockx, B.; Sidorov, I.A.; et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc. Natl. Acad. Sci. USA 2007, 104, 12123–12128. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.L.; Suscovich, T.J.; Fortune, S.M.; Alter, G. Beyond binding: Antibody effector functions in infectious diseases. Nat. Rev. Immunol. 2018, 18, 46–61. [Google Scholar] [CrossRef]
- Chan, K.R.; Ong, E.Z.; Mok, D.Z.; Ooi, E.E. Fc receptors and their influence on efficacy of therapeutic antibodies for treatment of viral diseases. Expert. Rev. Anti. Infect. Ther. 2015, 13, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Novel mechanisms and functions of complement. Nat. Immunol. 2017, 18, 1288–1298. [Google Scholar] [CrossRef]
- Junker, F.; Gordon, J.; Qureshi, O. Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front. Immunol. 2020, 11, 1393. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Palese, P.; Wilson, P.C.; Ravetch, J.V. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J. Clin. Investig. 2016, 126, 605–610. [Google Scholar] [CrossRef]
- Earnest, J.T.; Holmes, A.C.; Basore, K.; Mack, M.; Fremont, D.H.; Diamond, M.S. The mechanistic basis of protection by non-neutralizing anti-alphavirus antibodies. Cell Rep. 2021, 35, 108962. [Google Scholar] [CrossRef]
- Gunn, B.M.; Yu, W.; Karim, M.M.; Brannan, J.M.; Herbert, A.S.; Wec, A.Z.; Halfmann, P.J.; Fusco, M.L.; Schendel, S.L.; Gangavarapu, K.; et al. A Role for Fc Function in Therapeutic Monoclonal Antibody-Mediated Protection against Ebola Virus. Cell Host Microbe 2018, 24, 221–233.e5. [Google Scholar] [CrossRef]
- Liu, Y.; Tan, H.X.; Koutsakos, M.; Jegaskanda, S.; Esterbauer, R.; Tilmanis, D.; Aban, M.; Kedzierska, K.; Hurt, A.C.; Kent, S.J.; et al. Cross-lineage protection by human antibodies binding the influenza B hemagglutinin. Nat. Commun. 2019, 10, 324. [Google Scholar] [CrossRef]
- Parsons, M.S.; Lee, W.S.; Kristensen, A.B.; Amarasena, T.; Khoury, G.; Wheatley, A.K.; Reynaldi, A.; Wines, B.D.; Hogarth, M.; Davenport, M.P.; et al. Fc-dependent functions are redundant to efficacy of anti-HIV antibody PGT121 in macaques. J. Clin. Investig. 2019, 129, 182–191. [Google Scholar] [CrossRef]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Weinreich, D.M.; Sivapalasingam, S.; Norton, T.; Ali, S.; Gao, H.; Bhore, R.; Musser, B.J.; Soo, Y.; Rofail, D.; Im, J.; et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with Covid-19. N. Engl. J. Med. 2021, 384, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, P.J.M.; Caniels, T.G.; van der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.M.A.; Claireaux, M.; Kerster, G.; et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 2020, 369, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Su, B.; Guo, X.; Sun, W.; Deng, Y.; Bao, L.; Zhu, Q.; Zhang, X.; Zheng, Y.; Geng, C.; et al. Potent Neutralizing Antibodies against SARS-CoV-2 Identified by High-Throughput Single-Cell Sequencing of Convalescent Patients’ B Cells. Cell 2020, 182, 73–84.e16. [Google Scholar] [CrossRef]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef]
- Noy-Porat, T.; Makdasi, E.; Alcalay, R.; Mechaly, A.; Levy, Y.; Bercovich-Kinori, A.; Zauberman, A.; Tamir, H.; Yahalom-Ronen, Y.; Israeli, M.; et al. A panel of human neutralizing mAbs targeting SARS-CoV-2 spike at multiple epitopes. Nat. Commun. 2020, 11, 4303. [Google Scholar] [CrossRef]
- Suryadevara, N.; Shrihari, S.; Gilchuk, P.; VanBlargan, L.A.; Binshtein, E.; Zost, S.J.; Nargi, R.S.; Sutton, R.E.; Winkler, E.S.; Chen, E.C.; et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell 2021, 184, 2316–2331.e15. [Google Scholar] [CrossRef] [PubMed]
- Atyeo, C.; Slein, M.D.; Fischinger, S.; Burke, J.; Schäfer, A.; Leist, S.R.; Kuzmina, N.A.; Mire, C.; Honko, A.; Johnson, R.; et al. Dissecting strategies to tune the therapeutic potential of SARS-CoV-2-specific monoclonal antibody CR3022. JCI Insight 2021, 6, e143129. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.; Muecksch, F.; Lorenzi, J.C.C.; Leist, S.R.; Cipolla, M.; Bournazos, S.; Schmidt, F.; Maison, R.M.; Gazumyan, A.; Martinez, D.R.; et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J. Exp. Med. 2021, 218, e20201993. [Google Scholar] [CrossRef]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.S.; Gilchuk, P.; Yu, J.; Bailey, A.L.; Chen, R.E.; Chong, Z.; Zost, S.J.; Jang, H.; Huang, Y.; Allen, J.D.; et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell 2021, 184, 1804–1820.e1816. [Google Scholar] [CrossRef]
- Chan, C.E.Z.; Seah, S.G.K.; Chye, H.; Massey, S.; Torres, M.; Lim, A.P.C.; Wong, S.K.K.; Neo, J.J.Y.; Wong, P.S.; Lim, J.H.; et al. The Fc-mediated effector functions of a potent SARS-CoV-2 neutralizing antibody, SC31, isolated from an early convalescent COVID-19 patient, are essential for the optimal therapeutic efficacy of the antibody. PLoS ONE 2021, 16, e0253487. [Google Scholar] [CrossRef]
- Eroshenko, N.; Gill, T.; Keaveney, M.K.; Church, G.M.; Trevejo, J.M.; Rajaniemi, H. Implications of antibody-dependent enhancement of infection for SARS-CoV-2 countermeasures. Nat. Biotechnol. 2020, 38, 789–791. [Google Scholar] [CrossRef]
- Makdasi, E.; Levy, Y.; Alcalay, R.; Noy-Porat, T.; Zahavy, E.; Mechaly, A.; Epstein, E.; Peretz, E.; Cohen, H.; Bar-On, L.; et al. Neutralizing Monoclonal Anti-SARS-CoV-2 Antibodies Isolated from Immunized Rabbits Define Novel Vulnerable Spike-Protein Epitope. Viruses 2021, 13, 566. [Google Scholar] [CrossRef]
- Noy-Porat, T.; Alcalay, R.; Mechaly, A.; Peretz, E.; Makdasi, E.; Rosenfeld, R.; Mazor, O. Characterization of antibody-antigen interactions using biolayer interferometry. STAR Protoc. 2021, 2, 100836. [Google Scholar] [CrossRef] [PubMed]
- Gaudinski, M.R.; Coates, E.E.; Houser, K.V.; Chen, G.L.; Yamshchikov, G.; Saunders, J.G.; Holman, L.A.; Gordon, I.; Plummer, S.; Hendel, C.S.; et al. Safety and pharmacokinetics of the Fc-modified HIV-1 human monoclonal antibody VRC01LS: A Phase 1 open-label clinical trial in healthy adults. PLoS Med. 2018, 15, e1002493. [Google Scholar] [CrossRef]
- Noy-Porat, T.; Mechaly, A.; Levy, Y.; Makdasi, E.; Alcalay, R.; Gur, D.; Aftalion, M.; Falach, R.; Leviatan Ben-Arye, S.; Lazar, S.; et al. Therapeutic antibodies, targeting the SARS-CoV-2 spike N-terminal domain, protect lethally infected K18-hACE2 mice. Iscience 2021, 24, 102479. [Google Scholar] [CrossRef]
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Morgenstern, D.; Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Stein, D.; Israeli, O.; et al. The coding capacity of SARS-CoV-2. Nature 2021, 589, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.T.; Kang, T.H.; Kelton, W.; Georgiou, G. Bypassing glycosylation: Engineering aglycosylated full-length IgG antibodies for human therapy. Curr. Opin. Biotechnol. 2011, 22, 858–867. [Google Scholar] [CrossRef]
- Quast, I.; Peschke, B.; Lunemann, J.D. Regulation of antibody effector functions through IgG Fc N-glycosylation. Cell Mol. Life Sci. 2017, 74, 837–847. [Google Scholar] [CrossRef]
- Tao, M.H.; Morrison, S.L. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. 1989, 143, 2595–2601. [Google Scholar]
- Wang, X.; Mathieu, M.; Brezski, R.J. IgG Fc engineering to modulate antibody effector functions. Protein Cell 2018, 9, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Sazinsky, S.L.; Ott, R.G.; Silver, N.W.; Tidor, B.; Ravetch, J.V.; Wittrup, K.D. Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 20167–20172. [Google Scholar] [CrossRef] [Green Version]
- Falach, R.; Bar-On, L.; Lazar, S.; Kadar, T.; Mazor, O.; Aftalion, M.; Gur, D.; Evgy, Y.; Shifman, O.; Aminov, T.; et al. Mice with induced pulmonary morbidities display severe lung inflammation and mortality following exposure to SARS-CoV-2. JCI Insight 2021, 6, e145916. [Google Scholar] [CrossRef]
- Makdasi, E.; Zvi, A.; Alcalay, R.; Noy-Porat, T.; Peretz, E.; Mechaly, A.; Levy, Y.; Epstein, E.; Chitlaru, T.; Tennenhouse, A.; et al. The neutralization potency of anti-SARS-CoV-2 therapeutic human monoclonal antibodies is retained against viral variants. Cell Rep. 2021, 36, 109679. [Google Scholar] [CrossRef]
- Oganesyan, V.; Damschroder, M.M.; Cook, K.E.; Li, Q.; Gao, C.; Wu, H.; Dall’Acqua, W.F. Structural insights into neonatal Fc receptor-based recycling mechanism. J. Biol. Chem. 2014, 289, 7812–7824. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.Q.; Robbie, G.J.; Wu, Y.; Esser, M.T.; Jensen, K.; Schwartz, H.I.; Bellamy, T.; Hernandez-Illas, M.; Jafri, H.S. Safety, Tolerability, and Pharmacokinetics of MEDI4893, an Investigational, Extended-Half-Life, Anti-Staphylococcus aureus Alpha-Toxin Human Monoclonal Antibody, in Healthy Adults. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Dall’Acqua, W.F.; Kiener, P.A.; Wu, H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor. J. Biol. Chem. 2006, 281, 23514–23524. [Google Scholar] [CrossRef] [Green Version]
- Saunders, K.O. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front. Immunol. 2019, 10, 1296. [Google Scholar] [CrossRef] [PubMed]
- Edri, A.; Shemesh, A.; Iraqi, M.; Matalon, O.; Brusilovsky, M.; Hadad, U.; Radinsky, O.; Gershoni-Yahalom, O.; Dye, J.M.; Mandelboim, O.; et al. The Ebola-Glycoprotein Modulates the Function of Natural Killer Cells. Front. Immunol. 2018, 9, 1428. [Google Scholar] [CrossRef]
- Radinsky, O.; Edri, A.; Brusilovsky, M.; Fedida-Metula, S.; Sobarzo, A.; Gershoni-Yahalom, O.; Lutwama, J.; Dye, J.; Lobel, L.; Porgador, A. Sudan ebolavirus long recovered survivors produce GP-specific Abs that are of the IgG1 subclass and preferentially bind FcgammaRI. Sci. Rep. 2017, 7, 6054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golden, J.W.; Cline, C.R.; Zeng, X.; Garrison, A.R.; Carey, B.D.; Mucker, E.M.; White, L.E.; Shamblin, J.D.; Brocato, R.L.; Liu, J.; et al. Human angiotensin-converting enzyme 2 transgenic mice infected with SARS-CoV-2 develop severe and fatal respiratory disease. JCI Insight 2020, 5, e142032. [Google Scholar] [CrossRef]
- Jia, H.; Yue, X.; Lazartigues, E. ACE2 mouse models: A toolbox for cardiovascular and pulmonary research. Nat. Commun. 2020, 11, 5165. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.S.; Bailey, A.L.; Kafai, N.M.; Nair, S.; McCune, B.T.; Yu, J.; Fox, J.M.; Chen, R.E.; Earnest, J.T.; Keeler, S.P.; et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020, 21, 1327–1335. [Google Scholar] [CrossRef]
- Zheng, J.; Wong, L.R.; Li, K.; Verma, A.K.; Ortiz, M.E.; Wohlford-Lenane, C.; Leidinger, M.R.; Knudson, C.M.; Meyerholz, D.K.; McCray, P.B., Jr.; et al. COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature 2021, 589, 603–607. [Google Scholar] [CrossRef] [PubMed]
- McCray, P.B., Jr.; Pewe, L.; Wohlford-Lenane, C.; Hickey, M.; Manzel, L.; Shi, L.; Netland, J.; Jia, H.P.; Halabi, C.; Sigmund, C.D.; et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J. Virol. 2007, 81, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Yinda, C.K.; Port, J.R.; Bushmaker, T.; Offei Owusu, I.; Purushotham, J.N.; Avanzato, V.A.; Fischer, R.J.; Schulz, J.E.; Holbrook, M.G.; Hebner, M.J.; et al. K18-hACE2 mice develop respiratory disease resembling severe COVID-19. PLoS Pathog. 2021, 17, e1009195. [Google Scholar] [CrossRef]
- Andreano, E.; Nicastri, E.; Paciello, I.; Pileri, P.; Manganaro, N.; Piccini, G.; Manenti, A.; Pantano, E.; Kabanova, A.; Troisi, M.; et al. Extremely potent human monoclonal antibodies from COVID-19 convalescent patients. Cell 2021, 184, 1821–1835.e16. [Google Scholar] [CrossRef] [PubMed]
- Oladunni, F.S.; Park, J.G.; Pino, P.A.; Gonzalez, O.; Akhter, A.; Allué-Guardia, A.; Olmo-Fontánez, A.; Gautam, S.; Garcia-Vilanova, A.; Ye, C.; et al. Lethality of SARS-CoV-2 infection in K18 human angiotensin-converting enzyme 2 transgenic mice. Nat. Commun. 2020, 11, 6122. [Google Scholar] [CrossRef]
- Idusogie, E.E.; Wong, P.Y.; Presta, L.G.; Gazzano-Santoro, H.; Totpal, K.; Ultsch, M.; Mulkerrin, M.G. Engineered antibodies with increased activity to recruit complement. J. Immunol. 2001, 166, 2571–2575. [Google Scholar] [CrossRef] [Green Version]
- Lund, J.; Winter, G.; Jones, P.T.; Pound, J.D.; Tanaka, T.; Walker, M.R.; Artymiuk, P.J.; Arata, Y.; Burton, D.R.; Jefferis, R.; et al. Human Fc gamma RI and Fc gamma RII interact with distinct but overlapping sites on human IgG. J. Immunol. 1991, 147, 2657–2662. [Google Scholar]
- Oganesyan, V.; Damschroder, M.M.; Leach, W.; Wu, H.; Dall’Acqua, W.F. Structural characterization of a mutated, ADCC-enhanced human Fc fragment. Mol. Immunol. 2008, 45, 1872–1882. [Google Scholar] [CrossRef]
- Sarmay, G.; Lund, J.; Rozsnyay, Z.; Gergely, J.; Jefferis, R. Mapping and comparison of the interaction sites on the Fc region of IgG responsible for triggering antibody dependent cellular cytotoxicity ADCC) through different types of human Fc gamma receptor. Mol. Immunol. 1992, 29, 633–639. [Google Scholar] [CrossRef]
- Vafa, O.; Gilliland, G.L.; Brezski, R.J.; Strake, B.; Wilkinson, T.; Lacy, E.R.; Scallon, B.; Teplyakov, A.; Malia, T.J.; Strohl, W.R. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods 2014, 65, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Wessels, U.; Poehler, A.; Moheysen-Zadeh, M.; Zadak, M.; Staack, R.F.; Umana, P.; Heinrich, J.; Stubenrauch, K. Detection of antidrug antibodies against human therapeutic antibodies lacking Fc-effector functions by usage of soluble Fcgamma receptor I. Bioanalysis 2016, 8, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Alegre, M.L.; Varga, S.S.; Rothermel, A.L.; Collins, A.M.; Pulito, V.L.; Hanna, L.S.; Dolan, K.P.; Parren, P.W.; Bluestone, J.A.; et al. In vitro characterization of five humanized OKT3 effector function variant antibodies. Cell Immunol. 2000, 200, 16–26. [Google Scholar] [CrossRef]
- Hezareh, M.; Hessell, A.J.; Jensen, R.C.; van de Winkel, J.G.; Parren, P.W. Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J. Virol. 2001, 75, 12161–12168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, M.; Kim, H.S.; Tong, R.K.; Bainbridge, T.W.; Vernes, J.M.; Zhang, Y.; Lin, Y.L.; Chung, S.; Dennis, M.S.; Zuchero, Y.J.; et al. Effector-attenuating Substitutions That Maintain Antibody Stability and Reduce Toxicity in Mice. J. Biol. Chem. 2017, 292, 3900–3908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pincetic, A.; Bournazos, S.; DiLillo, D.J.; Maamary, J.; Wang, T.T.; Dahan, R.; Fiebiger, B.M.; Ravetch, J.V. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat. Immunol. 2014, 15, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Ravetch, J.V. Fcgamma Receptor Function and the Design of Vaccination Strategies. Immunity 2017, 47, 224–233. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Verploegen, S.; Ortiz Buijsse, A.; Vink, T.; Leusen, J.H.; Bleeker, W.K.; Parren, P.W. Crosstalk between human IgG isotypes and murine effector cells. J. Immunol. 2012, 189, 3430–3438. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noy-Porat, T.; Edri, A.; Alcalay, R.; Makdasi, E.; Gur, D.; Aftalion, M.; Evgy, Y.; Beth-Din, A.; Levy, Y.; Epstein, E.; et al. Fc-Independent Protection from SARS-CoV-2 Infection by Recombinant Human Monoclonal Antibodies. Antibodies 2021, 10, 45. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10040045
Noy-Porat T, Edri A, Alcalay R, Makdasi E, Gur D, Aftalion M, Evgy Y, Beth-Din A, Levy Y, Epstein E, et al. Fc-Independent Protection from SARS-CoV-2 Infection by Recombinant Human Monoclonal Antibodies. Antibodies. 2021; 10(4):45. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10040045
Chicago/Turabian StyleNoy-Porat, Tal, Avishay Edri, Ron Alcalay, Efi Makdasi, David Gur, Moshe Aftalion, Yentl Evgy, Adi Beth-Din, Yinon Levy, Eyal Epstein, and et al. 2021. "Fc-Independent Protection from SARS-CoV-2 Infection by Recombinant Human Monoclonal Antibodies" Antibodies 10, no. 4: 45. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10040045