Central Nervous System Delivery of Antibodies and Their Single-Domain Antibodies and Variable Fragment Derivatives with Focus on Intranasal Nose to Brain Administration


Abstract
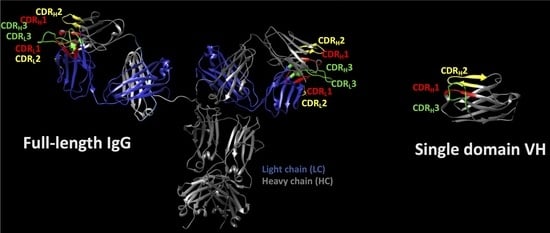
:
{kind=link}
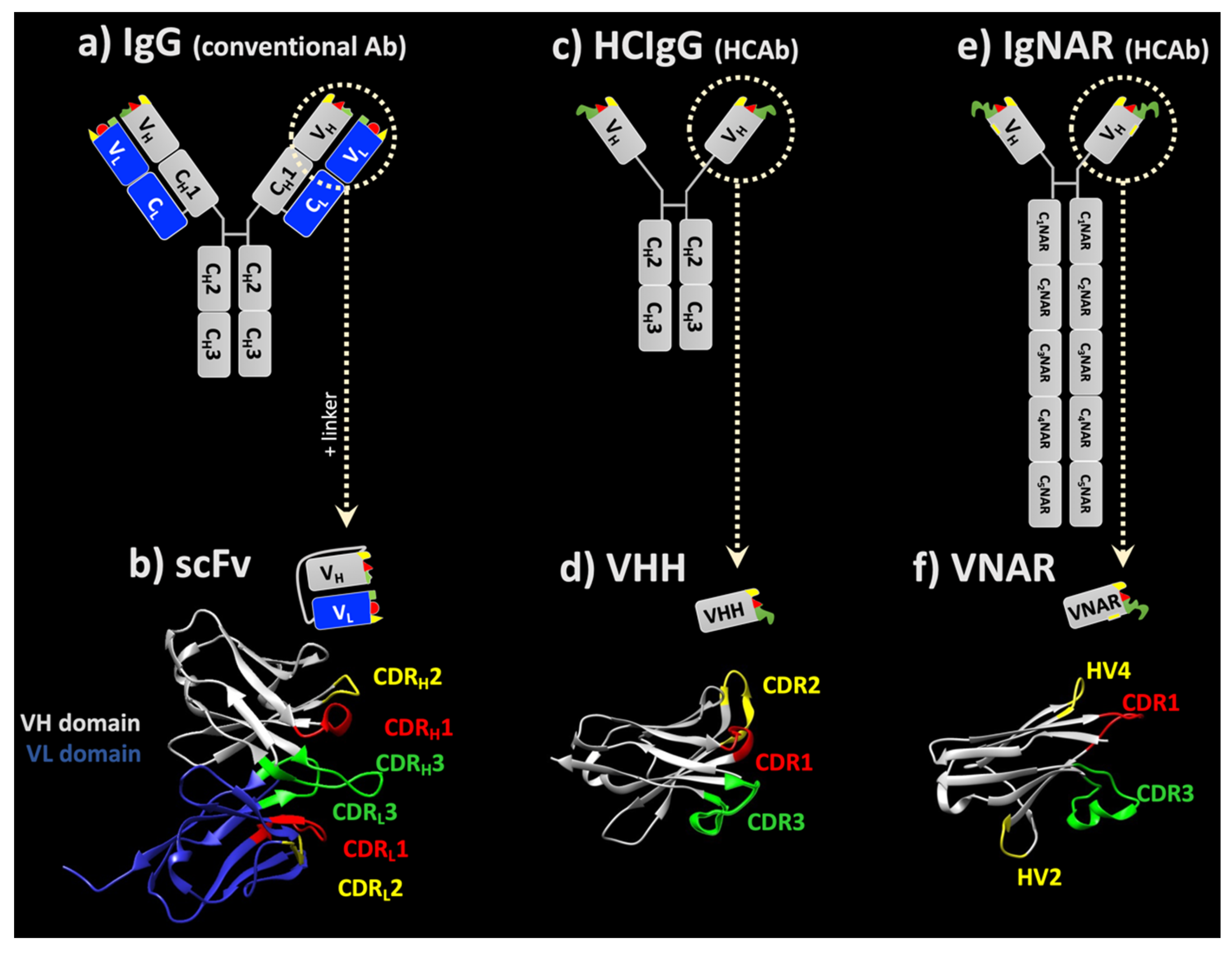{kind=link}
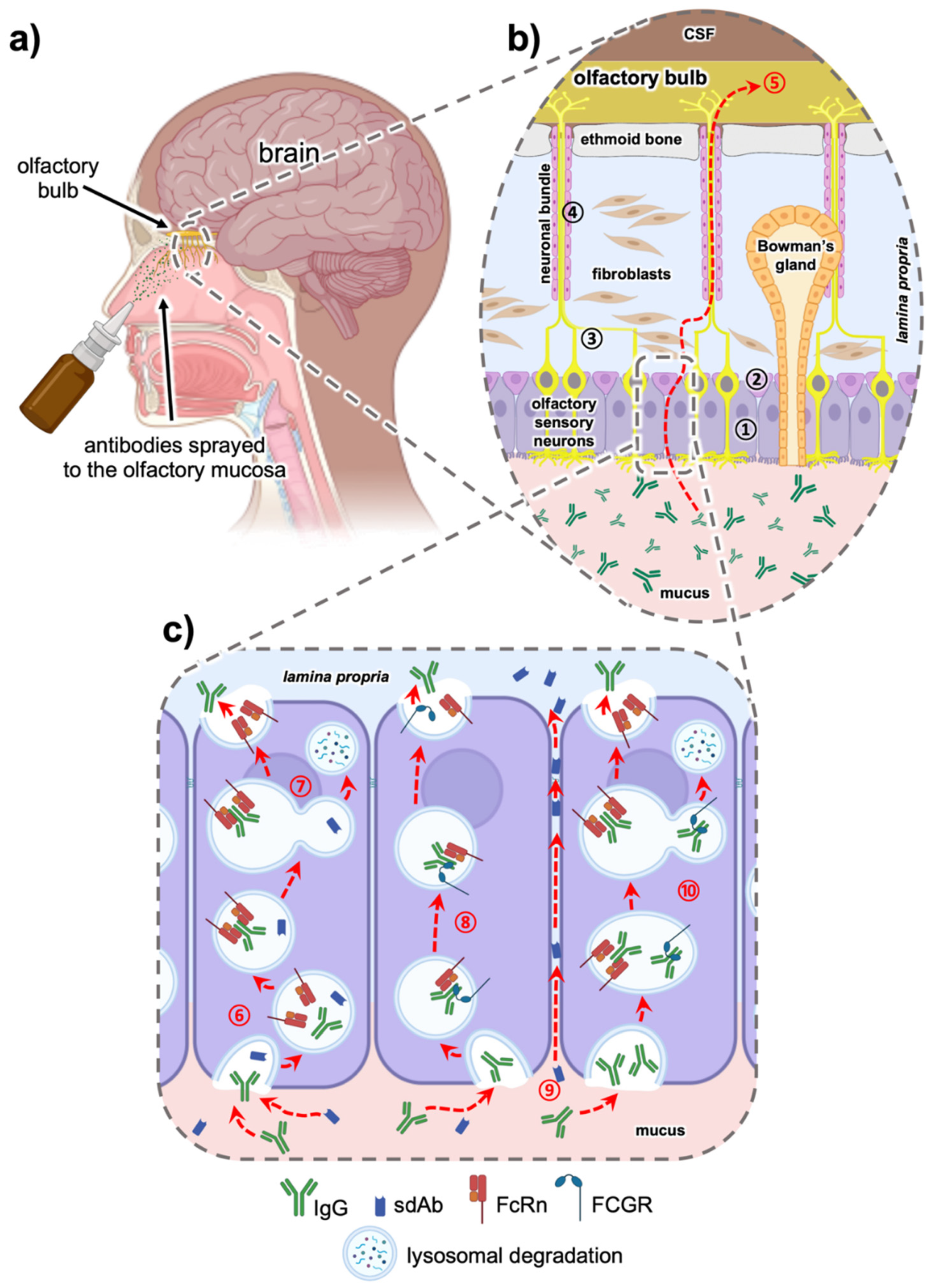{kind=link}
1. General Overview on Immunoglobulin Structures
2. General Considerations Relating to the Distribution and Tissue Penetration of Full-Length IgG Antibodies
2.1. Limitation to the Intravascular Compartment
2.2. Limited Tissue Penetration
3. How to Overcome Biodistribution Limitations of Full-Length IgGs
3.1. ScFv
3.2. Single-Domain Antibodies
3.2.1. sdAbs from Camelids
3.2.2. sdAbs from Sharks
- Type I has two or four Cys residues in CDR3 and two Cys residues in FR2 and FR4. This results in the formation of disulfide bonds between FR2-CDR3 and CDR3-FR4. In addition to increasing the stability of this type of domain VNAR, it is assumed that the extra disulfide bonds also stabilize the structure of the loop–antigen complex [79].
- In Type II (or Type IIa), in addition to a canonical disulfide bond, there is a single Cys residue in CDR3 and also a Cys in CDR1 [80]. These Cys residues form an extra disulfide bond within the molecule, which brings CDR1 and CDR3 loops close to each other, specific to this type of VNAR.
- Type III is the most dominating antibody type in neonatal sharks but is rarely found in adult sharks [81,82]. Type III VNARs have the same disulfide bonds as Type II. Contrary to a Type II VNAR, however, they have less diverse CDR3s and a conserved tryptophan residue in the vicinity of Cys in CDR1 [79].
- Type IV (or Type IIb) VNAR hold only the canonical conserved Cys in their structure. Thus, the topology structure of this antibody domain is more flexible compared to other types of VNAR, except type III. Moreover, another type of IV VNAR which lacks the non-canonical cysteine residues but has an invariant tryptophan residue in CDR1 has been identified. This type of VNAR is referred to as Type IIIb [83,84].

3.3. Human-Derived sdAbs
4. General Considerations on Therapy with Antibodies and Their Derivatives
4.1. Molecular Size and Size of the Paratope—Size Does Matter
4.2. Elimination and Half-Life Are also Important Criteria for Drug Delivery
4.3. Higher Stability Enables Aerosolization and Airway Delivery
5. CNS Drug Delivery with Full-Length IgG, scFv and sdAb
5.1. Intranasal Nose to Brain Delivery
5.2. The Role of the Fc Domain in Intranasal Transmucosal Delivery—Is it a Friend or a Foe?
6. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Adams, G.P.; Weiner, L.M. Monoclonal antibody therapy of cancer. Nat. Biotechnol. 2005, 23, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Caucheteur, D.; Robin, G.; Parez, V.; Martineau, P. Construction of Synthetic Antibody Libraries. In Advanced Structural Safety Studies; Springer: Singapore, 2018; Volume 1827, pp. 93–108. [Google Scholar]
- Pande, J.; Szewczyk, M.M.; Grover, A.K. Phage display: Concept, innovations, applications and future. Biotechnol. Adv. 2010, 28, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Feng, N.; Simanski, S.; Islam, K.; Hynan, L.S.; Kodadek, T.; German, D.C. Antibody biomarker for de novo Parkinson disease: Attempted validation. NPJ Park. Dis. 2018, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Di Pauli, F.; Berger, T. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disorders: Toward a New Spectrum of Inflammatory Demyelinating CNS Disorders? Front. Immunol. 2018, 9, 2753. [Google Scholar] [CrossRef]
- Coban, A.; Ulusoy, C.; Giris, M.; Turan, S.; Turkoglu, R.; Tüzün, E.; Idrisoğlu, H.A. Serum anti-neuronal antibodies in amyotrophic lateral sclerosis. Int. J. Neurosci. 2013, 123, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Moreth, J.; Mavoungou, C.; Schindowski, K.Z. Is abeta a sufficient biomarker for monitoring anti-abeta clinical studies? A critical review. Front. Aging Neurosci. 2013, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Delves, P.J.; Roitt, I.M. The Immune System. N. Engl. J. Med. 2000, 343, 37–49. [Google Scholar] [CrossRef]
- Johnson, G. Kabat Database and its applications: Future directions. Nucleic Acids Res. 2001, 29, 205–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilschmann, N.; Craig, L.C. Amino acid sequence studies with Bence-Jones proteins. Proc. Natl. Acad. Sci. USA 1965, 53, 1403–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.N.; Milstein, C. Structure of Antibody Molecules. Nat. Cell Biol. 1967, 214, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Chailyan, A.; Marcatili, P.; Tramontano, A. The association of heavy and light chain variable domains in antibodies: Implications for antigen specificity. FEBS J. 2011, 278, 2858–2866. [Google Scholar] [CrossRef] [Green Version]
- Ganten, D.; Ruckpaul, K. Immunoglobulin-Fold. In Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine; Springer: Berlin/Heidelberg, Germany, 2006; p. 867. [Google Scholar] [CrossRef]
- Barnard, E.; Timson, D.J. Split-EGFP Screens for the Detection and Localisation of Protein–Protein Interactions in Living Yeast Cells. In Advanced Structural Safety Studies; Springer: Singapore, 2010; Volume 638, pp. 303–317. [Google Scholar]
- David, M.P.C.; Asprer, J.J.T.; Ibana, J.S.A.; Concepcion, G.P.; Padlan, E.A. A study of the structural correlates of affinity maturation: Antibody affinity as a function of chemical interactions, structural plasticity and stability. Mol. Immunol. 2007, 44, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Padlan, E.A. Anatomy of the antibody molecule. Mol. Immunol. 1994, 31, 169–217. [Google Scholar] [CrossRef] [Green Version]
- Vauquelin, G.; Charlton, S. Exploring avidity: Understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. Br. J. Pharmacol. 2013, 168, 1771–1785. [Google Scholar] [CrossRef] [Green Version]
- Padlan, E.A.; Abergel, C.; Tipper, J.P. Identification of specificity-determining residues in antibodies. FASEB J. 1995, 9, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Collis, A.V.; Brouwer, A.; Martin, A.C. Analysis of the Antigen Combining Site: Correlations between Length and Sequence Composition of the Hypervariable Loops and the Nature of the Antigen. J. Mol. Biol. 2003, 325, 337–354. [Google Scholar] [CrossRef]
- Richards, D.A. Exploring alternative antibody scaffolds: Antibody fragments and antibody mimics for targeted drug delivery. Drug Discov. Today Technol. 2018, 30, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, M.; Skerra, A. Engineered protein scaffolds as next-generation antibody therapeutics. Curr. Opin. Chem. Biol. 2009, 13, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Pham, J.V.; Yilma, M.A.; Feliz, A.; Majid, M.T.; Maffetone, N.; Walker, J.R.; Kim, E.; Cho, H.J.; Reynolds, J.M.; Song, M.C.; et al. A Review of the Microbial Production of Bioactive Natural Products and Biologics. Front. Microbiol. 2019, 10, 1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Jeong, K.J. Challenges to production of antibodies in bacteria and yeast. J. Biosci. Bioeng. 2015, 120, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Schneier, M.; Razdan, S.; Miller, A.M.; Briceno, M.E.; Barua, S. Current technologies to endotoxin detection and removal for biopharmaceutical purification. Biotechnol. Bioeng. 2020, 117, 2588–2609. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Bellone, M.; Rumio, C.; Corti, A. Approaches to improve tumor accumulation and interactions between monoclonal antibodies and immune cells. mAbs 2013, 5, 34–46. [Google Scholar] [CrossRef] [Green Version]
- Thurber, G.; Schmidt, M.M.; Wittrup, K.D. Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Adv. Drug Deliv. Rev. 2008, 60, 1421–1434. [Google Scholar] [CrossRef] [Green Version]
- Shin, T.-H.; Sung, E.-S.; Kim, Y.-J.; Kim, K.-S.; Kim, S.-H.; Kim, S.-K.; Lee, Y.-D.; Kim, Y.-S. Enhancement of the Tumor Penetration of Monoclonal Antibody by Fusion of a Neuropilin-Targeting Peptide Improves the Antitumor Efficacy. Mol. Cancer Ther. 2014, 13, 651–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röhm, M.; Carle, S.; Maigler, F.; Flamm, J.; Kramer, V.; Mavoungou, C.; Schmid, O.; Schindowski, K. A comprehensive screening platform for aerosolizable protein formulations for intranasal and pulmonary drug delivery. Int. J. Pharm. 2017, 532, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a Tumor-Penetrating Peptide Enhances the Efficacy of Cancer Drugs. Science 2010, 328, 1031–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omidi, Y.; Barar, J. Impacts of Blood-Brain Barrier in Drug Delivery and Targeting of Brain Tumors. BioImpacts 2012, 2, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Rojko, J.L.; Price-Schiavi, S. Physiologic IgG Biodistribution, Transport, and Clearance: Implications for Monoclonal Antibody Products. In Pharmaceutical Sciences Encyclopedia; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 1–36. [Google Scholar] [CrossRef]
- Giasuddin, A.S.; Shembesh, N.M.; El-Bargathy, S.M.; Kashbur, I.M.; Rao, B.N. Levels of serum immunoglobulin G, CSF IgG and IgG index in acute bacterial meningitis. Br. J. Biomed. Sci. 1998, 55, 253–257. [Google Scholar] [PubMed]
- Pepinsky, R.B.; Shao, Z.; Ji, B.; Wang, Q.; Meng, G.; Walus, L.; Lee, X.; Hu, Y.; Graff, C.; Garber, E.; et al. Exposure Levels of Anti-LINGO-1 Li81 Antibody in the Central Nervous System and Dose-Efficacy Relationships in Rat Spinal Cord Remyelination Models after Systemic Administration. J. Pharmacol. Exp. Ther. 2011, 339, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultqvist, G.; Syvänen, S.; Fang, X.T.; Lannfelt, L.; Sehlin, D. Bivalent Brain Shuttle Increases Antibody Uptake by Monovalent Binding to the Transferrin Receptor. Theranostics 2017, 7, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, J.M.; Shusta, E.V. Targeting Receptor-Mediated Transport for Delivery of Biologics across the Blood-Brain Barrier. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 613–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aderibigbe, B.A.; Naki, T. Design and Efficacy of Nanogels Formulations for Intranasal Administration. Molecules 2018, 23, 1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gänger, S.; Schindowski, K. Tailoring Formulations for Intranasal Nose-to-Brain Delivery: A Review on Architecture, Physico-Chemical Characteristics and Mucociliary Clearance of the Nasal Olfactory Mucosa. Pharmaceutics 2018, 10, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohra, P.K. An in-silico approach to identify avian IgY as potential inhibitor of HIV envelope glycoprotein gp120. Int. Res. J. Eng. Technol. 2017, 4, 1671–1675. [Google Scholar] [CrossRef] [Green Version]
- Xenaki, K.T.; Oliveira, S.; van Bergen en Henegouwen, P.M.P. Antibody or Antibody Fragments: Implications for Molecular Imaging and Targeted Therapy of Solid Tumors. Front. Immunol. 2017, 8, 1287. [Google Scholar] [CrossRef] [PubMed]
- Garg, T.; Singh, O.; Arora, S.; Murthy, R.S.R. Scaffold: A Novel Carrier for Cell and Drug Delivery. Crit. Rev. Ther. Drug Carr. Syst. 2012, 29, 1–63. [Google Scholar] [CrossRef] [Green Version]
- Calori, I.R.; Braga, G.; de Jesus, P.D.C.C.; Bi, H.; Tedesco, A.C. Polymer scaffolds as drug delivery systems. Eur. Polym. J. 2020, 129, 109621. [Google Scholar] [CrossRef]
- Awwad, S.; Angkawinitwong, U. Overview of Antibody Drug Delivery. Pharmaceutics 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrak, K. Essential properties of drug-targeting delivery systems. Drug Discov. Today 2005, 10, 1667–1673. [Google Scholar] [CrossRef]
- Wörn, A.; Plückthun, A. Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 2001, 305, 989–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muyldermans, S.; Ghassabeh, G.H.; Saerens, D. Single-domain antibodies. In Recombinant Antibodies for Immunotherapy, 1st ed.; Little, M., Ed.; Cambridge University Press: Cambridge, UK, 2009; pp. 216–230. [Google Scholar]
- Tsianakas, A.; Brunner, P.M.; Ghoreschi, K.; Berger, C.; Loser, K.; Röcken, M.; Stingl, G.; Luger, T.; Jung, T. The single-chain anti-TNF-αantibody DLX105 induces clinical and biomarker responses upon local administration in patients with chronic plaque-type psoriasis. Exp. Dermatol. 2016, 25, 428–433. [Google Scholar] [CrossRef]
- Nguyen, Q.D.; Das, A.; Do, D.V.; Dugel, P.U.; Gomes, A.; Holz, F.G.; Koh, A.; Pan, C.K.; Sepah, Y.J.; Patel, N.; et al. Brolucizumab: Evolution through Preclinical and Clinical Studies and the Implications for the Management of Neovascular Age-Related Macular Degeneration. Ophthalmology 2020, 127, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Gökbuget, N.; Stein, A.S.; Zugmaier, G.; O’Brien, S.; Bargou, R.C.; Dombret, H.; Fielding, A.K.; Heffner, L.; Larson, R.; et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: A multicentre, single-arm, phase 2 study. Lancet Oncol. 2015, 16, 57–66. [Google Scholar] [CrossRef]
- Kowalski, M.; Guindon, J.; Brazas, L.; Moore, C.; Entwistle, J.; Cizeau, J.; Jewett, M.A.; Macdonald, G.C. A Phase II Study of Oportuzumab Monatox: An Immunotoxin Therapy for Patients with Noninvasive Urothelial Carcinoma In Situ Previously Treated with Bacillus Calmette-Guérin. J. Urol. 2012, 188, 1712–1718. [Google Scholar] [CrossRef]
- Weide, B.; Eigentler, T.; Catania, C.; Ascierto, P.A.; Cascinu, S.; Becker, J.C.; Hauschild, A.; Romanini, A.; Danielli, R.; Dummer, R.; et al. A phase II study of the L19IL2 immunocytokine in combination with dacarbazine in advanced metastatic melanoma patients. Cancer Immunol. Immunother. 2019, 68, 1547–1559. [Google Scholar] [CrossRef]
- Albinger, N.; Hartmann, J.; Ullrich, E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 2021, 28, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Conrath, K.E.; Lauwereys, M.; Wyns, L.; Muyldermans, S. Camel Single-domain Antibodies as Modular Building Units in Bispecific and Bivalent Antibody Constructs. J. Biol. Chem. 2001, 276, 7346–7350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.-H.; Pardon, E.; Menzer, L.; De Genst, E.; Kumita, J.R.; Christodoulou, J.; Saerens, D.; Brans, A.; Bouillenne, F.; Archer, D.B.; et al. Engineering a Camelid Antibody Fragment That Binds to the Active Site of Human Lysozyme and Inhibits Its Conversion into Amyloid Fibrils. Biochemistry 2008, 47, 11041–11054. [Google Scholar] [CrossRef]
- Ghahroudi, M.A.; Desmyter, A.; Wyns, L.; Hamers, R.; Muyldermans, S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997, 414, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Jovcevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef] [Green Version]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar] [CrossRef] [Green Version]
- Zadeh, A.S.; Grässer, A.; Dinter, H.; Hermes, M.; Schindowski, K. Efficient Construction and Effective Screening of Synthetic Domain Antibody Libraries. Methods Protoc. 2019, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Sargentini-Maier, M.L.; De Decker, P.; Tersteeg, C.; Canvin, J.; Callewaert, F.; De Winter, H. Clinical pharmacology of caplacizumab for the treatment of patients with acquired thrombotic thrombocytopenic purpura. Expert Rev. Clin. Pharmacol. 2019, 12, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C. Nanobody approval gives domain antibodies a boost. Nat. Rev. Drug Discov. 2019, 18, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Duggan, S. Correction to: Caplacizumab: First Global Approval. Drugs 2018, 78, 1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackaert, C.; Smiejkowska, N.; Xavier, C.; Sterckx, Y.G.J.; Denies, S.; Stijlemans, B.; Elkrim, Y.; Devoogdt, N.; Caveliers, V.; Lahoutte, T.; et al. Immunogenicity Risk Profile of Nanobodies. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Avila, D.; Hughes, M.; Hughes, A.; McKinney, E.C.; Flajnik, M.F. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nat. Cell Biol. 1995, 374, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, S.D.; Humberstone, K.S.; Krishnan, U.V.; Carmichael, J.A.; Doughty, L.; Hattarki, M.; Coley, A.M.; Casey, J.L.; Anders, R.F.; Foley, M.; et al. Selection and affinity maturation of IgNAR variable domains targeting Plasmodium falciparum AMA. Proteins Struct. Funct. Bioinform. 2004, 55, 187–197. [Google Scholar] [CrossRef]
- Dooley, H.; Stanfield, R.L.; Brady, R.A.; Flajnik, M.F. First molecular and biochemical analysis of in vivo affinity maturation in an ectothermic vertebrate. Proc. Natl. Acad. Sci. USA 2006, 103, 1846–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.L.; Anderson, G.; Delehanty, J.B.; Baumann, R.; Hayhurst, A.; Goldman, E.R. Selection of cholera toxin specific IgNAR single-domain antibodies from a naïve shark library. Mol. Immunol. 2007, 44, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Crouch, K.; Smith, L.E.; Williams, R.; Cao, W.; Lee, M.; Jensen, A.; Dooley, H. Humoral immune response of the small-spotted catshark, Scyliorhinus canicula. Fish Shellfish. Immunol. 2013, 34, 1158–1169. [Google Scholar] [CrossRef]
- Zielonka, S.; Weber, N.; Becker, S.; Doerner, A.; Christmann, A.; Christmann, C.; Uth, C.; Fritz, J.; Schäfer, E.; Steinmann, B.; et al. Shark Attack: High affinity binding proteins derived from shark vNAR domains by stepwise in vitro affinity maturation. J. Biotechnol. 2014, 191, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Flajnik, M.F.; Deschacht, N.; Muyldermans, S. A Case Of Convergence: Why Did a Simple Alternative to Canonical Antibodies Arise in Sharks and Camels? PLoS Biol. 2011, 9, e1001120. [Google Scholar] [CrossRef]
- Roux, K.H.; Greenberg, A.S.; Greene, L.; Strelets, L.; Avila, D.; McKinney, E.C.; Flajnik, M.F. Structural analysis of the nurse shark (new) antigen receptor (NAR): Molecular convergence of NAR and unusual mammalian immunoglobulins. Proc. Natl. Acad. Sci. USA 1998, 95, 11804–11809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielonka, S.; Empting, M.; Grzeschik, J.; Könning, D.; Barelle, C.J.; Kolmar, H. Structural insights and biomedical potential of IgNAR scaffolds from sharks. mAbs 2015, 7, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, H. Selection and characterization of naturally occurring single-domain (IgNAR) antibody fragments from immunized sharks by phage display. Mol. Immunol. 2003, 40, 25–33. [Google Scholar] [CrossRef]
- Nuttall, S.D. Overview and Discovery of IgNARs and Generation of VNARs. In Springer Protocols Handbooks; Springer: Singapore, 2012; Volume 911, pp. 27–36. [Google Scholar]
- Juma, S.; Gong, X.; Hu, S.; Lv, Z.; Shao, J.; Liu, L.; Chen, G. Shark New Antigen Receptor (IgNAR): Structure, Characteristics and Potential Biomedical Applications. Cells 2021, 10, 1140. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, O.V.; Olland, A.; Piché-Nicholas, N.; Godbole, A.; King, D.; Svenson, K.; Calabro, V.; Müller, M.R.; Barelle, C.J.; Somers, W.; et al. Atypical Antigen Recognition Mode of a Shark Immunoglobulin New Antigen Receptor (IgNAR) Variable Domain Characterized by Humanization and Structural Analysis. J. Biol. Chem. 2013, 288, 17408–17419. [Google Scholar] [CrossRef] [Green Version]
- Streltsov, V.A.; Carmichael, J.A.; Nuttall, S.D. Structure of a shark IgNAR antibody variable domain and modeling of an early-developmental isotype. Protein Sci. 2005, 14, 2901–2909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fennell, B.; Darmanin-Sheehan, A.; Hufton, S.; Calabro, V.; Wu, L.; Müller, M.; Cao, W.; Gill, D.; Cunningham, O.; Finlay, W. Dissection of the IgNAR V Domain: Molecular Scanning and Orthologue Database Mining Define Novel IgNAR Hallmarks and Affinity Maturation Mechanisms. J. Mol. Biol. 2010, 400, 155–170. [Google Scholar] [CrossRef]
- Diaz, M.; Stanfield, R.L.; Greenberg, A.S.; Flajnik, M.F. Structural analysis, selection, and ontogeny of the shark new antigen receptor (IgNAR): Identification of a new locus preferentially expressed in early development. Immunogenetics 2002, 54, 501–512. [Google Scholar] [CrossRef]
- Shao, C.-Y.; Secombes, C.J.; Porter, A.J. Rapid isolation of IgNAR variable single-domain antibody fragments from a shark synthetic library. Mol. Immunol. 2007, 44, 656–665. [Google Scholar] [CrossRef]
- Rumfelt, L.L.; Diaz, M.; Lohr, R.L.; Mochon, E.; Flajnik, M.F. Unprecedented Multiplicity of Ig Transmembrane and Secretory mRNA Forms in the Cartilaginous Fish. J. Immunol. 2004, 173, 1129–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovaleva, M.; Ferguson, L.; Steven, J.; Porter, A.; Barelle, C. Shark variable new antigen receptor biologics—A novel technology platform for therapeutic drug development. Expert Opin. Biol. Ther. 2014, 14, 1527–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, W.S.; Leow, C.Y.; Majeed, A.B.A. Diagnostic and therapeutic potential of shark variable new antigen receptor (VNAR) single domain antibody. Int. J. Biol. Macromol. 2020, 147, 369–375. [Google Scholar] [CrossRef]
- Stanfield, R.L.; Dooley, H.; Flajnik, M.F.; Wilson, I.A. Crystal Structure of a Shark Single-Domain Antibody V Region in Complex with Lysozyme. Science 2004, 305, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, R.L.; Dooley, H.; Verdino, P.; Flajnik, M.F.; Wilson, I.A. Maturation of Shark Single-domain (IgNAR) Antibodies: Evidence for Induced-fit Binding. J. Mol. Biol. 2007, 367, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.R.; Saunders, K.; Grace, C.; Jin, M.; Piche-Nicholas, N.; Steven, J.; O’Dwyer, R.; Wu, L.; Khetemenee, L.; Vugmeyster, Y.; et al. Improving the pharmacokinetic properties of biologics by fusion to an anti-HSA shark VNAR domain. mAbs 2012, 4, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Nuttall, S.D.; Krishnan, U.V.; Hattarki, M.; De Gori, R.; Irving, R.A.; Hudson, P.J. Isolation of the new antigen receptor from wobbegong sharks, and use as a scaffold for the display of protein loop libraries. Mol. Immunol. 2001, 38, 313–326. [Google Scholar] [CrossRef]
- Ubah, O.C.; Buschhaus, M.J.; Ferguson, L.; Kovaleva, M.; Steven, J.; Porter, A.J.; Barelle, C.J. Next-generation flexible formats of VNAR domains expand the drug platform’s utility and developability. Biochem. Soc. Trans. 2018, 46, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, C.J.; Koglin, M.; Olson, W.C.; Marshall, F.H. Opportunities for therapeutic antibodies directed at G-protein-coupled receptors. Nat. Rev. Drug Discov. 2017, 16, 787–810. [Google Scholar] [CrossRef] [PubMed]
- Keown, A. These 2 Biotechs’ Shark Antibodies Have Caught Amgen’s Eye|BioSpace. Available online: https://www.biospace.com/article/these-2-biotechs-shark-antibodies-have-caught-amgen-s-eye-/ (accessed on 23 August 2021).
- Griffiths, K.; Habiel, D.M.; Jaffar, J.; Binder, U.; Darby, W.G.; Hosking, C.G.; Skerra, A.; Westall, G.; Hogaboam, C.M.; Foley, M. Anti-fibrotic Effects of CXCR4-Targeting i-body AD-114 in Preclinical Models of Pulmonary Fibrosis. Sci. Rep. 2018, 8, 3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjerregaard-Andersen, K.; Heggelund, J.E.; Krengel, U. 6S2I Anti-tumor antibody 14F7-derived scFv in complex with NeuGc Gm3. 2019. Available online: https://www.rcsb.org/structure/6S2I (accessed on 1 November 2021).
- Spinelli, S.; Tegoni, M.; Frenken, L.; van Vliet, C.; Cambillau, C. Lateral recognition of a dye hapten by a llama VHH domain. J. Mol. Biol. 2001, 311, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, S.; Miao, Z.; Liu, Y.; Liu, X.; Xiao, Z.; Cao, Y. AbRSA: A robust tool for antibody numbering. Protein Sci. 2019, 28, 1524–1531. [Google Scholar] [CrossRef]
- Davies, J.; Riechmann, L. Antibody VH Domains as Small Recognition Units. Nat. Biotechnol. 1995, 13, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Reiter, Y.; Schuck, P.; Boyd, L.F.; Plaksin, D. An antibody single-domain phage display library of a native heavy chain variable region: Isolation of functional single-domain VH molecules with a unique interface. J. Mol. Biol. 1999, 290, 685–698. [Google Scholar] [CrossRef]
- Tomlinson, I.M.; Winter, G. Method to Screen Phage Display Libraries with Different Ligands. U.S. Patent US6846634B1, 25 January 2005. Available online: https://patents.google.com/patent/US6846634B1/en (accessed on 1 November 2021).
- Barthelemy, P.A.; Raab, H.; Appleton, B.; Bond, C.J.; Wu, P.; Wiesmann, C.; Sidhu, S.S. Comprehensive Analysis of the Factors Contributing to the Stability and Solubility of Autonomous Human VH Domains. J. Biol. Chem. 2008, 283, 3639–3654. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; To, R.; Kandalaft, H.; Ding, W.; Van Faassen, H.; Luo, Y.; Schrag, J.D.; St-Amant, N.; Hefford, M.; Hirama, T.; et al. Antibody light chain variable domains and their biophysically improved versions for human immunotherapy. mAbs 2013, 6, 219–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, K.A.; Kandalaft, H.; Lowden, M.J.; Rossotti, M.A.; van Faassen, H.; Hussack, G.; Durocher, Y.; Kim, D.Y.; Tanha, J. A disulfide-stabilized human V L single-domain antibody library is a source of soluble and highly thermostable binders. Mol. Immunol. 2017, 90, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Henry, K.A.; Tanha, J. Performance evaluation of phage-displayed synthetic human single-domain antibody libraries: A retrospective analysis. J. Immunol. Methods 2018, 456, 81–86. [Google Scholar] [CrossRef]
- Su, N.C.; Smith, Z.P.; Freeman, B.D.; Urban, J.J. Size-Dependent Permeability Deviations from Maxwell’s Model in Hybrid Cross-Linked Poly(ethylene glycol)/Silica Nanoparticle Membranes. Chem. Mater. 2015, 27, 2421–2429. [Google Scholar] [CrossRef]
- Finlay, W.J.J.; Almagro, J.C. Natural and man-made V-gene repertoires for antibody discovery. Front. Immunol. 2012, 3, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soleimanizadeh, A.; Flamm, J.; Ladel, S.; Dinter, H.; Grässer, A.; Lang, M.; Schindowski, K. Can sharks be helpful in reaching the CNS? An invitro permeability analysis of shark domain antibodies (vNARs) through the nasal RPMI cell line. In Proceedings of the PEGS Europe|Protein & Antibody Engineering Summit, Lisbon, Portugal, 18–22 November 2019. [Google Scholar]
- Soleimanizadeh, A.; Martineau, P.; Schindowski, K. A universal design of fully synthetic shark-human antibody. In Naunyn-Schmiedeberg’s Archives of Pharmacology; Spring: New York, NY, USA, 2018; p. P72. [Google Scholar]
- Mitchell, L.S.; Colwell, L.J. Analysis of nanobody paratopes reveals greater diversity than classical antibodies. Protein Eng. Des. Sel. 2018, 31, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundberg, E.J.; Mariuzza, R.A. Molecular recognition in antibody-antigen complexes. Adv. Protein Chem. 2002, 61, 119–160. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmyter, A.; Spinelli, S.; Roussel, A.; Cambillau, C. Camelid nanobodies: Killing two birds with one stone. Curr. Opin. Struct. Biol. 2015, 32, 1–8. [Google Scholar] [CrossRef]
- Beghein, E.; Gettemans, J. Nanobody Technology: A Versatile Toolkit for Microscopic Imaging, Protein–Protein Interaction Analysis, and Protein Function Exploration. Front. Immunol. 2017, 8, 771. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Nuttall, S.; Revill, P.; Colledge, D.; Cabuang, L.; Soppe, S.; Dolezal, O.; Griffiths, K.; Bartholomeusz, A.; Locarnini, S. Targeting the hepatitis B virus precore antigen with a novel IgNAR single variable domain intrabody. Virology 2011, 411, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Fridy, P.; Li, Y.; Keegan, S.; Thompson, M.K.; Nudelman, I.; Scheid, J.F.; Oeffinger, M.; Nussenzweig, M.C.; Fenyö, D.; Chait, B.T.; et al. A robust pipeline for rapid production of versatile nanobody repertoires. Nat. Methods 2014, 11, 1253–1260. [Google Scholar] [CrossRef] [Green Version]
- Maccallum, R.M.; Martin, A.; Thornton, J. Antibody-antigen Interactions: Contact Analysis and Binding Site Topography. J. Mol. Biol. 1996, 262, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Kirchhofer, A.; Helma, J.; Schmidthals, K.; Frauer, C.; Cui, S.; Karcher, A.; Pellis, M.; Muyldermans, S.; Casas-Delucchi, C.S.; Cardoso, M.C.; et al. Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 2009, 17, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.H.; Rodriguez, C.; Remacle, A.G.; Strongin, A.Y.; Ge, X. Active-site MMP-selective antibody inhibitors discovered from convex paratope synthetic libraries. Proc. Natl. Acad. Sci. USA 2016, 113, 14970–14975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, K.A.; MacKenzie, C.R. Antigen recognition by single-domain antibodies: Structural latitudes and constraints. mAbs 2018, 10, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Ubah, O.C.; Steven, J.; Kovaleva, M.; Ferguson, L.; Barelle, C.; Porter, A.J.R.; Barelle, C.J. Novel, Anti-hTNF-α Variable New Antigen Receptor Formats with Enhanced Neutralizing Potency and Multifunctionality, Generated for Therapeutic Development. Front. Immunol. 2017, 8, 1780. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.; Duarte, J.N.; Ling, J.; Li, Z.; Guzman, J.S.; Ploegh, H.L. Structurally Defined αMHC-II Nanobody-Drug Conjugates: A Therapeutic and Imaging System for B-Cell Lymphoma. Angew. Chem. Int. Ed. 2016, 55, 2416–2420. [Google Scholar] [CrossRef] [PubMed]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies as Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef] [PubMed]
- Ben-Fillippo, K.; Krippendorff, B.-F.; Shah, D.K. Influence of Molecular size on the clearance of antibody fragments. Pharm. Res. 2017, 34, 2131–2141. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Shah, D.K. Pharmacokinetic and pharmacodynamic considerations for the next generation protein therapeutics. J. Pharmacokinet. Pharmacodyn. 2015, 42, 553–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uenaka, K.; Nakano, M.; Willis, B.; Friedrich, S.; Ferguson-Sells, L.; Dean, R.A.; Ieiri, I.; Siemers, E.R. Comparison of Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of the Amyloid β Monoclonal Antibody Solanezumab in Japanese and White Patients With Mild to Moderate Alzheimer Disease. Clin. Neuropharmacol. 2012, 35, 25–29. [Google Scholar] [CrossRef]
- Kubetzko, S.; Sarkar, C.A.; Plückthun, A. Protein PEGylation Decreases Observed Target Association Rates via a Dual Blocking Mechanism. Mol. Pharmacol. 2005, 68, 1439–1454. [Google Scholar] [CrossRef] [Green Version]
- Schlapschy, M.; Binder, U.; Börger, C.; Theobald, I.; Wachinger, K.; Kisling, S.; Haller, D.; Skerra, A. PASylation: A biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng. Des. Sel. 2013, 26, 489–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stork, R.; Zettlitz, K.A.; Müller, D.; Rether, M.; Hanisch, F.-G.; Kontermann, R.E. N-Glycosylation as Novel Strategy to Improve Pharmacokinetic Properties of Bispecific Single-chain Diabodies. J. Biol. Chem. 2008, 283, 7804–7812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebner, R.; Mathaes, R.; Meyer, M.; Hey, T.; Winter, G.; Besheer, A. Protein HESylation for half-life extension: Synthesis, characterization and pharmacokinetics of HESylated anakinra. Eur. J. Pharm. Biopharm. 2014, 87, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E. Half-life extended biotherapeutics. Expert Opin. Biol. Ther. 2016, 16, 903–915. [Google Scholar] [CrossRef]
- García-Quintanilla, L.; Luaces-Rodríguez, A.; Gil-Martínez, M.; Mondelo-García, C.; Maroñas, O.; Mangas-Sanjuan, V.; González-Barcia, M.; Zarra-Ferro, I.; Aguiar, P.; Otero-Espinar, F.J.; et al. Pharmacokinetics of Intravitreal Anti-VEGF Drugs in Age-Related Macular Degeneration. Pharmaceutics 2019, 11, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, C.; Liao, T.; Luz, A.; Rafique, A.; Patel, K.; Sun, D.; Liu, Y.; Cao, J. Intravitreal Half-lives of Aflibercept and Brolucizumab in Rabbit Measured Using In Vivo Fluorophotometry. Investig. Ophthalmol. Vis. Sci. 2020, 61, 4926. [Google Scholar]
- Bodier-Montagutelli, E.; Mayor, A.; Vecellio, L.; Respaud, R.; Heuzé-Vourc’H, N. Designing inhaled protein therapeutics for topical lung delivery: What are the next steps? Expert Opin. Drug Deliv. 2018, 15, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Sécher, T.; Mayor, A.; Heuzé-Vourc’H, N. Inhalation of Immuno-Therapeutics/-Prophylactics to Fight Respiratory Tract Infections: An Appropriate Drug at the Right Place! Front. Immunol. 2019, 10, 2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yep, A.T.; Takeuchi, Y.; Engelhardt, O.; Hufton, S. Broad Reactivity Single Domain Antibodies against Influenza Virus and Their Applications to Vaccine Potency Testing and Immunotherapy. Biomolecules 2021, 11, 407. [Google Scholar] [CrossRef]
- Schoof, M.; Faust, B.; Saunders, R.A.; Sangwan, S.; Rezelj, V.; Hoppe, N.; Boone, M.; Billesbølle, C.B.; Zimanyi, M.; Deshpande, I.; et al. An ultra-high affinity synthetic nanobody blocks SARS-CoV-2 infection by locking Spike into an inactive conformation. bioRxiv 2020, 370, 1473–1479. [Google Scholar] [CrossRef]
- Koenig, P.-A.; Das, H.; Liu, H.; Kümmerer, B.M.; Gohr, F.N.; Jenster, L.-M.; Schiffelers, L.D.J.; Tesfamariam, Y.M.; Uchima, M.; Wuerth, J.D.; et al. Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science 2021, 371. [Google Scholar] [CrossRef]
- Sasisekharan, R. Preparing for the Future—Nanobodies for COVID-19? N. Engl. J. Med. 2021, 384, 1568–1571. [Google Scholar] [CrossRef]
- Wu, S.; Huang, J.; Zhang, Z.; Wu, J.; Zhang, J.; Hu, H.; Zhu, T.; Zhang, J.; Luo, L.; Fan, P.; et al. Safety, tolerability, and immunogenicity of an aerosolised adenovirus type-5 vector-based COVID-19 vaccine (Ad5-nCoV) in adults: Preliminary report of an open-label and randomised phase 1 clinical trial. Lancet Infect. Dis. 2021, 21, 1654–1664. [Google Scholar] [CrossRef]
- Halwe, S.; Kupke, A.; Vanshylla, K.; Liberta, F.; Gruell, H.; Zehner, M.; Rohde, C.; Krähling, V.; Serra, M.G.; Kreer, C.; et al. Intranasal Administration of a Monoclonal Neutralizing Antibody Protects Mice against SARS-CoV-2 Infection. Viruses 2021, 13, 1498. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. Developing drugs that can cross the blood-brain barrier: Applications to Alzheimer’s disease. BMC Neurosci. 2008, 9, S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrov, D.S. Therapeutic antibodies, vaccines and antibodyomes. mAbs 2010, 2, 347–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Vandesquille, M.; Koukouli, F.; Dudeffant, C.; Youssef, I.; Lenormand, P.; Ganneau, C.; Maskos, U.; Czech, C.; Grueninger, F.; et al. Camelid single-domain antibodies: A versatile tool for in vivo imaging of extracellular and intracellular brain targets. J. Control. Release 2016, 243, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-López, E.; Schuhmacher, A. Transportation of Single-Domain Antibodies through the Blood–Brain Barrier. Biomolecules 2021, 11, 1131. [Google Scholar] [CrossRef]
- Pothin, E.; Lesuisse, D.; Lafaye, P. Brain Delivery of Single-Domain Antibodies: A Focus on VHH and VNAR. Pharmaceutics 2020, 12, 937. [Google Scholar] [CrossRef]
- Hersh, D.; Wadajkar, A.S.; Roberts, N.B.; Perez, J.G.; Connolly, N.P.; Frenkel, V.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Evolving Drug Delivery Strategies to Overcome the Blood Brain Barrier. Curr. Pharm. Des. 2016, 22, 1177–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Bourgeois, J.; Celli, S.; Glacial, F.; Le Sourd, A.; Mecheri, S.; Weksler, B.; Romero, I.; Couraud, P.; Rougeon, F.; et al. Cell-penetrating anti-GFAP VHH and corresponding fluorescent fusion protein VHH-GFP spontaneously cross the blood-brain barrier and specifically recognize astrocytes: Application to brain imaging. FASEB J. 2012, 26, 3969–3979. [Google Scholar] [CrossRef]
- Miyashita, S.-I.; Zhang, J.; Zhang, S.; Shoemaker, C.B.; Dong, M. Delivery of single-domain antibodies into neurons using a chimeric toxin–based platform is therapeutic in mouse models of botulism. Sci. Transl. Med. 2021, 13, 4197. [Google Scholar] [CrossRef]
- Wei, X.; Chen, X.; Ying, M.; Lu, W. Brain tumor-targeted drug delivery strategies. Acta Pharm. Sin. B 2014, 4, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boado, R.J. A new generation of neurobiological drugs engineered to overcome the challenges of brain drug delivery. Drug News Perspect. 2008, 21, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, K.; Knobloch, T.; Kreuter, J. Targeting the insulin receptor: Nanoparticles for drug delivery across the blood–brain barrier (BBB). J. Drug Target. 2010, 19, 125–132. [Google Scholar] [CrossRef]
- Rhea, E.M.; Rask-Madsen, C.; Banks, W.A. Insulin transport across the blood-brain barrier can occur independently of the insulin receptor. J. Physiol. 2018, 596, 4753–4765. [Google Scholar] [CrossRef]
- Wong, K.H.; Riaz, M.K.; Xie, Y.; Zhang, X.; Liu, Q.; Chen, H.; Bian, Z.; Chen, X.; Lu, A.; Yang, Z. Review of Current Strategies for Delivering Alzheimer’s Disease Drugs across the Blood-Brain Barrier. Int. J. Mol. Sci. 2019, 20, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boado, R.J.; Zhang, Y.; Zhang, Y.; Pardridge, W.M. Humanization of anti-human insulin receptor antibody for drug targeting across the human blood–brain barrier. Biotechnol. Bioeng. 2007, 96, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Stanimirovic, D.B.; Kemmerich, K.; Haqqani, A.S.; Sulea, T.; Arbabi-Ghahroudi, M.; Massie, B.; Gilbert, R. Insulin-like Growth Factor 1 Receptor-Specific Antibodies and Uses Thereof. U.S. Patent US20170015748A1, 19 January 2017. [Google Scholar]
- Muruganandam, A.; Tanha, J.; Narang, S.; Stanimirovic, D. Selection of phage-displayed llama single-domain antibodies that transmigrate across human blood-brain barrier endothelium. FASEB J. 2002, 16, 1–22. [Google Scholar] [CrossRef]
- Abulrob, A.; Sprong, H.; Henegouwen, P.V.B.E.; Stanimirovic, D. The blood-brain barrier transmigrating single domain antibody: Mechanisms of transport and antigenic epitopes in human brain endothelial cells. J. Neurochem. 2005, 95, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Stanton, D. Lundbeck Banks on Shark Ab Delivery Tech to Cross Blood-Brain Barrier. Available online: https://www.biopharma-reporter.com/Article/2017/01/27/Lundbeck-banks-on-shark-Ab-delivery-tech-to-cross-blood-brain-barrier?utm_source=copyright&utm_medium=OnSite&utm_campaign=copyright (accessed on 23 August 2021).
- Glassman, P.M.; Walsh, L.R.; Villa, C.H.; Marcos-Contreras, O.A.; Hood, E.D.; Muzykantov, V.R.; Greineder, C.F. Molecularly Engineered Nanobodies for Tunable Pharmacokinetics and Drug Delivery. Bioconjugate Chem. 2020, 31, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Rotman, M.; Welling, M.; Bunschoten, A.; de Backer, M.E.; Rip, J.; Nabuurs, R.J.; Gaillard, P.J.; van Buchem, M.A.; van der Maarel, S.; van der Weerd, L. Enhanced glutathione PEGylated liposomal brain delivery of an anti-amyloid single domain antibody fragment in a mouse model for Alzheimer’s disease. J. Control. Release 2015, 203, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Ladel, S.; Flamm, J.; Zadeh, A.S.; Filzwieser, D.; Walter, J.-C.; Schlossbauer, P.; Kinscherf, R.; Lischka, K.; Luksch, H.; Schindowski, K. Allogenic Fc Domain-Facilitated Uptake of IgG in Nasal Lamina Propria: Friend or Foe for Intranasal CNS Delivery? Pharmaceutics 2018, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Ladel, S.; Maigler, F.; Flamm, J.; Schlossbauer, P.; Handl, A.; Hermann, R.; Herzog, H.; Hummel, T.; Mizaikoff, B.; Schindowski, K. Impact of Glycosylation and Species Origin on the Uptake and Permeation of IgGs through the Nasal Airway Mucosa. Pharmaceutics 2020, 12, 1014. [Google Scholar] [CrossRef] [PubMed]
- Erdő, F.; Bors, L.A.; Farkas, D.; Bajza, Á.; Gizurarson, S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Bernocchi, B.; Carpentier, R.; Lantier, I.; Ducournau, C.; Dimier-Poisson, I.; Betbeder, D. Mechanisms allowing protein delivery in nasal mucosa using NPL nanoparticles. J. Control. Release 2016, 232, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Pardeshi, C.V.; Belgamwar, V.S. Direct nose to brain drug deliveryviaintegrated nerve pathways bypassing the blood–brain barrier: An excellent platform for brain targeting. Expert Opin. Drug Deliv. 2013, 10, 957–972. [Google Scholar] [CrossRef]
- Ghandehari, H.; Smith, P.L.; Ellens, H.; Yeh, P.Y.; Kopecek, J. Size-dependent permeability of hydrophilic probes across rabbit colonic epithelium. J. Pharmacol. Exp. Ther. 1997, 280, 747–753. [Google Scholar]
- Renner, D.B.; Svitak, A.L.; Gallus, N.J.; Ericson, M.E.; Frey, W.H.; Hanson, L.R. Intranasal delivery of insulin via the olfactory nerve pathway. J. Pharm. Pharmacol. 2012, 64, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- Furrer, E.; Hulmann, V.; Urech, D.M. Intranasal delivery of ESBA105, a TNF-alpha-inhibitory scFv antibody fragment to the brain. J. Neuroimmunol. 2009, 215, 65–72. [Google Scholar] [CrossRef]
- Gomes, J.R.; Cabrito, I.; Soares, H.R.; Costelha, S.; Teixeira, A.; Wittelsberger, A.; Stortelers, C.; Vanlandschoot, P.; Saraiva, M.J. Delivery of an anti-transthyretin Nanobody to the brain through intranasal administration reveals transthyretin expression and secretion by motor neurons. J. Neurochem. 2018, 145, 393–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freiherr, J.; Hallschmid, M.; Frey, W.H.; Brünner, Y.F.; Chapman, C.D.; Hölscher, C.; Craft, S.; De Felice, F.G.; Benedict, C. Intranasal Insulin as a Treatment for Alzheimer’s Disease: A Review of Basic Research and Clinical Evidence. CNS Drugs 2013, 27, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Benedict, C.; Hallschmid, M.; Schmitz, K.; Schultes, B.; Ratter, F.; Fehm, H.L.; Born, J.; Kern, W. Intranasal Insulin Improves Memory in Humans: Superiority of Insulin Aspart. Neuropsychopharmacol. 2006, 32, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kullmann, S.; Heni, M.; Veit, R.; Scheffler, K.; Machann, J.; Häring, H.-U.; Fritsche, A.; Preissl, H. Intranasal insulin enhances brain functional connectivity mediating the relationship between adiposity and subjective feeling of hunger. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Kumar, N.N.; Lochhead, J.; Pizzo, M.; Nehra, G.; Boroumand, S.; Greene, G.; Thorne, R.G. Delivery of immunoglobulin G antibodies to the rat nervous system following intranasal administration: Distribution, dose-response, and mechanisms of delivery. J. Control. Release 2018, 286, 467–484. [Google Scholar] [CrossRef]
- Bourgeois, C.; Bour, J.B.; Aho, L.S.; Pothier, P. Prophylactic Administration of a Complementarity-Determining Region Derived from a Neutralizing Monoclonal Antibody Is Effective against Respiratory Syncytial Virus Infection in BALB/c Mice. J. Virol. 1998, 72, 807–810. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Davis, F.J.; Chauhan, B.C.; Viola, K.L.; Lacor, P.N.; Velasco, P.T.; Klein, W.L.; Chauhan, N.B. Brain Transit and Ameliorative Effects of Intranasally Delivered Anti-Amyloid-β Oligomer Antibody in 5XFAD Mice. J. Alzheimer’s Dis. 2013, 35, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Kozlovskaya, L.; Abou-Kaoud, M.; Stepensky, D. Quantitative analysis of drug delivery to the brain via nasal route. J. Control. Release 2014, 189, 133–140. [Google Scholar] [CrossRef]
- Pardridge, W.M. Biologic TNFα-inhibitors that cross the human blood-brain barrier. Bioeng. Bugs 2010, 1, 231–234. [Google Scholar] [CrossRef] [Green Version]
- Ladel, S.; Schlossbauer, P.; Flamm, J.; Luksch, H.; Mizaikoff, B.; Schindowski, K. Improved In Vitro Model for Intranasal Mucosal Drug Delivery: Primary Olfactory and Respiratory Epithelial Cells Compared with the Permanent Nasal Cell Line RPMI. Pharmaceutics 2019, 11, 367. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef]
- Guilleminault, L.; Azzopardi, N.; Arnoult, C.; Sobilo, J.; Hervé, V.; Montharu, J.; Guillon, A.; Andres, C.; Herault, O.; Le Pape, A.; et al. Fate of inhaled monoclonal antibodies after the deposition of aerosolized particles in the respiratory system. J. Control. Release 2014, 196, 344–354. [Google Scholar] [CrossRef]
- Günaydın, G.; Yu, S.; Gräslund, T.; Hammarström, L.; Marcotte, H. Fusion of the mouse IgG1 Fc domain to the VHH fragment (ARP1) enhances protection in a mouse model of rotavirus. Sci. Rep. 2016, 6, 30171. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soleimanizadeh, A.; Dinter, H.; Schindowski, K. Central Nervous System Delivery of Antibodies and Their Single-Domain Antibodies and Variable Fragment Derivatives with Focus on Intranasal Nose to Brain Administration. Antibodies 2021, 10, 47. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10040047
Soleimanizadeh A, Dinter H, Schindowski K. Central Nervous System Delivery of Antibodies and Their Single-Domain Antibodies and Variable Fragment Derivatives with Focus on Intranasal Nose to Brain Administration. Antibodies. 2021; 10(4):47. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10040047
Chicago/Turabian StyleSoleimanizadeh, Arghavan, Heiko Dinter, and Katharina Schindowski. 2021. "Central Nervous System Delivery of Antibodies and Their Single-Domain Antibodies and Variable Fragment Derivatives with Focus on Intranasal Nose to Brain Administration" Antibodies 10, no. 4: 47. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10040047