
MALDI-TOF-MS-Based Identification of Monoclonal Murine Anti-SARS-CoV-2 Antibodies within One Hour

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Determination of Intact Masses (IgG)
2.2. Determination of Light Chain Masses (IgG)
2.3. Cleavage with Diluted Sulfuric Acid
2.4. Enzymatic Cleavage with Trypsin
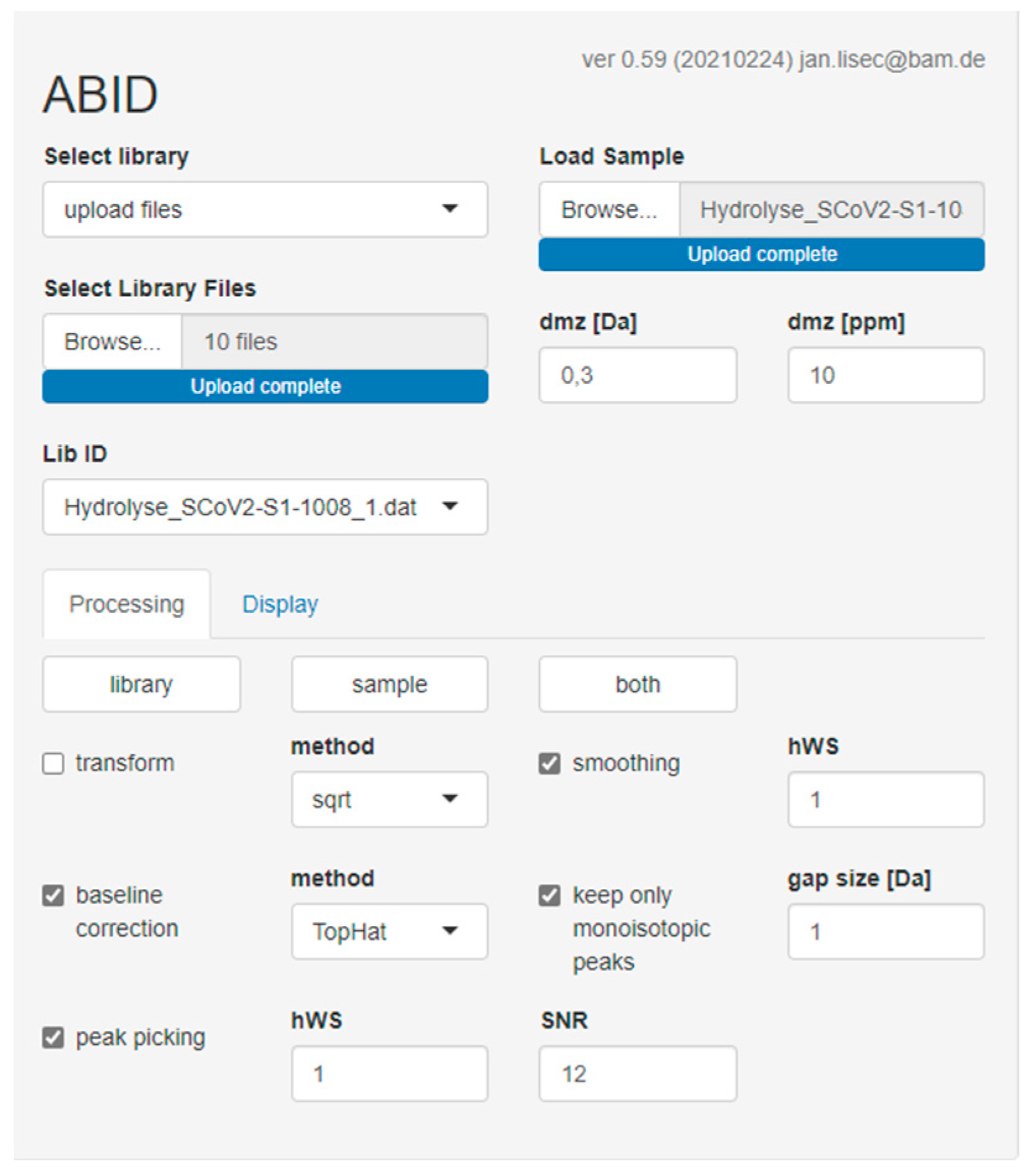
2.5. Software-Assisted Antibody Identification with ABID 2.0
3. Results
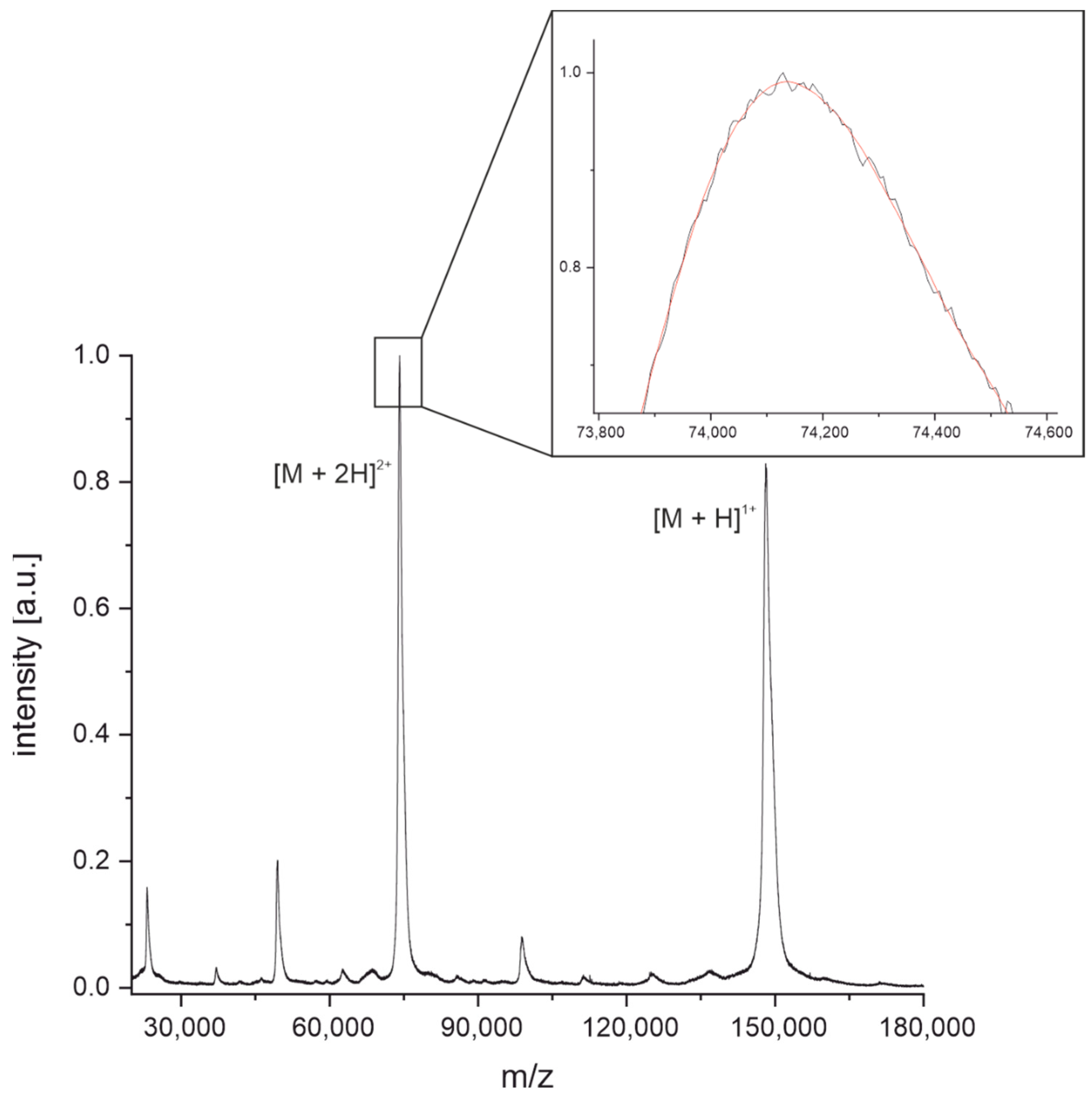
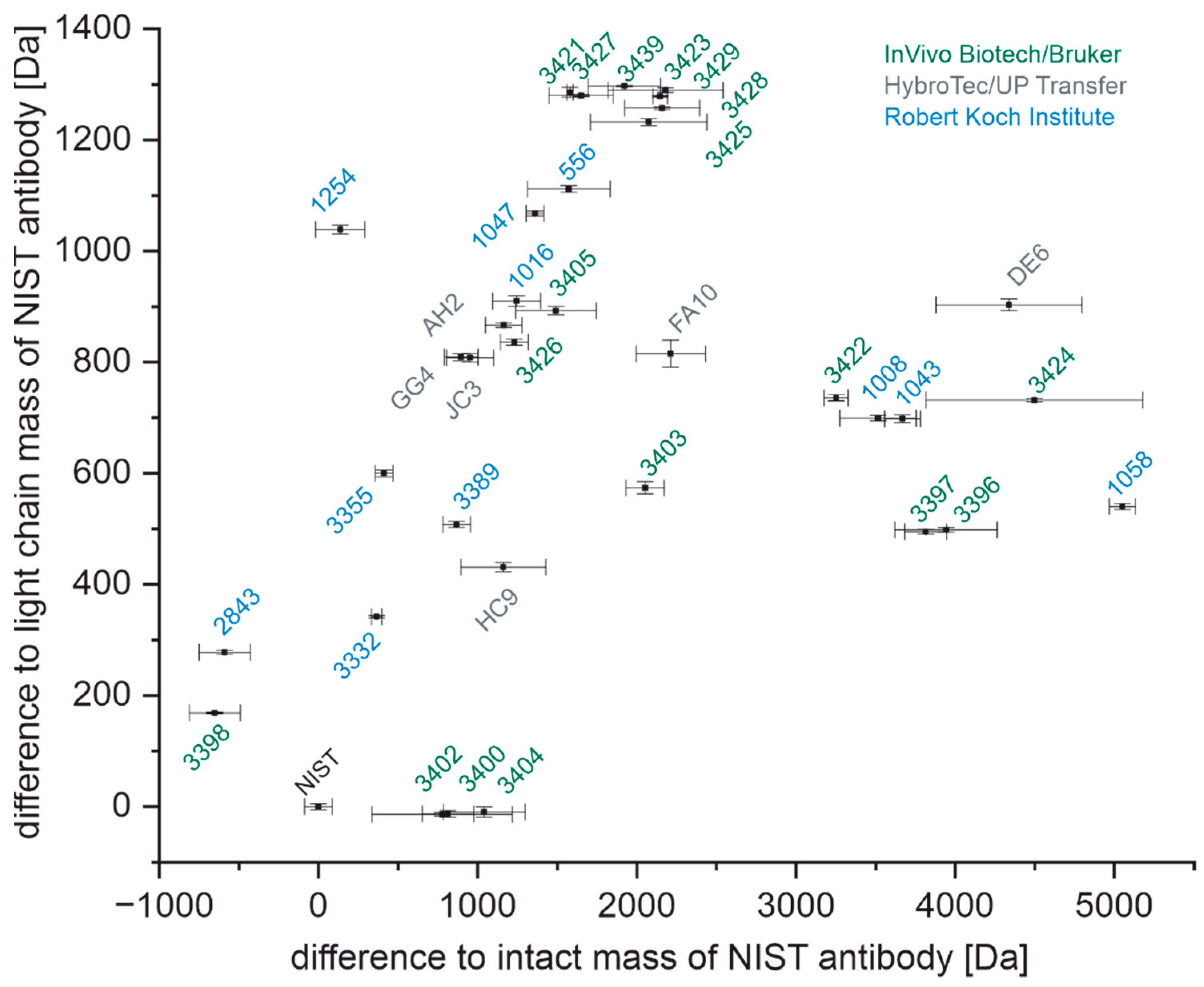
3.1. Determination of Intact Masses
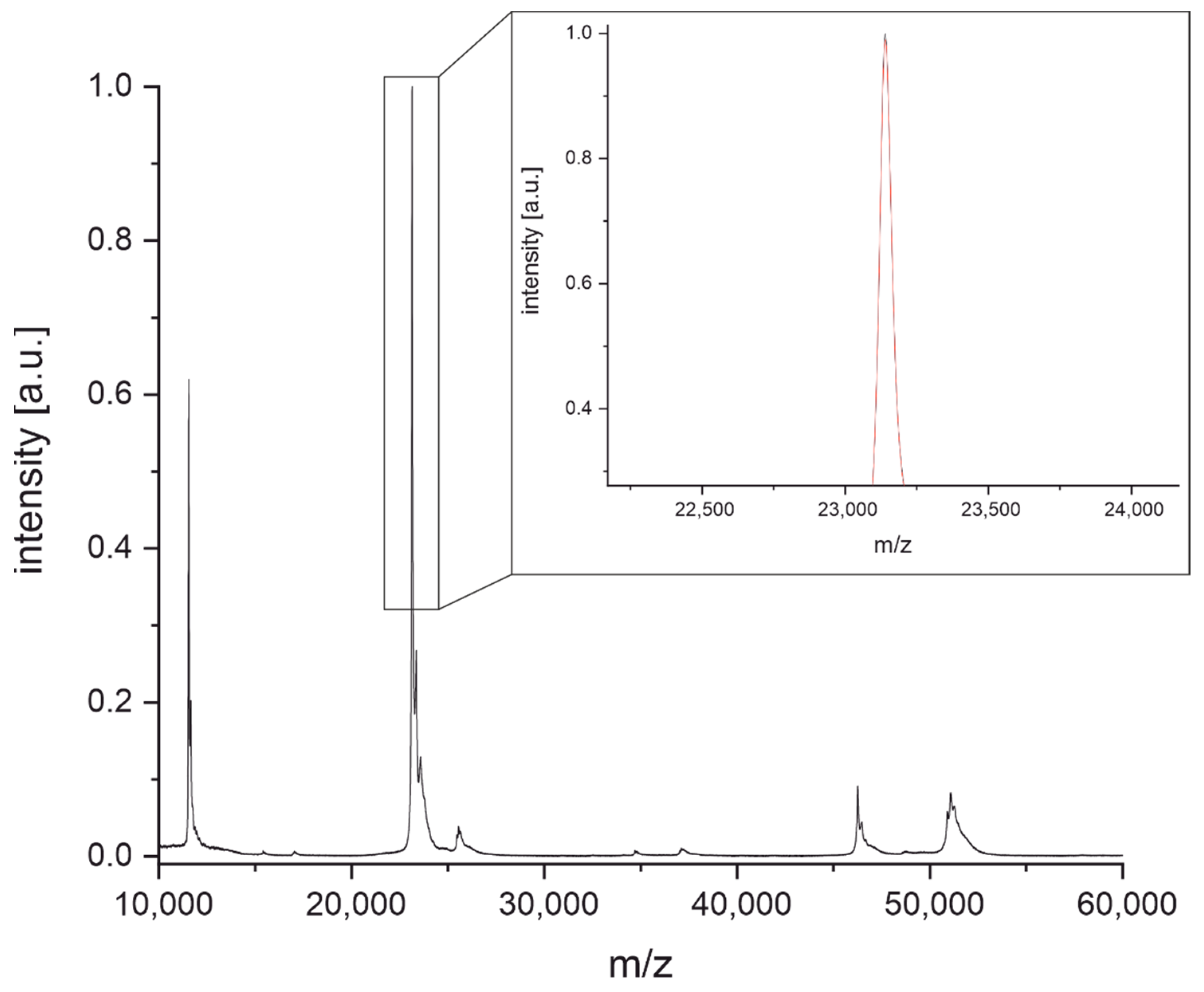
3.2. Determination of Light Chain Masses
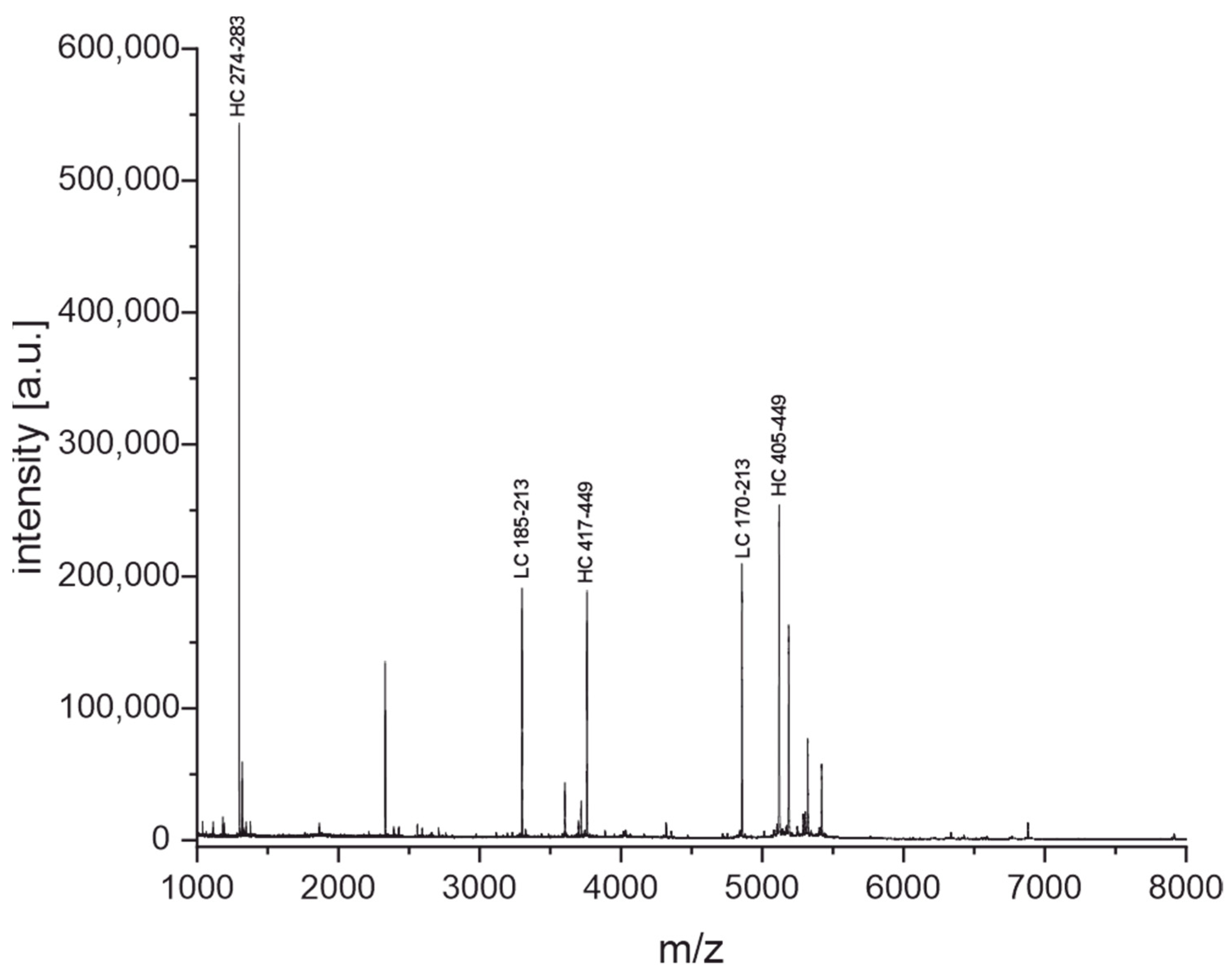
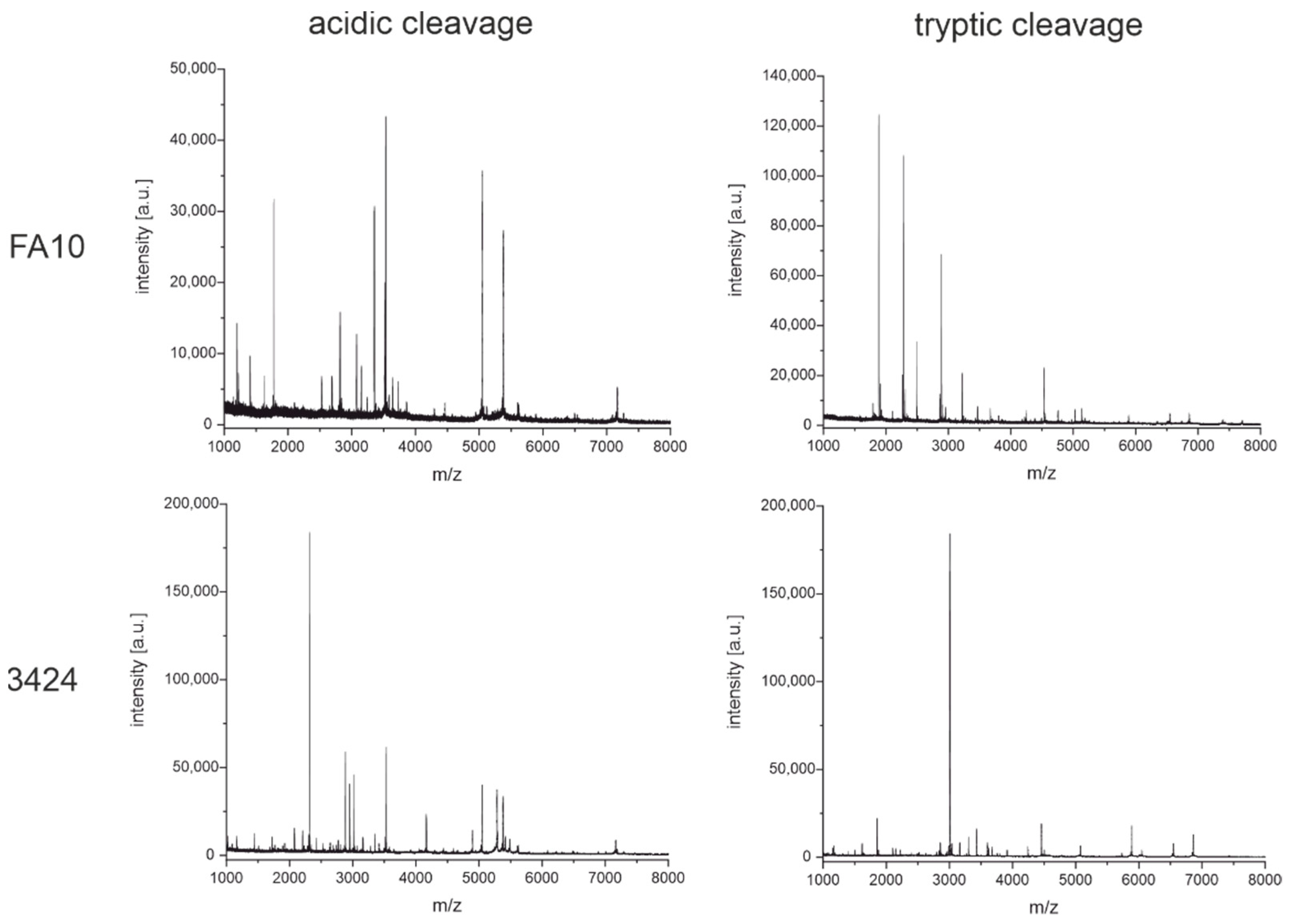
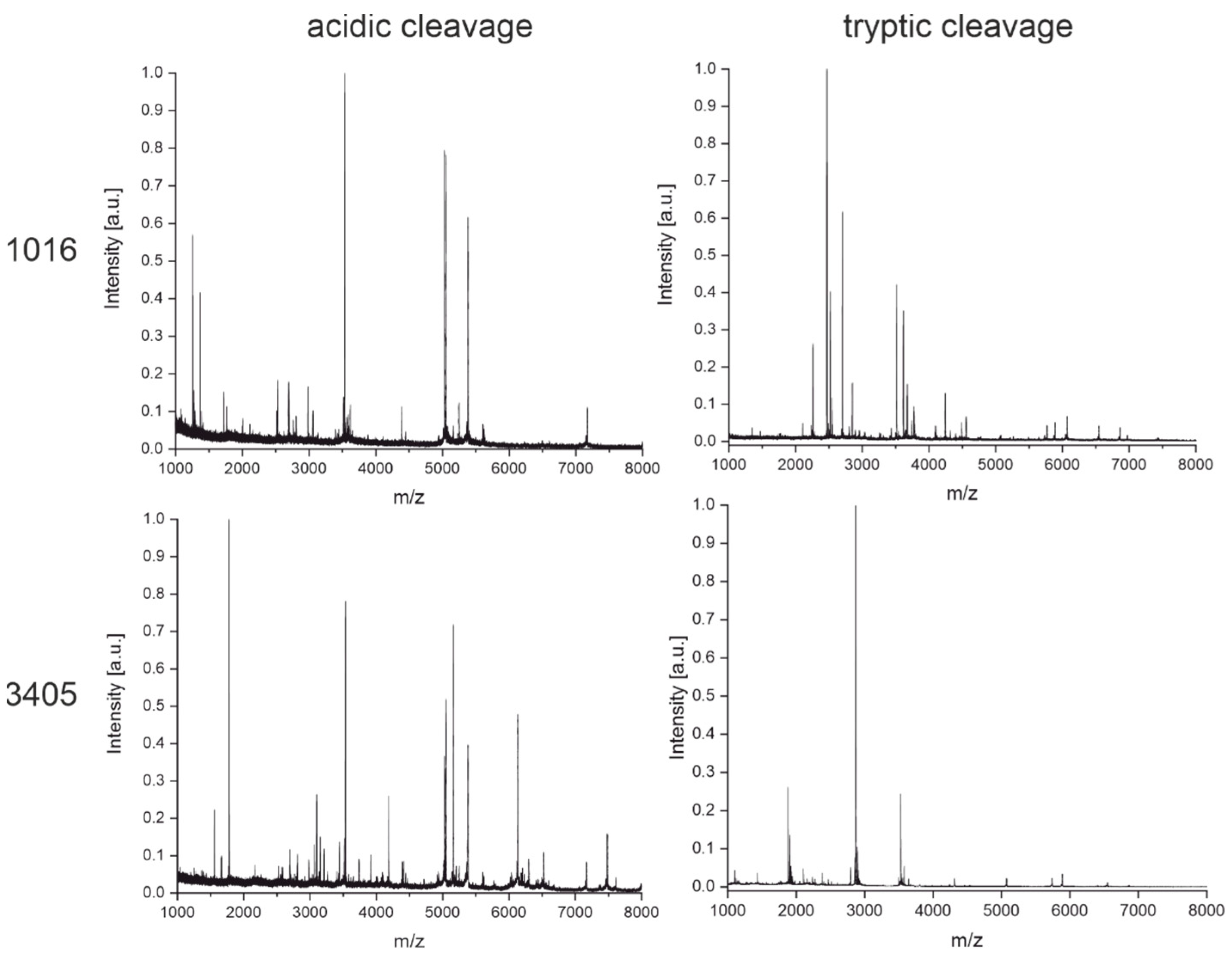
3.3. Peptide Mass Fingerprinting
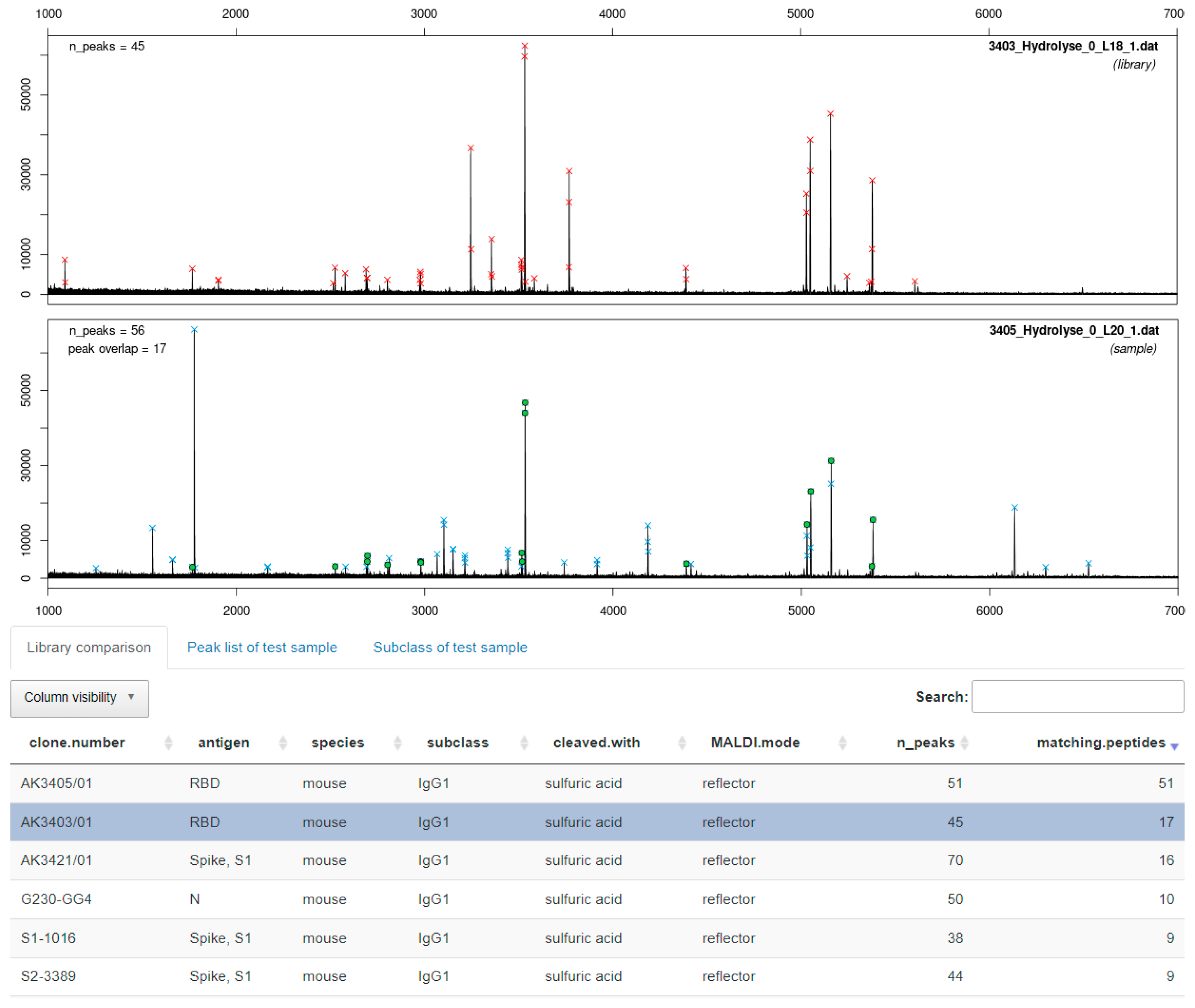
3.3.1. Cleavage with Diluted Sulfuric Acid
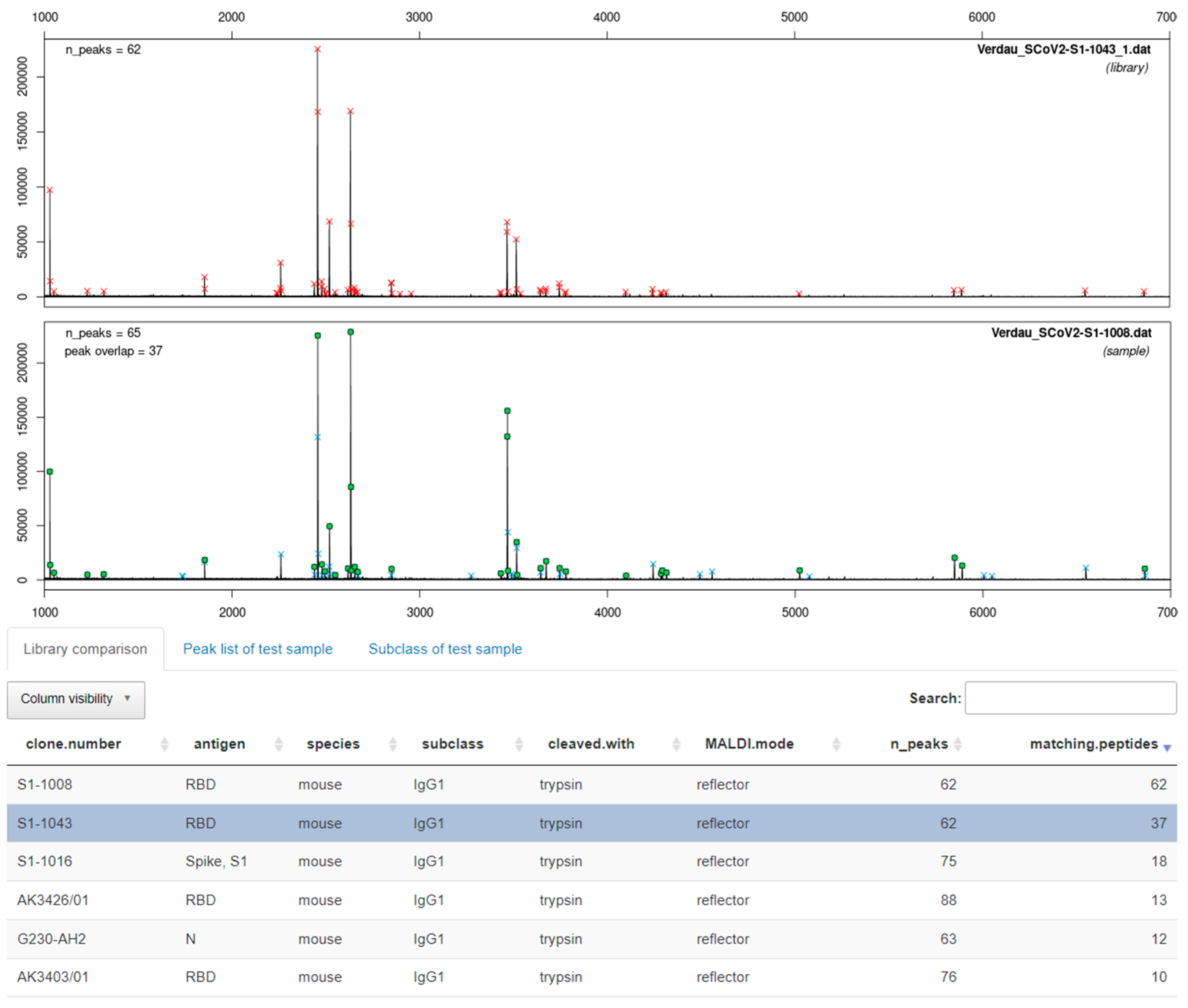
3.3.2. Enzymatic Cleavage with Trypsin
3.4. Software-Assisted Antibody Identification with ABID 2.0
4. Discussion
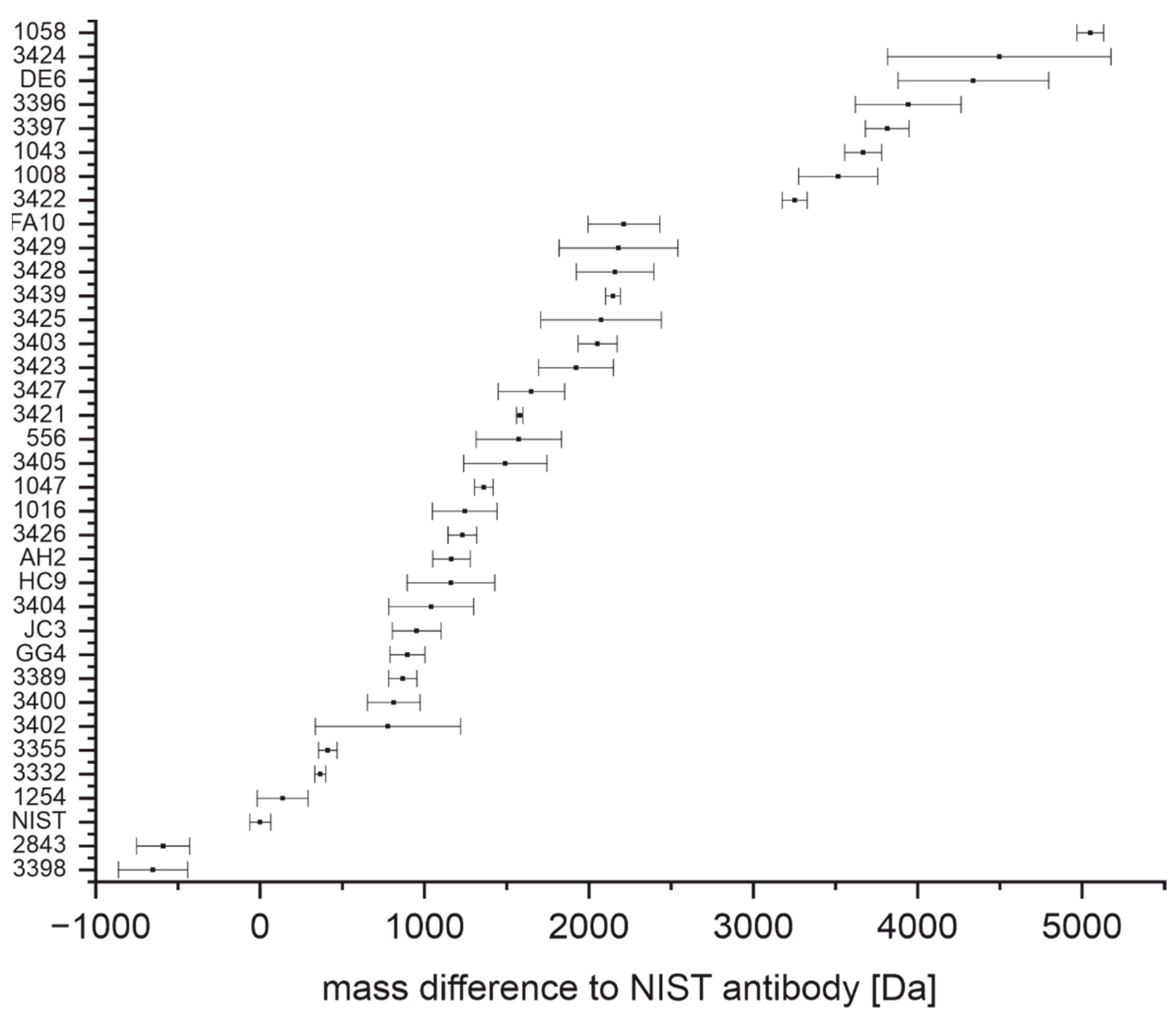
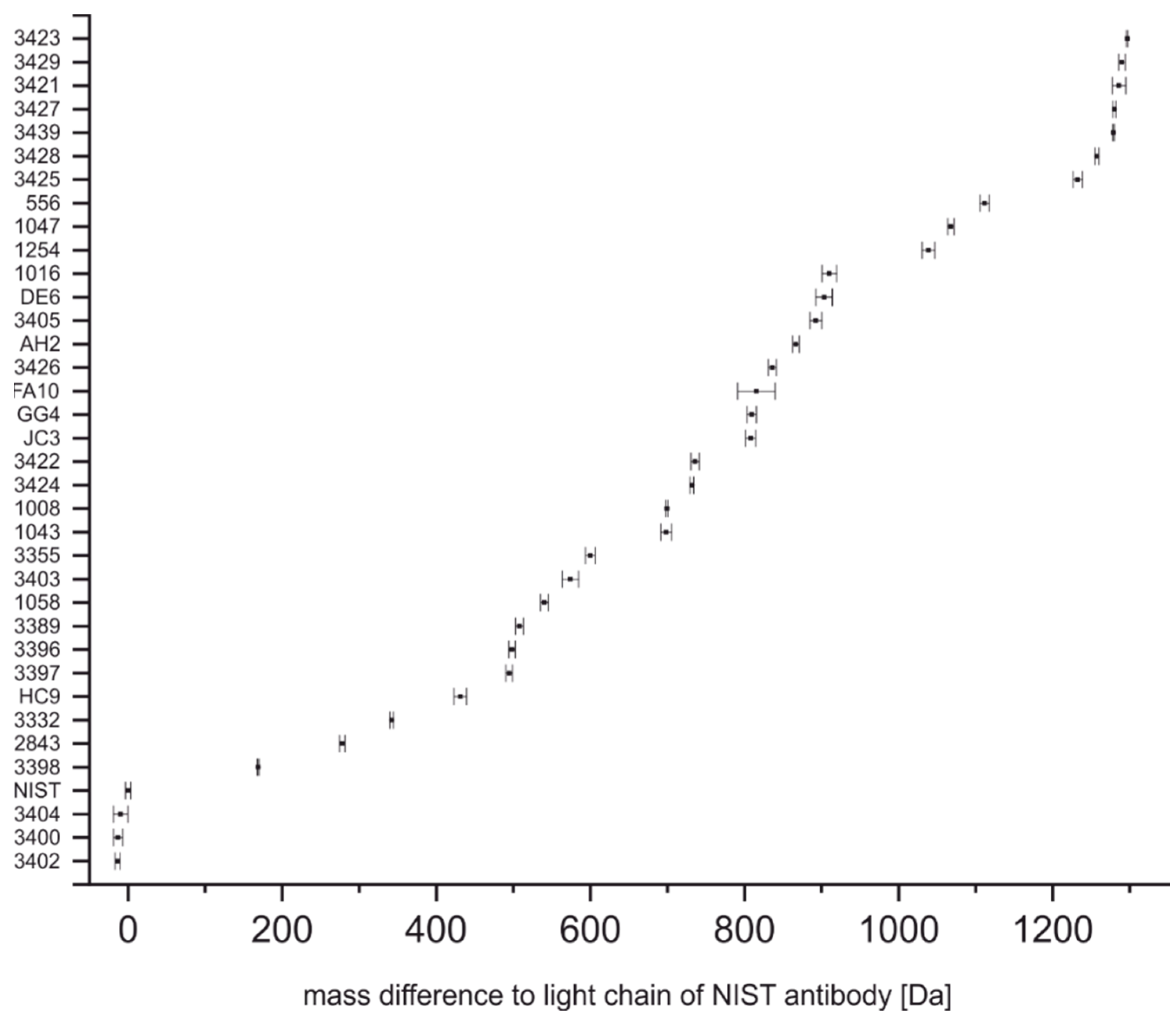
4.1. Determination of Light Chain and Intact Mass
4.2. Peptide Mass Fingerprinting
4.3. Software-Assisted Antibody Identification with ABID 2.0
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kaplon, H.; Chenoweth, A.; Crescioli, S.; Reichert, J.M. Antibodies to watch in 2022. mAbs 2022, 14, 2014296. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, H.; Valedkarimi, Z.; Aghebati-Maleki, L.; Majidi, J. Antibody-drug conjugates: Promising and efficient tools for targeted cancer therapy. J. Cell. Physiol. 2018, 233, 6441–6457. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voskuil, J.L.; Bandrowski, A.; Begley, C.G.; Bradbury, A.R.; Chalmers, A.D.; Gomes, A.V.; Hardcastle, T.; Lund-Johansen, F.; Plückthun, A.; Roncador, G.; et al. The Antibody Society’s antibody validation webinar series. mAbs 2020, 12, 1794421. [Google Scholar] [CrossRef] [PubMed]
- Wozniak-Knopp, G. Antibody Validation. In Introduction to Antibody Engineering; Springer: Berlin/Heidelberg, Germany, 2021; pp. 363–382. [Google Scholar]
- Vasilevsky, N.A.; Brush, M.H.; Paddock, H.; Ponting, L.; Tripathy, S.J.; LaRocca, G.M.; Haendel, M.A. On the reproducibility of science: Unique identification of research resources in the biomedical literature. PeerJ 2013, 1, e148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M. Reproducibility crisis: Blame it on the antibodies. Nature 2015, 521, 274–276. [Google Scholar] [CrossRef] [Green Version]
- Freedman, L.P.; Cockburn, I.M.; Simcoe, T.S. The Economics of Reproducibility in Preclinical Research. PLoS Biol. 2015, 13, e1002165. [Google Scholar] [CrossRef]
- Lima, W.C.; Gasteiger, E.; Marcatili, P.; Duek, P.; Bairoch, A.; Cosson, P. The ABCD database: A repository for chemically defined antibodies. Nucleic Acids Res. 2020, 48, D261–D264. [Google Scholar] [CrossRef]
- Bradbury, A.R.; Plückthun, A. Reproducibility: Standardize antibodies used in research. Nat. News 2015, 518, 27–29. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, A.R.; Plückthun, A. Getting to reproducible antibodies: The rationale for sequenced recombinant characterized reagents. Protein Eng. Des. Sel. 2015, 28, 303–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groff, K.; Brown, J.; Clippinger, A.J. Modern affinity reagents: Recombinant antibodies and aptamers. Biotechnol. Adv. 2015, 33, 1787–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, A.M.; Plückthun, A. Antibodies: Validate recombinants once. Nature 2015, 520, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman, L.P. Antibodies: Validate recombinants too. Nature 2015, 518, 483. [Google Scholar] [CrossRef] [Green Version]
- Voskuil, J.L. The challenges with the validation of research antibodies. F1000Research 2017, 6, 161. [Google Scholar] [CrossRef] [Green Version]
- Goodman, S.L. The antibody horror show: An introductory guide for the perplexed. New Biotechnol. 2018, 45, 9–13. [Google Scholar] [CrossRef]
- Uhlen, M.; Bandrowski, A.; Carr, S.; Edwards, A.; Ellenberg, J.; Lundberg, E.; Rimm, D.L.; Rodriguez, H.; Hiltke, T.; Snyder, M.; et al. A proposal for validation of antibodies. Nat. Methods 2016, 13, 823–827. [Google Scholar] [CrossRef]
- Weller, M.G. Quality Issues of Research Antibodies. Anal. Chem. Insights 2016, 11, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.G. Ten Basic Rules of Antibody Validation. Anal. Chem. Insights 2018, 13, 1177390118757462. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.G. The Protocol Gap. Methods Protoc. 2021, 4, 12. [Google Scholar] [CrossRef]
- Akashi, S.; Noguchi, K.; Yuji, R.; Tagami, U.; Hirayama, K.; Kato, K.; Kim, H.; Tokioka, K.; Shimada, I.; Arata, Y. Characterization of mouse switch variant antibodies by matrix-assisted laser desorption ionization mass spectrometry and electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 1996, 7, 707–721. [Google Scholar] [CrossRef]
- Dick, L.W., Jr.; Mahon, D.; Qiu, D.; Cheng, K.-C. Peptide mapping of therapeutic monoclonal antibodies: Improvements for increased speed and fewer artifacts. J. Chromatogr. B 2009, 877, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.E.M.; Kalume, D.E.; Calvo, L.; Mallo, M.F.; Vallin, A.; Roepstorff, P. Characterization of a recombinant monoclonal antibody by mass spectrometry combined with liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 2001, 752, 247–261. [Google Scholar] [CrossRef]
- Tscheuschner, G.; Schwaar, T.; Weller, M.G. Fast Confirmation of Antibody Identity by MALDI-TOF MS Fingerprints. Antibodies 2020, 9, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, L.; Swiderek, K.M.; Shively, J.E. Comparison of ESI-MS, LSIMS and MALDI-TOF-MS for the primary structure analysis of a monoclonal antibody. In Techniques in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 1995; Volume 6, pp. 21–30. [Google Scholar] [CrossRef]
- Li, A.; Sowder, I.R.C.; Henderson, L.E.; Moore, S.P.; Garfinkel, A.D.J.; Fisher, R.J. Chemical Cleavage at Aspartyl Residues for Protein Identification. Anal. Chem. 2001, 73, 5395–5402. [Google Scholar] [CrossRef]
- Swatkoski, S.; Gutierrez, P.; Ginter, J.; Petrov, A.; Dinman, J.D.; Edwards, N.; Fenselau, C. Integration of Residue-Specific Acid Cleavage into Proteomic Workflows. J. Proteome Res. 2007, 6, 4525–4527. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Chen, S.-T.; Chiou, S.-H.; Wang, K.-T. Specific peptide-bond cleavage by microwave irradiation in weak acid solution. J. Protein Chem. 1992, 11, 45–50. [Google Scholar] [CrossRef]
- Hua, L.; Low, T.Y.; Sze, S.K. Microwave-assisted specific chemical digestion for rapid protein identification. Proteomics 2006, 6, 586–591. [Google Scholar] [CrossRef]
- Hauser, N.J.; Basile, F. Online Microwave D-Cleavage LC-ESI-MS/MS of Intact Proteins: Site-Specific Cleavages at Aspartic Acid Residues and Disulfide Bonds. J. Proteome Res. 2008, 7, 1012–1026. [Google Scholar] [CrossRef]
- Swatkoski, S.; Gutierrez, P.; Wynne, C.; Petrov, A.; Dinman, J.D.; Edwards, N.; Fenselau, C. Evaluation of Microwave-Accelerated Residue-Specific Acid Cleavage for Proteomic Applications. J. Proteome Res. 2008, 7, 579–586. [Google Scholar] [CrossRef]
- Pappin, D.J.; Højrup, P.; Bleasby, A.J. Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 1993, 3, 327–332. [Google Scholar] [CrossRef]
- Henzel, W.J.; Watanabe, C.; Stults, J.T. Protein identification: The origins of peptide mass fingerprinting. J. Am. Soc. Mass Spectrom. 2003, 14, 931–942. [Google Scholar] [CrossRef] [Green Version]
- Schiel, J.E.; Turner, A. The NISTmAb Reference Material 8671 lifecycle management and quality plan. Anal. Bioanal. Chem. 2018, 410, 2067–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www-s.nist.gov/srmors/certificates/8671.pdf (accessed on 5 April 2022).
- Mouchahoir, T.; Schiel, J.E. Development of an LC-MS/MS peptide mapping protocol for the NISTmAb. Anal. Bioanal. Chem. 2018, 410, 2111–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Huang, Y.; Ito, A.; Kawakami, T.; Hojo, H.; Murata, M. Trifluoroethanol-containing RP-HPLC mobile phases for the separation of transmembrane peptides human glycophorin-A, integrin alpha-1, and p24: Analysis and prevention of potential side reactions due to formic acid. J. Pept. Sci. 2015, 21, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Colleary, C.; Little, N.C.; Cleland, T.P. Microwave-assisted acid hydrolysis for whole-bone proteomics and paleoproteomics. Rapid Commun. Mass Spectrom. 2020, 34, e8568. [Google Scholar] [CrossRef]
- Park, Z.-Y.; Russell, D.H. Thermal Denaturation: A Useful Technique in Peptide Mass Mapping. Anal. Chem. 2000, 72, 2667–2670. [Google Scholar] [CrossRef]
- Chen, C.-H.; Feng, H.T.; Guo, R.; Li, P.J.; Laserna, A.K.C.; Ji, Y.; Ng, B.H.; Li, S.F.Y.; Khan, S.H.; Paulus, A.; et al. Intact NIST monoclonal antibody characterization—Proteoforms, glycoforms—Using CE-MS and CE-LIF. Cogent Chem. 2018, 4, 1480455. [Google Scholar] [CrossRef]
- Gstöttner, C.; Reusch, D.; Haberger, M.; Dragan, I.; Van Veelen, P.; Kilgour, D.P.A.; Tsybin, Y.O.; van der Burgt, Y.E.M.; Wuhrer, M.; Nicolardi, S. Monitoring glycation levels of a bispecific monoclonal antibody at subunit level by ultrahigh-resolution MALDI FT-ICR mass spectrometry. mAbs 2020, 12, 1682403. [Google Scholar] [CrossRef] [Green Version]
- van der Burgt, Y.E.; Kilgour, D.P.A.; Tsybin, Y.O.; Srzentić, K.; Fornelli, L.; Beck, A.; Wuhrer, M.; Nicolardi, S. Structural Analysis of Monoclonal Antibodies by Ultrahigh Resolution MALDI In-Source Decay FT-ICR Mass Spectrometry. Anal. Chem. 2018, 91, 2079–2085. [Google Scholar] [CrossRef] [Green Version]
- McMurry, J.A.; Juty, N.; Blomberg, N.; Burdett, T.; Conlin, T.; Conte, N.; Courtot, M.; Deck, J.; Dumontier, M.; Fellows, D.K.; et al. Identifiers for the 21st century: How to design, provision, and reuse persistent identifiers to maximize utility and impact of life science data. PLoS Biol. 2017, 15, e2001414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roncador, G.; Engel, P.; Maestre, L.; Anderson, A.P.; Cordell, J.L.; Cragg, M.S.; Šerbec, V.Č.; Jones, M.; Lisnic, V.J.; Kremer, L. The European antibody network’s practical guide to finding and validating suitable antibodies for research. mAbs 2016, 8, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandrowski, A.; Brush, M.; Grethe, J.S.; Haendel, M.A.; Kennedy, D.N.; Hill, S.; Hof, P.R.; Martone, M.E.; Pols, M.; Tan, S.C.; et al. The Resource Identification Initiative: A cultural shift in publishing. Neuroinformatics 2016, 14, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helsby, M.A.; Leader, P.M.; Fenn, J.R.; Gulsen, T.; Bryant, C.; Doughton, G.; Sharpe, B.; Whitley, P.; Caunt, C.J.; James, K.; et al. CiteAb: A searchable antibody database that ranks antibodies by the number of times they have been cited. BMC Cell Biol. 2014, 15, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clone | Abbreviation | Isotype | Concentration (g/L) | Antigen |
|---|---|---|---|---|
| S1-556 | 556 | IgG1 | 1.84 | S1 |
| S1-1008 | 1008 | IgG1 | 1.81 | RBD |
| S1-1016 | 1016 | IgG1 | 1.58 | S1 |
| S1-1043 | 1043 | IgG1 | 1.74 | RBD |
| S1-1047 | 1047 | IgG1 | 1.91 | S1 |
| S1-1058 | 1058 | IgG2a | 1.65 | S1 |
| S2-1254 | 1254 | IgG1 | 1.84 | S2 |
| S2-2843 | 2843 | IgG1 | 1.81 | S2 |
| S1-3332 | 3332 | IgG1 | 1.58 | S1 |
| S1-3355 | 3355 | IgG1 | 1.77 | RBD |
| S2-3389 | 3389 | IgG1 | 1.75 | S2 |
| AK3396/01 | 3396 | IgG2b | 1.00 | RBD |
| AK3397/01 | 3397 | IgG2b | 1.40 | RBD |
| AK3398/02 | 3398 | IgG1 | 1.50 | RBD |
| AK3400/01 | 3400 | IgG2b | 1.10 | RBD |
| AK3402/01 | 3402 | IgG2b | 1.15 | RBD |
| AK3403/01 | 3403 | IgG1 | 0.70 | RBD |
| AK3404/02 | 3404 | IgG1 | 1.45 | RBD |
| AK3405/01 | 3405 | IgG1 | 0.75 | RBD |
| AK3421/01 | 3421 | IgG1 | 2.35 | S1 |
| AK3422/01 | 3422 | IgG2a | 2.05 | S1 |
| AK3423/01 | 3423 | IgG1 | 1.15 | S1 |
| AK3424/01 | 3424 | IgG2a | 1.25 | S1 |
| AK3425/01 | 3425 | IgG1 | 0.95 | S1 |
| AK3426/01 | 3426 | IgG1 | 1.00 | RBD |
| AK3427/01 | 3427 | IgG1 | 1.15 | RBD |
| AK3428/01 | 3428 | IgG1 | 1.05 | S1 |
| AK3429/01 | 3429 | IgG1 | 1.30 | RBD |
| AK3439/01 | 3439 | IgG1 | 1.25 | S1 |
| G230-AH2 | AH2 | IgG1 | 2.00 | N |
| G230-HC9 | HC9 | IgG2a | 1.05 | N |
| G230-DE6 | DE6 | IgG2b | 2.10 | N |
| G230-GG4 | GG4 | IgG1 | 3.10 | N |
| G230-JC3 | JC3 | IgG2b | 1.75 | N |
| G229-FA10 | FA10 | IgG1 | 0.75 | N |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tscheuschner, G.; Kaiser, M.N.; Lisec, J.; Beslic, D.; Muth, T.; Krüger, M.; Mages, H.W.; Dorner, B.G.; Knospe, J.; Schenk, J.A.; et al. MALDI-TOF-MS-Based Identification of Monoclonal Murine Anti-SARS-CoV-2 Antibodies within One Hour. Antibodies 2022, 11, 27. https://0-doi-org.brum.beds.ac.uk/10.3390/antib11020027
Tscheuschner G, Kaiser MN, Lisec J, Beslic D, Muth T, Krüger M, Mages HW, Dorner BG, Knospe J, Schenk JA, et al. MALDI-TOF-MS-Based Identification of Monoclonal Murine Anti-SARS-CoV-2 Antibodies within One Hour. Antibodies. 2022; 11(2):27. https://0-doi-org.brum.beds.ac.uk/10.3390/antib11020027
Chicago/Turabian StyleTscheuschner, Georg, Melanie N. Kaiser, Jan Lisec, Denis Beslic, Thilo Muth, Maren Krüger, Hans Werner Mages, Brigitte G. Dorner, Julia Knospe, Jörg A. Schenk, and et al. 2022. "MALDI-TOF-MS-Based Identification of Monoclonal Murine Anti-SARS-CoV-2 Antibodies within One Hour" Antibodies 11, no. 2: 27. https://0-doi-org.brum.beds.ac.uk/10.3390/antib11020027