3.1. Recognition of HA1 by Antibody HC19.
Antibody HC19 has been developed against H3N2 influenza virus [
22]. The crystal structure indicates that the antibody recognizes the receptor binding domain, thus blocking the entry to the host cell [
19] (PDB code 2VIR).
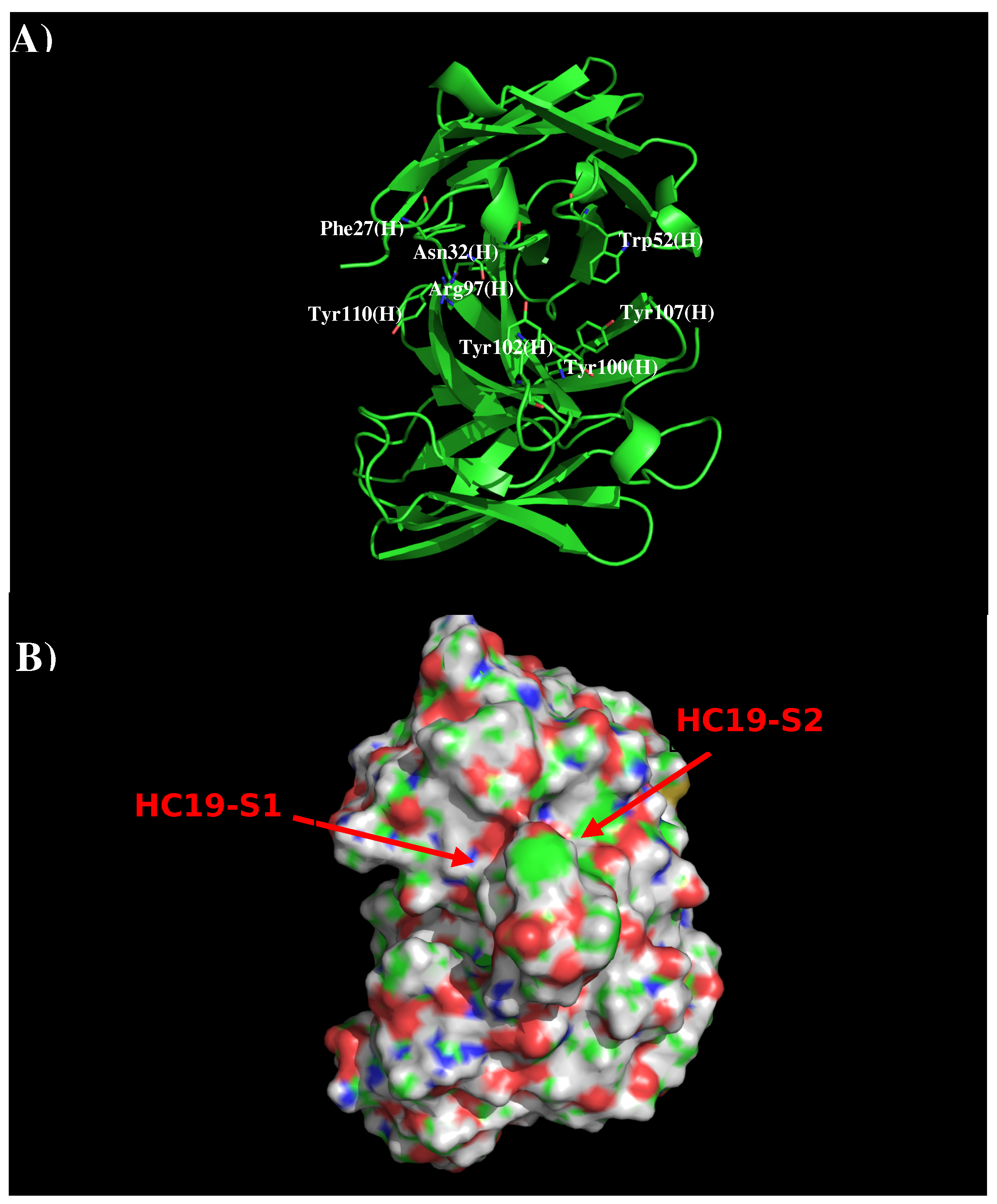
Figure 1 shows the structure and surface of AbHC19. There are two distinct regions around the CDR3 loop which are potentially significant to binding. The first region HC19-S1 is formed by residues Phe27, Asn32, Arg97, Tyr102, and Tyr110 of the H chain, with these residues contributing towards a polar charged surface. The second region HC19-S2 is formed by residues Trp52, Arg97, Tyr100, Tyr102, and Tyr107 also of the H chain. These residues form a surface with both polar and hydrophobic properties. These two binding surfaces (S1 and S2) are separated by ca. 9.0 Å.
Figure 2 shows the distribution of the MCSS minima of functional groups on the surface of AbHC19 occupying the two binding sites, S1 and S2. Because the binding pockets are quite deep, the minima are often buried deep inside the binding sites.
Table S1 (Supplementary Data) shows the number of minima in each cluster, their interaction energies and any specific interactions with residues. Overall, a strong correlation was observed between the distribution of the MCSS minima and the physical properties of the antibody surface at S1 and S2. Generally, minima were only observed for functional groups that are charged or aromatic polar functional groups, apart from benzene. We can see from
Table S1 (Supplementary Data) that most of the clusters occurred at the S2 binding site. Of the seven functional groups that formed minima, two formed clusters at S1, and six formed clusters at S2. Minima for both PHEN (16) and ACET (5) functional groups were observed, as we would expect forming polar interactions with the positively charged surface of S1, notably with Arg97(H) of the antibody. Generally, larger clusters consisting of minima of BENZ (112), IMIA (55), PHEN (7), INDO (33), MAMM (66), and MGUA (49) groups were observed at S2. Here we see a combination of both polar and hydrophobic interactions in keeping with the properties of the S2 surface. These interactions include hydrophobic interactions between groups (BENZ, IMIA, PHEN, INDO, MGUA) and Trp52(H), Arg97(H) of the antibody. Polar interactions were also observed between groups (INDO, MAMM, MGUA) and Asp98(H), Arg 97(H) of the antibody. The interaction energies of the minima are shown in
Table S1 (Supplementary Data). These values are generally as expected, with the polar and non-polar groups showing interaction energies of around −10.00 kcal/mol, and the charged and IMIA groups of around −15.00 kcal/mol, in line with the defined cut-offs. There are some exceptions, PHEN and MGUA groups both show stronger interaction energies, with Arg97(H) (−17.60 kcal/mol) and Tyr107(H) (−18.70 kcal/mol), respectively. The higher concentration of interactions that we observe between the functional groups and the S2 surface are probably because S2 has the properties of being both charged and hydrophobic. This allows the site to interact with a greater variety of functional groups.
Using the minima on the two surfaces S1 and S2, we constructed a sequence pattern for the peptides that could potentially bind to the antibody. The maximum distance between the two binding surfaces S1 and S2 is approximately 9.0 Å, corresponding to a separation by one amino acid between potential residues of antigen bind to residue at S1 and S2 of antibody. Our results showed most of the functional group minima were located at S2, with only PHEN and ACET groups found at surface S1. Therefore, the key sequence pattern for the binding epitope peptides can be defined as “X-J”, in which X = R, K, Y, W, H, F, “-” = any amino acid and J = D/E, Y. Note that the sequence is aligned from S2 to S1 as the negatively charged minima ACET is only found on S1, thus corresponding to the C-terminal of peptides.
Table 3 lists the distribution of key MCSS minima and the derived amino acid sequence pattern.
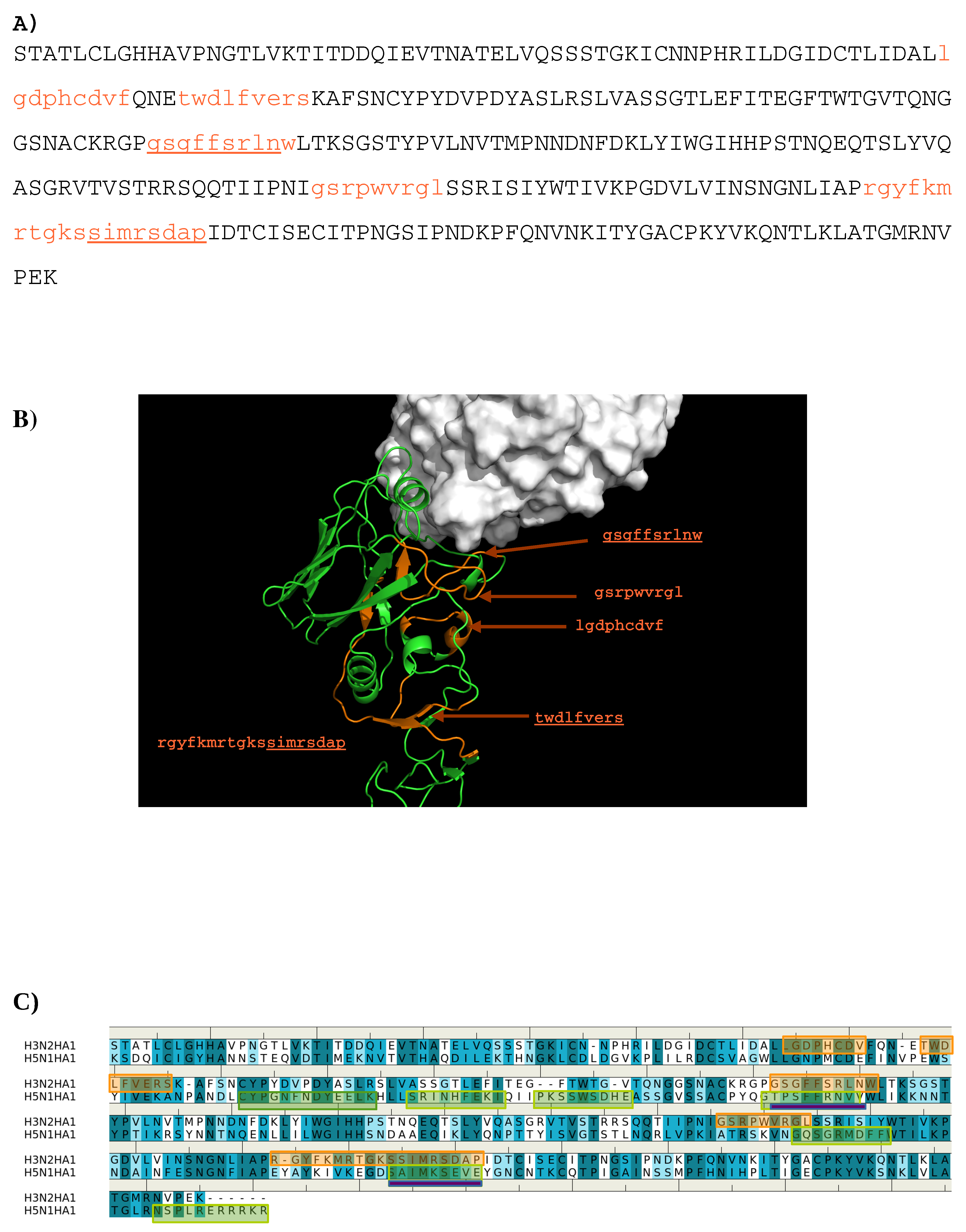
The sequence pattern was subsequently used to search for “binders” from the peptide libraries derived from the sequence of HA1 from the H3N2 virus (see Methods section). The seven libraries were searched for peptides matching the calculated sequence pattern of the binding epitope. Five peptides of HA1 were predicted to bind to the antibody (
1- “lgdphcdvf”;
2- “twdlfvers”;
3- “
gsgffsrlnw”;
4- “gsrpwvrgl”;
5- “rgyfkmrtgks
simrsdap”).
Figure 3 shows the amino acid sequence of HA1 with the predicted epitopes highlighted in lower case and colored in orange (
Figure 3A), and their location in the protein structure (
Figure 3B). For comparison, the predicted epitopes of HA1 from the H5N1 virus are also shown (
Figure 3C).
Figure 3B reveals that peptide
3 is located in the interaction site, and is clearly involved in the HA1-antibody binding. Close inspection show residues Ser145 and Trp153 of peptide
3 interacting with Asp101 and Tyr102 of the H chain, respectively. These direct contacts between the antigen and antibody are located in the regions of residues 134–144, residues 162–168 and residues 173–203 of HA1 of H3N2. The Tyr102, of the above-mentioned Trp153-Tyr102(H) interaction, is one of the residues involved in specific interactions identified from our MCSS calculations. This interaction occurs at the center of the interface, potentially playing a significant role in the antibody-antigen recognition. Peptide
1 is also of interest as a potential epitope. This peptide is located at the vestigial esterase domain, another known antibody recognition region.
Epitope mapping was also performed for HA1 from the H5N1 virus.
Figure 3C shows the comparison of the predicted epitopes between two viruses. Only two regions are identified to be overlay between the two antigens (“gssffsrlnw” vs. “gtpsffrnvv”, “sstmrsdap” vs. “satmkseve”). These are underlined in
Figure 3A. The epitopes from H5N1 do not include the important Trp153-Tyr102(H) interaction observed between H3N2 and the antibody as Trp153 of H5N1 is outside of the identified epitope “gtpsffrnvv”. This is in keeping with the selectivity shown experimentally of AbHC19 towards H3N2 [
34,
35,
44].
3.2. Recognition of HA1 by Antibodies CR9114 and FI6V3.
Antibodies CR9114 and FI6V3 have been developed against H3N2 influenza virus [
21,
36]. The crystal structures of their complexes to H3N2 indicated that both antibodies bind in similar fashion to the membrane fusion part of HA. Therefore, we only present the results of AbCR9114 here.
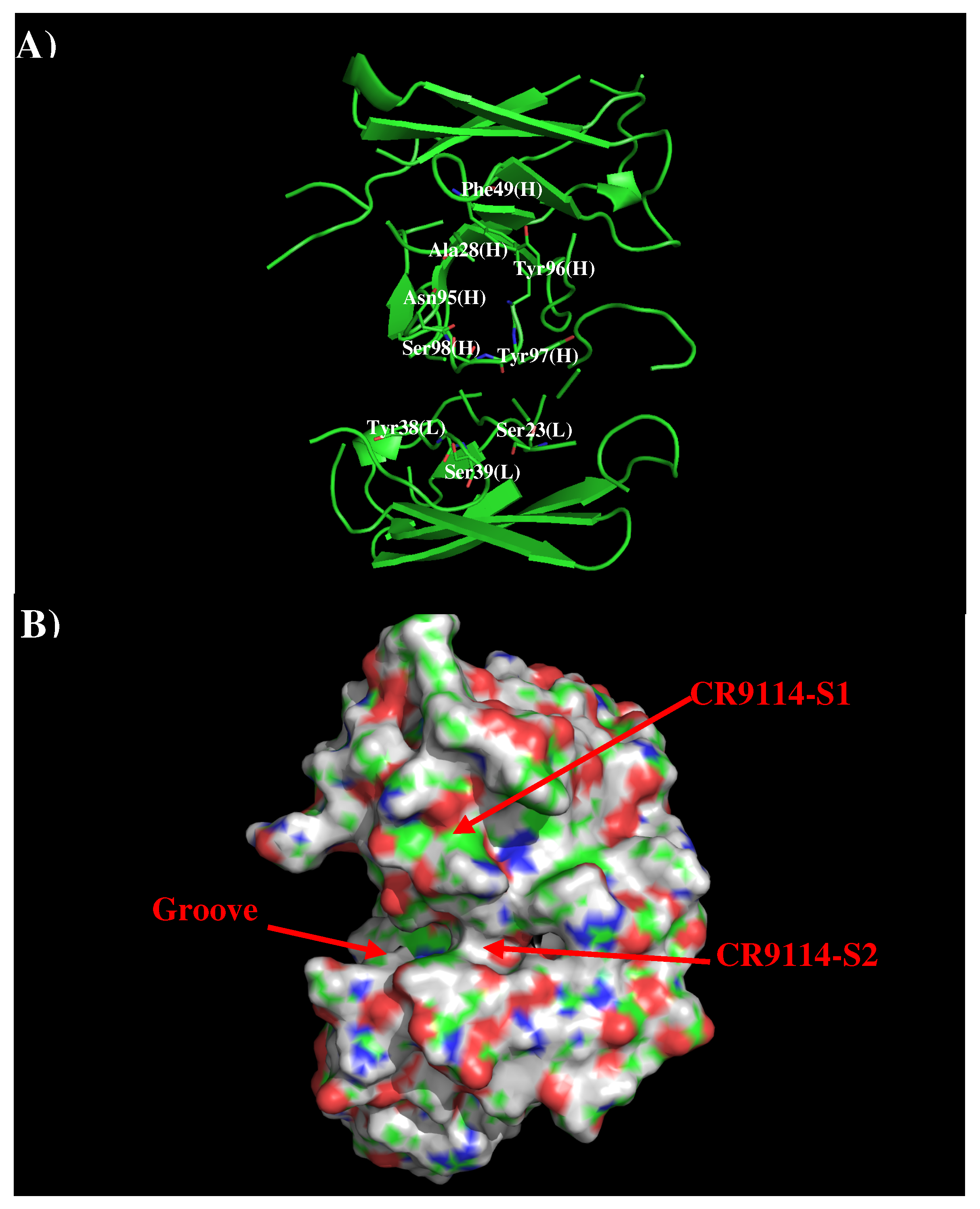
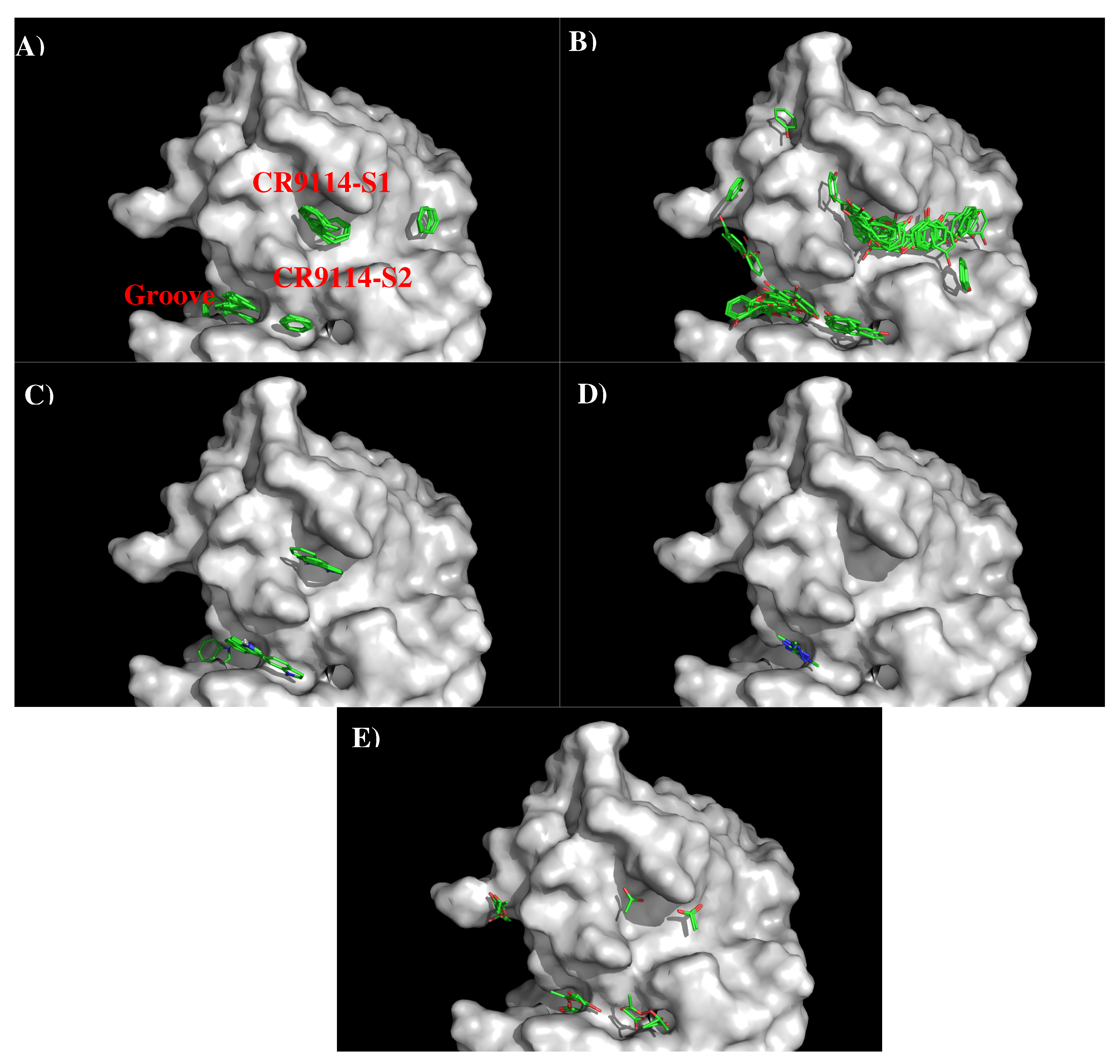
Figure 4 shows that structure and surface of AbCR9114. There are two distinctive regions around the CDR3 loop. The first site (CR9114-S1) is a hydrophobic surface formed by residues Ala28, Phe49, Asn95, Tyr96, and Tyr97 of H chain while the second polar surface (CR9114-S2) is formed by residues Ser23, Tyr38, and Ser39 of L chain and Ser98 of H chain. These two binding surfaces are separated by ca. 11.50 Å.
Figure 5 shows the locations of the minima of functional groups on the surface of AbCR9114.
Table S2 (Supplementary Data) shows the number of minima in each cluster and their interaction energies with residues. The minima are mainly clustered around the S1 and S2, as well as a groove nearby. No minima of small groups such as MEOH, MESH, and IBUT were found on the surfaces around the CDR3 loop. For the apolar groups, 29 and 21 BENZ minima were found at the S1 and S2 with favorable interaction energy values of −11.10 kcal/mol and −10.40 kcal/mol, respectively. The stronger interaction at S1 is due to the hydrophobicity of the binding site formed by two Tyrosine residues (Tyr96 and Tyr97 of H chain). For PHEN group, the minima is more spread out. 42 PHEN minima were located at S1 and nearby the surface, half of which concentrated at S1 with strong interaction energies up to −13.30 kcal/mol. These minima form interaction to Phe49 of H chain. Only seven minima were found at S2 with the binding energy range of (−10.00, −11.30) kcal/mol. For the INDO group, four minima were found to overlay each other at S1 with binding energies of ca. −15.40 kcal/mol. These minima form interaction to Phe49(H) and are oriented perpendicularly to Tyr96(H). Three INDO minima were found at S2, one of which formed a hydrogen bond to Asn25(L) with energy of −15.50 kcal/mol and the other two minima forming similar interaction to Tyr38 of L chain (−15.20 kcal/mol). For the charged groups, no MAMM minima were found around CDR3 loop while four MGUA minima were found only at S2 with energy range of (−15.00, −17.20) kcal/mol arising from its electrostatic interaction to Asp101(H). For the ACET group, four minima were found at S1, one of which interacts with Ser46(H) and three interacting with Ser51(H) showing energy range of −17.20 kcal/mol and (−15.20, −15.40) kcal/mol, respectively. At S2 six ACET minima were found, two of which interact in a similar fashion with Ser39(L) with energy of −16.10 kcal/mol, while other four minima interact with Arg22(L) showing energy range of (−15.10, −18.10) kcal/mol.
Based on the distribution of the important minima as shown in
Figure 6, a sequence pattern for peptides that bind to AbCR9114 was derived. The MCSS minima at the binding sites S1 and S2 are separated by ca 11.50 Å, a distance that could accommodate two amino acids. While most of the apolar and negatively charged group (i.e., BENZ, PHEN, INDO and AET) are located at both surfaces, only positively charged group MGUA was identified at S2 by interacting to residue Asp101(H). Therefore, the key sequence pattern for the binders was derived as “X-J”, in which X = R,F,W,Y,D/E, and J = F,W,Y,D/E.
Table 4 lists the distribution of key MCSS minima and the derived sequence pattern.
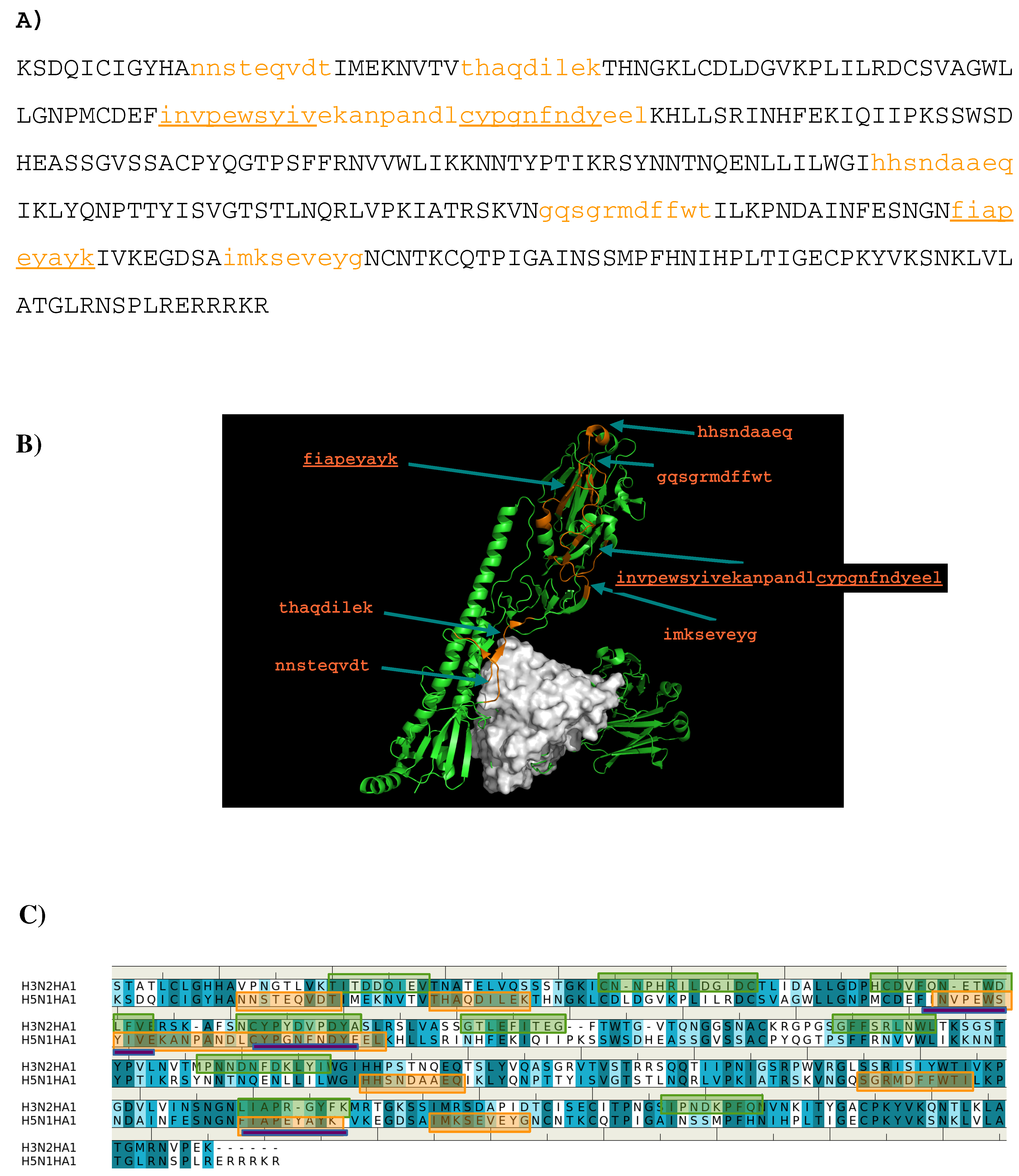
Seven peptides (I- “nnsteqvdt”, II- “thaqdilek”, III-”invpewsyivekanpandlcypgnfndyeel”, IV- ”hhsndaaeq”, V- “gqsgrmdffwt”, VI- ”fiapeyayk”, VII- “imkseveyg”) are predicted to bind to the antibody (
Figure 6A) and
Figure 6B) shows the location of these peptide in the crystal structure of AbCR9114 complexed with HA of H5N1. Peptide VI is buried inside the protein so as to be discarded as epitope. Of all the peptides, peptide II is located at the interface between the antibody and HA1 of H5N1 virus, together with peptide I forms a recognition surface. Two more regions were also predicted and are formed by the C-terminal portion of peptide III and peptide VII, and by peptide IV and N-terminal portion of peptide V, respectively. These two regions are located close to the vestigial esterate domain and the receptor binding domain, respectively.
Figure 6C shows the comparison between the epitopes predicted for the two viruses binding AbCR9114. Only the region of peptide III is consistently observed in both antigens as the other overlay region at the peptide VI is buried in the protein. The epitope “h
cdvfqnetwdlfv” of subtype H3 is located at the vestigial esterate domain.
3.3. Recognition of HA1 to Antibody BH151
AbBH151 is developed to against H3N2 [
18]. The crystal structure revealed that AbBH151 binds to a vestigial esterase domain.
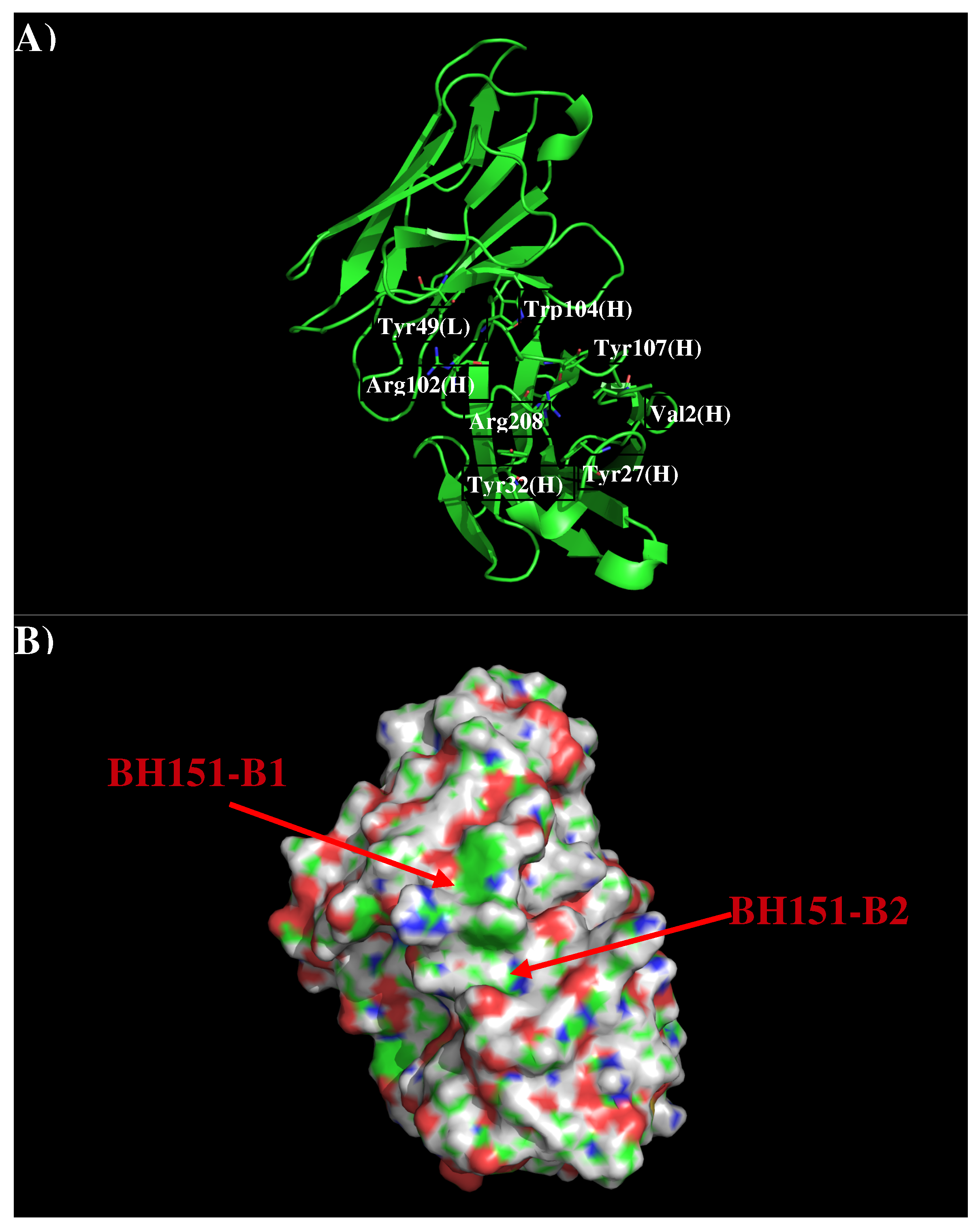
Figure 7 shows that structure and surface of AbBH151. The surface around the CDR3 loop is highly charged with a binding site (BH151-S1) formed by residues Val2, Tyr27, Tyr32, Arg98, Arg102, Phe105 and Tyr107 of H chain, and a relatively flat surface (BH151-S2) formed by residues Tyr49 of L chain and Arg102, Tyr104 of H chain. The two surfaces are separated by 9.50 Å.
Figure 8 shows the locations of the minima for the functional groups on the surface of AbBH151.
Table S3 (Supplementary Data) shows the number of minima in each cluster and their interaction energies with residues. A relatively small number of minima are clustered around CDR3 loop, including BENZ, PHEN, and ACET. While two BENZ minima, 11 PHEN minima and 30 ACET minima were found at the binding site B2, only 9 ACET minima were located at the B1. For the BENZ group, two minima are separated by 8.00 Å along the site B2 and interact with Phe105(H) with energies of −10.10 kcal/mol and −10.20 kcal/mol, respectively. The same positions were occupied by five and six PHEN minima with interaction energy range of (−10.00, −10.40) kcal/mol and (−10.00, −11.70) kcal/mol, respectively. The second group forms stronger interactions with Arg98(H) at the bottom of the binding site. For the ACET group, 30 minima at B2 are split into two clusters, one interacts with Arg102(H) measuring energies up to −17.70 kcal/mol, and the other one interacts with Arg98(H) showing energies up to −18.50 kcal/mol. At the binding site B1, nine ACET minima mainly interact with Arg102(H) (energy up to −16.80 kcal/mol).
Based on the distribution of the important minima as shown in
Figure 8, a sequence pattern for peptides that bind to AbBH151 was derived. The MCSS minima at the binding sites B1 and B2 are separated by ca 9.50 Å, implying one amino acid insertion. As only ACET minima were obtained at B1 while ACET, BENZ and PHEN minima were found at B2, the key sequence pattern for the binders was defined as “X-J”, in which X = D/E, and J = F,Y,D/E. The sequence is aligned from B1 to B2 as more ACET minima were located at B2, corresponding to the C-terminal of peptides.
Table 5 lists the distribution of key MCSS minima and the derived sequence pattern.
Overall, only three peptides (A: “
dphcdvfqnetwdlfve”, B: ”efitegftw”, C: ”mpnndnfdk”) from H3N2 HA1 are predicted to bind to the antibody.
Figure 9 shows the location of these epitopes. Out of these, the first half (“dphcdv”) of the peptide A forms a helical conformation and interact to the CDR3 loop of the antibody (residues 208–217 of H chain). This epitope is also obtained for the H5N1 HA1 as shown in
Figure 9C.
3.4. Prediction of Epitopes of HA1 to Antibody 4F5
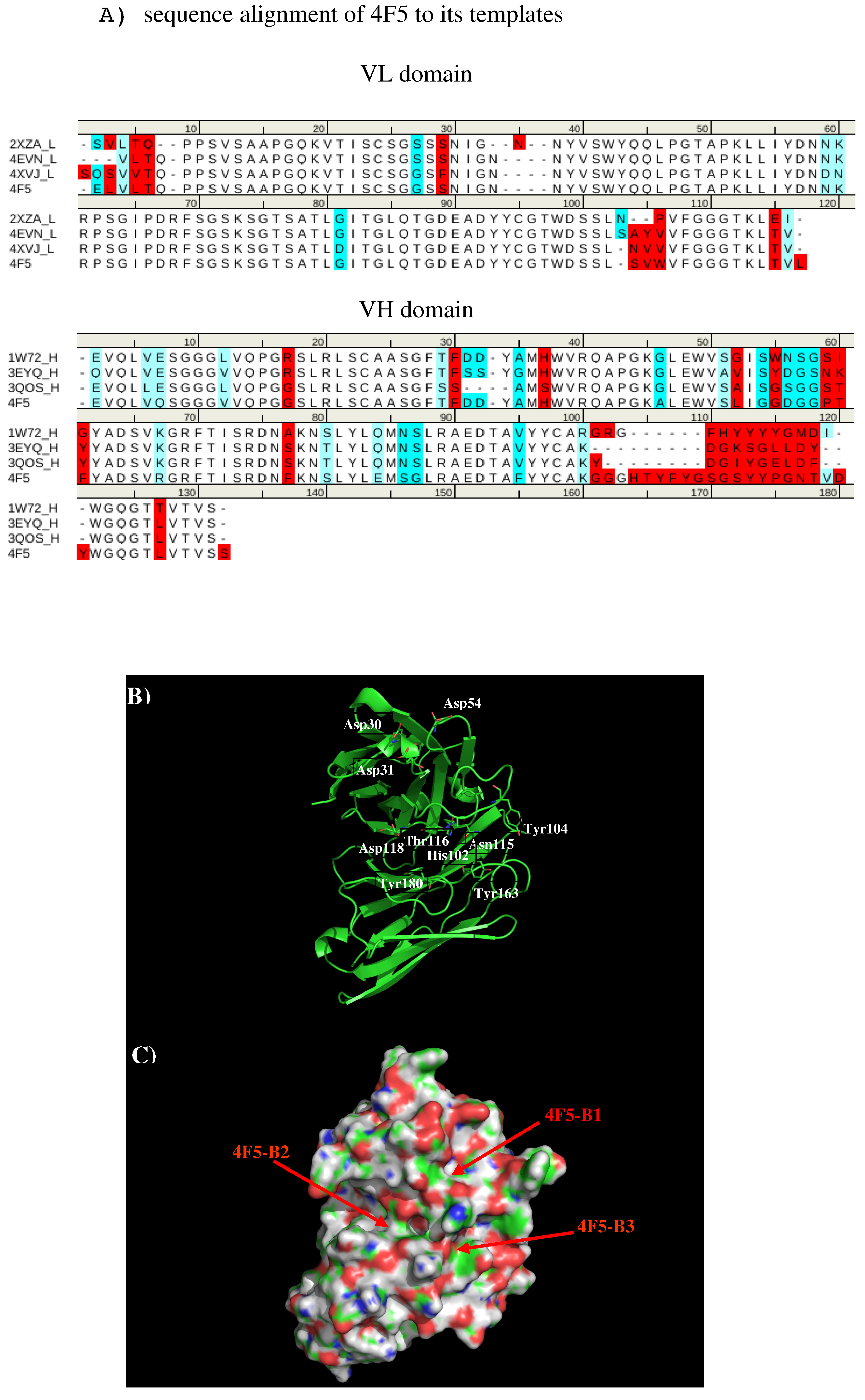
We created a 3D model of the VL and VH domains of antibody 4F5 [
23] using the X-ray structures with PDB [
33] identifiers 2XZA [
34], 4EVN [
44], and 4XVJ [
36] as templates for VL domain, and identifiers 1W72 [
37], 3EYQ [
38] and 3QOS [
39] for VH domain.
Figure 10 shows the sequence alignment of 4F5 to its templates, model structure and surface representation of Ab4F5 with the important residues highlighted. Compared to other antibodies, the surface around the CDR3 loop are highly charged with one site (4F5-B1) close to a cluster of negatively charged residues Asp31, Asp40, and Asp54 of L chain, and another site (4F5-B2) formed by residues His102 of L chain, residues Thr30, Thr50, and Asp51 of H chain, as well as a more hydrophobic site formed by residues His102, Tyr104, and Asn115 of L chain and Tyr33 of H chain. These three sites are separated by ca. 13.00 Å from each other.
Figure 11 shows the locations of the minima of functional groups on the surface of antibody 4F5. Overall, the minima are clustered around the three binding sites B1, B2, and B3 as defined above.
Table S4 (Supplementary Data) shows the number of minima in each cluster and their interaction energies with residues. For the aromatic group, clusters of 37, 6, and 11 BENZ minima were found at the three sites with favorable energies of −11.00 kcal/mol (B1), −10.00 kcal/mol (B2) and −11.20 kcal/mol (B3), respectively. Minima at these sites form hydrophobic interactions with Leu47(H), to Tyr50(H) and with both Tyr33(H) and Tyr104(L). For the IMIA group, 10, 3, and 3 minima were found at three sites with favorable energies of −12.90 kcal/mol (B1), −10.70 kcal/mol (B2), and −10.90 kcal/mol (B3), respectively. These minima interact with Leu47(H), Asp108(L), and with both Tyr33(H) and Tyr104(L), respectively. For the PHEN group, 37, 20, and 18 minima were found with energies up to −13.40 kcal/mol (B1), −13.80 kcal/mol (B2) and −13.40 kcal/mol (B3). While the best minimum at the site B3 forms hydrogen bonds with Asn115(L) and Asn32(H), the best minimum at B2 forms pi-pi interaction with both Tyr104(L) and Tyr33(H). For the INDO group, 11 minima were found at the B1 with favorable energies of −16.10 kcal/mol by hydrogen bonding to CO group of Gly101(L). Only one minimum was found at B3 with energy of −15.20 kcal/mol. For the case of polar groups, 16, 4, and 2 ACEM minima were found at three sites with favorable energies of −11.70 kcal/mol, −10.00 kcal/mol, and −10.10 kcal/mol, respectively. At these three sites, the best minimum interacts with Asp54(L), Asp51(H), and Asn32(H). The positively charged minima MAMM and MGUA were found only at B1 by interacting with Asp54 of L chain. While three MAMM minima have the interaction energies of (−15.20, −16.00) kcal/mol, the best minima consisting of 60 MGUA fragments has the strongest interaction energy of −20.80 kcal/mol.
Based on the distribution of the important minima as shown in
Figure 11, a sequence pattern for peptides that bind to Ab4F5 was derived. As the binding B2 is ca. 15.00 Å distant from the CDR3 loop, we disregard the minima at this binding site. The MCSS minima at the binding sites B1 and B2 are separated by ca 13.00 Å, a distance that could accommodate two amino acids. Based on the distribution of MCSS minima, the key sequence pattern for the binders was defined as “X-J”, in which X = R/K,Q/N,W,F/Y/H, J = F/Y/H,W.
Table 6 lists the distribution of key MCSS minima and the derived sequence pattern.
The predicted epitopes of the H5N1 HA1 using the above-mentioned protocol are shown in
Figure 12A) with the epitope shown in orange lower case characters. Six binding peptides (E1: “ypgnfndyeelkhllsrinhfekiqiipksswsdhe”, E2: ”sffrnvvwlikknntyptikrsyn”, E3: ”llilwgihh”, E4: ”gqsgrmdff”, E5: ”nfesngnfi”, E6: ”smpfhnih”) are predicted from H5N1 HA1 to bind to the antibody (
Figure 12B). While the peptide E1 and E6 are binding to the HA2 in a similar fashion as observed in the crystal structure of AbCR9114 complexed with H5N1 (
Figure 12C), peptides E3 and E4 are buried inside the protein. Therefore, the epitopes for the antibody recognition are considered to be peptides E2 and E5 which are locate at the receptor binding domain, similar to those that bind to antibody HC19 [
18,
22]. This is in contrast to recent phase display experiments in which a conserved epitope (WLLGNP) amongst different clades was identified; this peptide is located at the vestigial esterase domain where the antibody BH151 binds [
23].
Figure 12C) also shows the epitopes predicted from H3N2 HA1 and its comparison to H5N1 HA1. While the overlay region
E3 (“llilwgihh” vs. “lyiwgihh”) is buried in protein, the other region at
E2 (“sffrnvvw” vs. “gffsrlnwl”) is close to the second binder peptide “dvfqnetwdlfv” which is located at the vestigial esterase region and next to the position of 76-WLLGNP-81 identified as epitope in H5N1. Therefore, Ab4F5 could recognize the H3N2 by binding to the vestigial esterase domain.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}