Enhancing CDC and ADCC of CD19 Antibodies by Combining Fc Protein-Engineering with Fc Glyco-Engineering

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Antibodies
2.3. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE), Lectin Blot Analysis, WESTERN Transfer Experiments and Size Exclusion Chromatography (SEC)
2.4. Flow Cytometry
2.5. Cytotoxicity Assay
2.6. Statistical Analysis
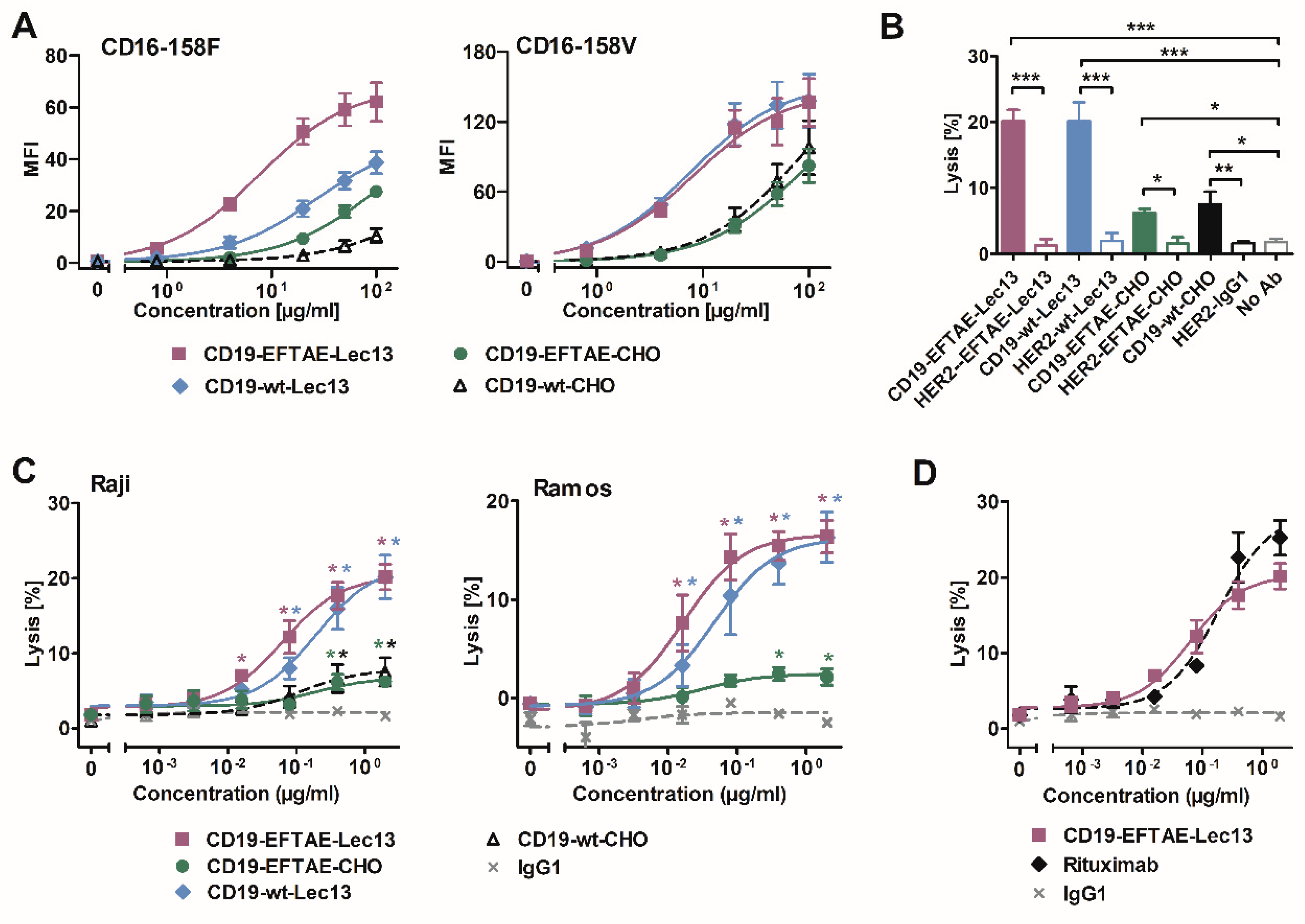
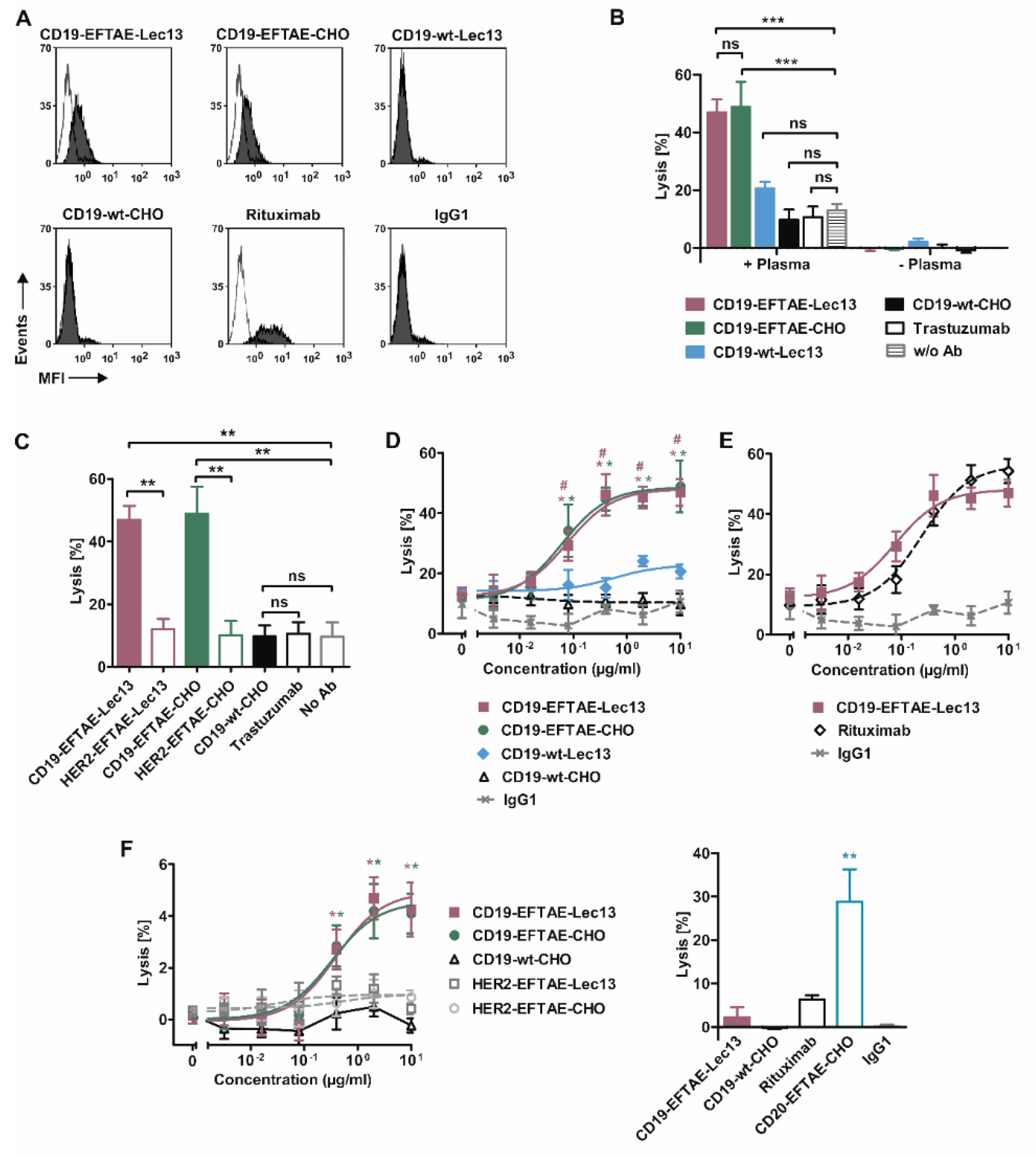
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Hammer, O. CD19 as an attractive target for antibody-based therapy. mAbs 2012, 4, 571–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazawa, N.; Hamaguchi, Y.; Poe, J.C.; Tedder, T.F. Immunotherapy using unconjugated CD19 monoclonal antibodies in animal models for B lymphocyte malignancies and autoimmune disease. Proc. Natl. Acad. Sci. USA 2005, 102, 15178–15183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellner, C.; Peipp, M.; Gramatzki, M.; Schrappe, M.; Schewe, D.M. Perspectives of Fc engineered antibodies in CD19 targeting immunotherapies in pediatric B-cell precursor acute lymphoblastic leukemia. Oncoimmunology 2018, 7, e1448331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frigault, M.J.; Maus, M.V. State of the art in CAR T cell therapy for CD19+ B cell malignancies. J. Clin. Invest. 2020, 130, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foa, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Horton, H.M.; Bernett, M.J.; Pong, E.; Peipp, M.; Karki, S.; Chu, S.Y.; Richards, J.O.; Vostiar, I.; Joyce, P.F.; Repp, R.; et al. Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res. 2008, 68, 8049–8057. [Google Scholar] [CrossRef] [Green Version]
- Salles, G.; Duell, J.; Gonzalez Barca, E.; Tournilhac, O.; Jurczak, W.; Liberati, A.M.; Nagy, Z.; Obr, A.; Gaidano, G.; Andre, M.; et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): A multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020, 21, 978–988. [Google Scholar] [CrossRef]
- Kellner, C.; Otte, A.; Cappuzzello, E.; Klausz, K.; Peipp, M. Modulating Cytotoxic Effector Functions by Fc Engineering to Improve Cancer Therapy. Transfus. Med. Hemother. 2017, 44, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Di Gaetano, N.; Cittera, E.; Nota, R.; Vecchi, A.; Grieco, V.; Scanziani, E.; Botto, M.; Introna, M.; Golay, J. Complement activation determines the therapeutic activity of rituximab in vivo. J. Immunol. 2003, 171, 1581–1587. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, A.D.; Beum, P.V.; Solga, M.D.; DiLillo, D.J.; Lindorfer, M.A.; Hess, C.E.; Densmore, J.J.; Williams, M.E.; Taylor, R.P. Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J. Immunol. 2004, 172, 3280–3288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klepfish, A.; Schattner, A.; Ghoti, H.; Rachmilewitz, E.A. Addition of fresh frozen plasma as a source of complement to rituximab in advanced chronic lymphocytic leukaemia. Lancet Oncol. 2007, 8, 361–362. [Google Scholar] [CrossRef]
- Bannerji, R.; Kitada, S.; Flinn, I.W.; Pearson, M.; Young, D.; Reed, J.C.; Byrd, J.C. Apoptotic-regulatory and complement-protecting protein expression in chronic lymphocytic leukemia: Relationship to in vivo rituximab resistance. J. Clin. Oncol. 2003, 21, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- De Haij, S.; Jansen, J.H.; Boross, P.; Beurskens, F.J.; Bakema, J.E.; Bos, D.L.; Martens, A.; Verbeek, J.S.; Parren, P.W.; van de Winkel, J.G.; et al. In vivo cytotoxicity of type I CD20 antibodies critically depends on Fc receptor ITAM signaling. Cancer Res. 2010, 70, 3209–3217. [Google Scholar] [CrossRef] [Green Version]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002, 99, 754–758. [Google Scholar] [CrossRef] [Green Version]
- Weng, W.K.; Levy, R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 2003, 21, 3940–3947. [Google Scholar] [CrossRef]
- Persky, D.O.; Dornan, D.; Goldman, B.H.; Braziel, R.M.; Fisher, R.I.; Leblanc, M.; Maloney, D.G.; Press, O.W.; Miller, T.P.; Rimsza, L.M. Fc gamma receptor 3a genotype predicts overall survival in follicular lymphoma patients treated on SWOG trials with combined monoclonal antibody plus chemotherapy but not chemotherapy alone. Haematologica 2012, 97, 937–942. [Google Scholar] [CrossRef]
- Dahal, L.N.; Roghanian, A.; Beers, S.A.; Cragg, M.S. FcgammaR requirements leading to successful immunotherapy. Immunol. Rev. 2015, 268, 104–122. [Google Scholar] [CrossRef]
- Cartron, G.; Houot, R.; Kohrt, H.E. Scientific Significance of Clinically Insignificant FcgammaRIIIa-V158F Polymorphism. Clin. Cancer Res. 2016, 22, 787–789. [Google Scholar] [CrossRef] [Green Version]
- Moore, G.L.; Chen, H.; Karki, S.; Lazar, G.A. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. mAbs 2010, 2, 181–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idusogie, E.E.; Wong, P.Y.; Presta, L.G.; Gazzano-Santoro, H.; Totpal, K.; Ultsch, M.; Mulkerrin, M.G. Engineered antibodies with increased activity to recruit complement. J. Immunol. 2001, 166, 2571–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diebolder, C.A.; Beurskens, F.J.; de Jong, R.N.; Koning, R.I.; Strumane, K.; Lindorfer, M.A.; Voorhorst, M.; Ugurlar, D.; Rosati, S.; Heck, A.J.; et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014, 343, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oostindie, S.C.; van der Horst, H.J.; Lindorfer, M.A.; Cook, E.M.; Tupitza, J.C.; Zent, C.S.; Burack, R.; VanDerMeid, K.R.; Strumane, K.; Chamuleau, M.E.D.; et al. CD20 and CD37 antibodies synergize to activate complement by Fc-mediated clustering. Haematologica 2019, 104, 1841–1852. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Zacharias, M. Atomic resolution model of the antibody Fc interaction with the complement C1q component. Mol. Immunol. 2012, 51, 66–72. [Google Scholar] [CrossRef]
- Radaev, S.; Motyka, S.; Fridman, W.H.; Sautes-Fridman, C.; Sun, P.D. The structure of a human type III Fcgamma receptor in complex with Fc. J. Biol. Chem. 2001, 276, 16469–16477. [Google Scholar] [CrossRef] [Green Version]
- Sondermann, P.; Huber, R.; Oosthuizen, V.; Jacob, U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature 2000, 406, 267–273. [Google Scholar] [CrossRef]
- Schewe, D.M.; Alsadeq, A.; Sattler, C.; Lenk, L.; Vogiatzi, F.; Cario, G.; Vieth, S.; Valerius, T.; Rosskopf, S.; Meyersieck, F.; et al. An Fc-engineered CD19 antibody eradicates MRD in patient-derived MLL-rearranged acute lymphoblastic leukemia xenografts. Blood 2017, 130, 1543–1552. [Google Scholar] [CrossRef] [Green Version]
- Kellner, C.; Zhukovsky, E.A.; Potzke, A.; Bruggemann, M.; Schrauder, A.; Schrappe, M.; Kneba, M.; Repp, R.; Humpe, A.; Gramatzki, M.; et al. The Fc-engineered CD19 antibody MOR208 (XmAb5574) induces natural killer cell-mediated lysis of acute lymphoblastic leukemia cells from pediatric and adult patients. Leukemia 2013, 27, 1595–1598. [Google Scholar] [CrossRef] [Green Version]
- Zalevsky, J.; Leung, I.W.; Karki, S.; Chu, S.Y.; Zhukovsky, E.A.; Desjarlais, J.R.; Carmichael, D.F.; Lawrence, C.E. The impact of Fc engineering on an anti-CD19 antibody: Increased Fcgamma receptor affinity enhances B-cell clearing in nonhuman primates. Blood 2009, 113, 3735–3743. [Google Scholar] [CrossRef]
- Jurczak, W.; Zinzani, P.L.; Gaidano, G.; Goy, A.; Provencio, M.; Nagy, Z.; Robak, T.; Maddocks, K.; Buske, C.; Ambarkhane, S.; et al. Phase IIa study of the CD19 antibody MOR208 in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma. Ann. Oncol. 2018, 29, 1266–1272. [Google Scholar] [CrossRef]
- Dalziel, M.; Beers, S.A.; Cragg, M.S.; Crispin, M. Through the barricades: Overcoming the barriers to effective antibody-based cancer therapeutics. Glycobiology 2018, 28, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Matlawska-Wasowska, K.; Ward, E.; Stevens, S.; Wang, Y.; Herbst, R.; Winter, S.S.; Wilson, B.S. Macrophage and NK-mediated killing of precursor-B acute lymphoblastic leukemia cells targeted with a-fucosylated anti-CD19 humanized antibodies. Leukemia 2013, 27, 1263–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, P.; Barbin, K.; Feuchtinger, T.; Greil, J.; Peipp, M.; Zunino, S.J.; Pfeiffer, M.; Handgretinger, R.; Niethammer, D.; Fey, G.H. Chimeric CD19 antibody mediates cytotoxic activity against leukemic blasts with effector cells from pediatric patients who received T-cell-depleted allografts. Blood 2004, 103, 3982–3985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohmachi, K.; Ogura, M.; Suehiro, Y.; Ando, K.; Uchida, T.; Choi, I.; Ogawa, Y.; Kobayashi, M.; Fukino, K.; Yokoi, Y.; et al. A multicenter phase I study of inebilizumab, a humanized anti-CD19 monoclonal antibody, in Japanese patients with relapsed or refractory B-cell lymphoma and multiple myeloma. Int. J. Hematol. 2019, 109, 657–664. [Google Scholar] [CrossRef]
- Cree, B.A.C.; Bennett, J.L.; Kim, H.J.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; Fujihara, K.; Paul, F.; Cutter, G.R.; Marignier, R.; et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): A double-blind, randomised placebo-controlled phase 2/3 trial. Lancet 2019, 394, 1352–1363. [Google Scholar] [CrossRef]
- Wirt, T.; Rosskopf, S.; Rosner, T.; Eichholz, K.M.; Kahrs, A.; Lutz, S.; Kretschmer, A.; Valerius, T.; Klausz, K.; Otte, A.; et al. An Fc Double-Engineered CD20 Antibody with Enhanced Ability to Trigger Complement-Dependent Cytotoxicity and Antibody-Dependent Cell-Mediated Cytotoxicity. Transfus. Med. Hemother. 2017, 44, 292–300. [Google Scholar] [CrossRef]
- Glorius, P.; Baerenwaldt, A.; Kellner, C.; Staudinger, M.; Dechant, M.; Stauch, M.; Beurskens, F.J.; Parren, P.W.; Winkel, J.G.; Valerius, T.; et al. The novel tribody [(CD20)(2)xCD16] efficiently triggers effector cell-mediated lysis of malignant B cells. Leukemia 2013, 27, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Ripka, J.; Adamany, A.; Stanley, P. Two Chinese hamster ovary glycosylation mutants affected in the conversion of GDP-mannose to GDP-fucose. Arch. Biochem. Biophys. 1986, 249, 533–545. [Google Scholar] [CrossRef]
- Patnaik, S.K.; Stanley, P. Lectin-resistant CHO glycosylation mutants. Methods Enzymol. 2006, 416, 159–182. [Google Scholar] [CrossRef]
- Repp, R.; Kellner, C.; Muskulus, A.; Staudinger, M.; Nodehi, S.M.; Glorius, P.; Akramiene, D.; Dechant, M.; Fey, G.H.; van Berkel, P.H.; et al. Combined Fc-protein- and Fc-glyco-engineering of scFv-Fc fusion proteins synergistically enhances CD16a binding but does not further enhance NK-cell mediated ADCC. J. Immunol. Methods 2011, 373, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.R. IgG effector mechanisms. Chem. Immunol. 1997, 65, 88–110. [Google Scholar] [PubMed]
- Cleary, K.L.S.; Chan, H.T.C.; James, S.; Glennie, M.J.; Cragg, M.S. Antibody Distance from the Cell Membrane Regulates Antibody Effector Mechanisms. J. Immunol. 2017, 198, 3999–4011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boross, P.; Jansen, J.H.; de Haij, S.; Beurskens, F.J.; van der Poel, C.E.; Bevaart, L.; Nederend, M.; Golay, J.; van de Winkel, J.G.; Parren, P.W.; et al. The in vivo mechanism of action of CD20 monoclonal antibodies depends on local tumor burden. Haematologica 2011, 96, 1822–1830. [Google Scholar] [CrossRef]
- Gong, Q.; Ou, Q.; Ye, S.; Lee, W.P.; Cornelius, J.; Diehl, L.; Lin, W.Y.; Hu, Z.; Lu, Y.; Chen, Y.; et al. Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J. Immunol. 2005, 174, 817–826. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, A.; Ishida, T.; Yano, H.; Ishii, T.; Kusumoto, S.; Ito, A.; Ri, M.; Mori, F.; Ding, J.; Komatsu, H.; et al. Expression of the ULBP ligands for NKG2D by B-NHL cells plays an important role in determining their susceptibility to rituximab-induced ADCC. Int. J. Cancer 2009, 125, 212–221. [Google Scholar] [CrossRef]
- Meyer, S.; Leusen, J.H.; Boross, P. Regulation of complement and modulation of its activity in monoclonal antibody therapy of cancer. mAbs 2014, 6, 1133–1144. [Google Scholar] [CrossRef] [Green Version]
- Van Meerten, T.; van Rijn, R.S.; Hol, S.; Hagenbeek, A.; Ebeling, S.B. Complement-induced cell death by rituximab depends on CD20 expression level and acts complementary to antibody-dependent cellular cytotoxicity. Clin. Cancer Res. 2006, 12, 4027–4035. [Google Scholar] [CrossRef] [Green Version]
- Derer, S.; Glorius, P.; Schlaeth, M.; Lohse, S.; Klausz, K.; Muchhal, U.; Desjarlais, J.R.; Humpe, A.; Valerius, T.; Peipp, M. Increasing FcgammaRIIa affinity of an FcgammaRIII-optimized anti-EGFR antibody restores neutrophil-mediated cytotoxicity. mAbs 2014, 6, 409–421. [Google Scholar] [CrossRef]
- Peipp, M.; Lammerts van Bueren, J.J.; Schneider-Merck, T.; Bleeker, W.W.; Dechant, M.; Beyer, T.; Repp, R.; van Berkel, P.H.; Vink, T.; van de Winkel, J.G.; et al. Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood 2008, 112, 2390–2399. [Google Scholar] [CrossRef]
- Wang, S.Y.; Veeramani, S.; Racila, E.; Cagley, J.; Fritzinger, D.C.; Vogel, C.W.; St John, W.; Weiner, G.J. Depletion of the C3 component of complement enhances the ability of rituximab-coated target cells to activate human NK cells and improves the efficacy of monoclonal antibody therapy in an in vivo model. Blood 2009, 114, 5322–5330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junttila, T.T.; Parsons, K.; Olsson, C.; Lu, Y.; Xin, Y.; Theriault, J.; Crocker, L.; Pabonan, O.; Baginski, T.; Meng, G.; et al. Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2-amplified breast cancer. Cancer Res. 2010, 70, 4481–4489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmerjahn, F.; Ravetch, J.V. Antibodies, Fc receptors and cancer. Curr. Opin. Immunol. 2007, 19, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Casey, E.; Bournazos, S.; Mo, G.; Mondello, P.; Tan, K.S.; Ravetch, J.V.; Scheinberg, D.A. A new mouse expressing human Fcgamma receptors to better predict therapeutic efficacy of human anti-cancer antibodies. Leukemia 2018, 32, 547–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roßkopf, S.; Eichholz, K.M.; Winterberg, D.; Diemer, K.J.; Lutz, S.; Münnich, I.A.; Klausz, K.; Rösner, T.; Valerius, T.; Schewe, D.M.; et al. Enhancing CDC and ADCC of CD19 Antibodies by Combining Fc Protein-Engineering with Fc Glyco-Engineering. Antibodies 2020, 9, 63. https://0-doi-org.brum.beds.ac.uk/10.3390/antib9040063
Roßkopf S, Eichholz KM, Winterberg D, Diemer KJ, Lutz S, Münnich IA, Klausz K, Rösner T, Valerius T, Schewe DM, et al. Enhancing CDC and ADCC of CD19 Antibodies by Combining Fc Protein-Engineering with Fc Glyco-Engineering. Antibodies. 2020; 9(4):63. https://0-doi-org.brum.beds.ac.uk/10.3390/antib9040063
Chicago/Turabian StyleRoßkopf, Sophia, Klara Marie Eichholz, Dorothee Winterberg, Katarina Julia Diemer, Sebastian Lutz, Ira Alexandra Münnich, Katja Klausz, Thies Rösner, Thomas Valerius, Denis Martin Schewe, and et al. 2020. "Enhancing CDC and ADCC of CD19 Antibodies by Combining Fc Protein-Engineering with Fc Glyco-Engineering" Antibodies 9, no. 4: 63. https://0-doi-org.brum.beds.ac.uk/10.3390/antib9040063