Appendix A.3. Chemical Synthesis
3-O-Triphenylmethyl-dl-glycerol (4). To a stirred solution of glycerol (3a, 10.0 g, 109.6 mmol), DMAP (0.075 g, 0.6 mmol) and trityl chloride (7.5 g, 26.9 mmol) in 20 mL of anhydrous THF at 0 °C were added 4.5 mL of anhydrous triethylamine. The reaction mixture was stirred at r.t. overnight. A solution of NaHCO3 (2.0 g in 50 mL of H2O) was added followed by stirring for 15 min. The product was then extracted with EtOAc (2 × 35 mL). The combined organic phases were washed with brine (2 × 50 mL) and dried over anhydrous Na2SO4. The crude material obtained after evaporation of the solvent was crystallized from dichloromethane upon addition of pentane (1:10 v/v) to give 32.57 g (89.7%) of a white powder containing 4. Rf (hexane/EtOAc 1:1 v/v) 0.42; 1H NMR (400 MHz, CDCl3): δH = 7.49–7.12 (m, 15 H, 3 × Ph), 3.85–3.79 (m, 1 H, C(2)H), 3.63 (dd, J = 11.4, 3.8 Hz, 1 H, C(3)Ha), 3.55 (dd, J = 11.4, 5.7 Hz, 1 H, C(3)Hb), 3.26–3.16 (m, 2H, C(3)H2); 4c: 1H NMR (400 MHz, CDCl3): δH = 7.49–7.12 (m, 15 H, 3 × Ph), 3.97–3.89 (m, 1 H, C(2)H), 3.32–3.26 (m, 4H, C(1)H2 and C(3)H2); 13C NMR (100 MHz, CDCl3, selected signals): δC = 64.1 (C1, HSQC), 64.8 (C3, HSQC), 70.6 (C2, HSQC), 71.1 (C2, HSQC), 86.8 (CqAr), 127.2–129.9 (series of CHAr), 144.0 (OC(Ph)3), 147.1 (OC(Ph)3); [α]D25 = 0.00 (c 0.1, CHCl3); HRMS m/z: [M]+ calcd. for C22H22NaO3: 357.1466, found C22H22NaO3: 357.1461.
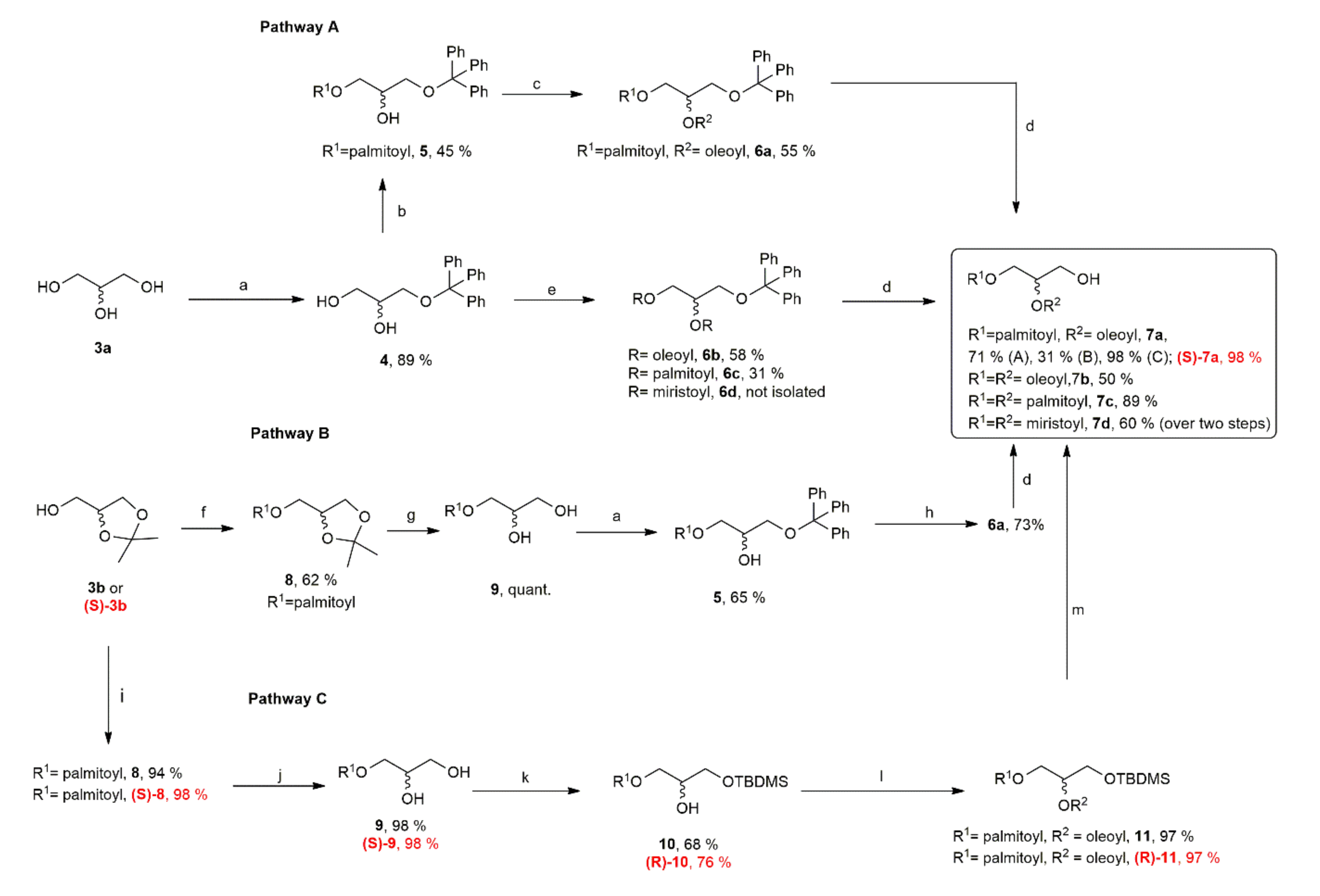
Compound 5, (Pathway A). To a cold (0 °C) solution of 4 (4 g, 12.2 mmol) in 100 mL of CHCl3 were added portion wise 6 g palmitic anhydride (12.1 mmol) and 1.6 g DMAP (13.1 mmol). The resulting solution was allowed to return to room temperature and left under vigorous stirring at r.t. for 48 h. The solution was cooled down to 0 °C (ice bath) and 100 mL of saturated NaHCO3 were slowly added until the excess of palmitic anhydride was hydrolyzed. The phases were separated, and the organic layers were washed with brine (4 × 50 mL) and dried over dry Na2SO4. The crude material obtained after evaporation of the solvent was purified over freshly activated SiO2 with PE:EtOAc (10:0 to 8:2 v/v) yielding 5 as a viscous oil (4.96 g, 55%). Rf (PE/EtOAc 3:1) 0.74. 1H NMR (300 MHz, CDCl3): δH = 7.44–7.39 (m, 5 H, 1 × Ph), 7.33–7.20 (m, 10 H, 2 × Ph), 4.22–4.08 (m, 2 H, C(1)H2), 3.92–3.73 (m, 1 H, C(1)H), 3.21 (d, J = 5.4 Hz, 2 H, C(3)H2), 2.45 (s, 1 H, OH), 2.26 (t, J = 7.4 Hz, 2 H, CH2CH2COOR), 1.62–1.41 (m, 2 H, CH2CH2COOR), 1.25 (s, 24 H, 12 × CH2), 0.87 (t, J = 6.7 Hz, 3H, CH3). 13C NMR: δC = 14.3 (CH3), 22.9 (CH2), 25.1 (CH2), 29.3 (CH2), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2), 29.8–29.9 (series of CH2), 31.2 (CH2), 34.4 (CH2), 64.4 (C3), 65.8 (C2), 69.1 (C1) 87.1 (CqAr), 126.9 (CHAr) 127.7(CHAr) 128.4 (CHAr), 143.8 (OC(Ph)3),173.8 (C=O); [α]D25 = 0.00 (c 0.1, CHCl3).
Compound 6a (Pathway A). 1.96 g of 5 (5.85 mmol) were dissolved in cold CHCl3 (50 mL, 0 °C) together with DMAP (0.8 g, 6.44 mmol) and oleic acid (3.18 g, 6.44 mmol). The resulting solution was slowly warmed to r.t. and the conversion of 5 into 6a was monitored periodically by TLC (PE:EtOAc 4:1 v/v). The starting material 5 was consumed after 16 h. To the cold mixture, 50 mL of a solution of NaHCO3 (3% w/w in water) were slowly added and the biphasic mixtures was stirred for 30 min until it went back to r.t. The organic layer was extracted by adding extra volumes of CHCl3 (3 × 20 mL) and the combined organic layers were washed with saturated solutions of citric acid (pH 6, 2 × 50 mL), NaHCO3 (3 × 25 mL) and brine (3 × 50 mL) and dried over Na2SO4. The crude material obtained after evaporation of the solvent was purified over freshly activated SiO2 eluting with PE:EtOAc (4:1 to 3:1 v/v) yielding 6a as white wax (1.57 g, 55%). 1H NMR (300 MHz, CDCl3): δH = 7.43–7.40 (m, 5 H, 1 × Ph), 7.33–7.18 (m, 10 H, 2 × Ph), 5.39–5.29 (m, s, 1 × Z-CH=CH), 5.28–5.20 (m, 1 H, C(2)H), 4.34 (dd, J = 11.8, 3.8 Hz, 1 H, C(1)Hb), (dd, J = 11.8, 6.6 Hz, 1H, C(1)Ha), 3.28–3.17 (m, 2H, C(3)H2), 2.33 (t, J = 7.5 Hz, 2 H, 1 × CH2COOR) 2.22 (t, J = 7.5 Hz, 2 H, 1 × CH2COOR), 2.10–1.80 (m, 4 H, 2 × CH2-CH=CH-CH2), 1.71–1.47 (m, 4 H, 2 × CH2CH2COOR), 1.25 (s, 42 H, 21 × CH2), 0.95–0.79 (m, superimposition of 2 × t, apparent J = 7.0 Hz, 6 H, 2 × CH3). 13C NMR (75 MHz, CDCl3): δC = 14.3 (CH3), 22.9 (CH2), 25.1 (CH2), 25.2 (CH2), 27.4 (CH2), 27.5 (CH2), 29.3–29.7 (series of CH2), 32.1 (CH2), 32.2 (CH2), 62.4 (C3), 63.0 (C1), 70.6 (C2), 86.9 (CqAr), 127.3 (CH), 128.1 (CH), 129.9 (CH), 130.2 (CH), 129.9 (Z-CH=CH), 130.2 (Z-CH=CH), 143.2 (OC(Ph)3), 173.2 (C=O), 173.6 (C=O). [α]D25 = 0.00 (c 0.1, CHCl3).
Compound 6a (Pathway B). To a cold solution (0 °C) prepared by dissolving 5 (1.6 g, 2.6 mmol) and DMAP (0.4 g, 3.3 mmol) in 10 mL anhydrous CHCl3, were slowly added by using a syringe pump 1.0 g oleoyl chloride (3.3 mmol) and the resulting solution was kept in the dark and under stirring for 18 h at r.t. After consumption of starting material 5 (TLC monitoring), the solution was cooled with an ice bath and 25 mL of saturated NaHCO3 solution were added. Organic layers were separated and then washed with additional saturated NaHCO3 solution (2 × 25 mL) and with brine (3 × 25 mL) and dried over Na2SO4. The crude material obtained after evaporation of the solvent was purified over freshly activated SiO2 with PE:EtOAc (10:0 to 9:1, v/v) yielding 6a as a white wax (1.60 g, 73%). Rf (PE/EtOAc 9:1) 0.75. Spectroscopic data were in agreement with those recorded for the compound obtained in route A.
Appendix A.3.1 Synthesis of Compounds 6b–6d
General method. To a stirred solution of 4 in 25–50 mL CHCl3 were added equimolar amounts of the corresponding acyl chlorides: oleoyl (for 6b), palmitoyl (for 6c) or myristoyl (for 6d) and DMAP (0.4 mol eq.). The resulting solutions were stirred overnight at r.t. The excess of acyl chlorides was decomposed by addition of 50 mL NaHCO3 (0.4 M) and the resulting biphasic solutions were stirred for 15 min. The biphasic solutions were extracted with CHCl3 (2 × 50 mL), and the combined organic phases were washed with 2 × 10 mL of brine and dried over Na2SO4. Evaporation of the solvent followed by chromatography over freshly activated SiO2 with CHCl3 gave the wished compounds 6b and 6c. Product 6d was not isolated, and the deprotection was carried out on crude mixture.
6b. White solid. Rf (hexane/EtOAc 4:1) 0.36; 1H NMR (300 MHz, CDCl3): δH = 7.40–7.11 (m, 15 H, 3 × Ph), 5.27 (m, 4 H, 2 × Z-CH=CH), 5.21–5.17 (m, 1 H, C(2)H), 4.33–4.01 (m, 2 H, C(1)H2), 3.16 (m, 2 H, C(3)H2), 2.34–2.21 (m, 4 H, 2 × CH2COOR), 2.02–1.85 (m, 8 H, 2 × CH2-CH=CH-CH2), 1.62–1.42 (2 × br, 4 H, 2 × CH2CH2COOR), 1.21 (s, 40 H, 20 × CH2), 0.81 (t, J = 8.0 Hz, 6 H, 2 × CH3). 13C NMR (75 MHz, CDCl3): δC = 14.3 (CH3), 22.9 (CH2), 25.0 (CH2), 25.3 (CH2), 27.3 (CH2), 27.4 (CH2), 29.4–29.9 (series of CH2), 32.1 (CH2), 34.3 (CH2), 34.4.7 (CH2), 62.2 (C3), 62.0 (C1), 72.2 (C2), 86.9 (CqAr), 127.1 (CH), 127.4 (CH), 127.9 (CH), 128.0 (CH), 128.1(CH),128.7 (CH), 128.8 (CH), 129.9 (Z-CH=CH), 130.4 (Z-CH=CH), 143.2 (OC(Ph)3), 173.5 (C=O), 173.9 (C=O). [α]D25 = 0.00 (c 0.1, CHCl3). ESI-MS m/z 885 as M+Na+.
6c. White wax. Rf (PE:EtOAc 4:1) 0.50; 1H NMR (400 MHz, CDCl3): δH = 7.45–7.39 (m, 5 H, 1 × Ph), 7.33–7.20 (m, 10 H, 2 × Ph), 5.30–5.22 (m, 1 H, C(2)H), 4.34 (dd, J = 11.8, 3.7 Hz, 1 H, C(1)H2), 4.23 (dd, J = 11.8, 6.7 Hz, 1 H, C(3)H2), 3.26–3.19 (m, 4 H, 2 × CH2CH2COOR), 2.33 (t, J = 9.0 Hz, 2 H, 1 × CH2CH2COOR), 2.23 (t, J = 9.0 Hz, 2 H, 1 × CH2CH2COOR), 1.69–1.50 (m, 4 H, 2 × CH2CH3), 1.25, (s 44 H, 22 × CH2), 0.88 (t, J = 9.0 Hz, 6 H, 2 × CH3); 13C (100 MHz) δC = 14.2 (CH3), 22.9 (CH2), 24.4 (CH2–COOR), 24.8 (CH2CH2COOR), 25.1 (CH2), 25.2 (CH2), 29.1 (CH2), 29.2 (CH2), 29.3 (CH2), 29.2 (CH2), 29.4 (CH2), 29.7 (CH2), 29.8–30.0 (series of CH2), 63.1 (C3), 62.7 (C1), 70.5 (C2), 86.9 (CqAr), 122.2 (Ar) 127.9 (Ar), 128.6 (Ar), 144.8 (C(Ph3)), 173.2 (C=O), 173.6 (C=O); [α]D25 = 0.00 (c 0.1, CHCl3); HRMS m/z: [M]+ calcd. for C54H82O5: 819.6162, found C54H82NaO5: 833.6054.
6d. The crude mixture containing 6d was directly treated for deprotection. ESI-MS m/z 777 as M+Na+;
Compound 8 (Pathway B). 3.0 g of α,β-isopropylidene-dl-glycerol (3b, 22.7 mmol) were dissolved in dry CH2Cl2 (80.0 mL) and the solution was cooled to 0 °C using a thermostatic bath. Palmitoyl chloride (7.80 g, 28.4 mmol) was added together with DMAP (3.5 g, 28.4 mmol) and the resulting solution was left under vigorous stirring at r.t. for 18 h. 25 mL of saturated NaHCO3 were added dropwise until the excess of palmitoyl chloride was consumed. The phases were separated, and the organic layers were washed with brine (3 × 25 mL) and dried over dry Na2SO4. The crude material obtained after evaporation of the solvent was purified over freshly activated SiO2 with Cy: EtOAc (9:1 v/v) yielding 8 as a viscous oil (5.23 g, 62%). Rf (Cy/EtOAc 1:1) 0.70. 1H NMR (300 MHz, CDCl3): δH = 4.35–4.30 (m, C(4)), 4.20–4.01 (m, 3 H, RCOOCHHb, RCOOCHaH, C(5)Hb), 3.72 (dd, J = 8.4, 6.2 Hz, 1 H; C(5)Ha), 2.32 (t, J = 6.0 Hz, 2 H, CH2COOR), 2.03 (s, 1H), 1.69–1.51 (m, 2 H, CH2CH2COOR), 1.42, 1.36 (2 × s, 6 H, (CH3)2C(2)), 1.24 (m, 20 H, 10 × CH2), 0.86 (t, J = 8.0 Hz, 3 H, CH3). 13C NMR: δC = 14.1 (CH3), 22.7 (CH2), 24.9 (CH2), 25.4 (CH2), 26.7 (CH2), 29.8–29.9 (series of CH2), 31.9 (CH2), 34.1 (CH2), 64.5 (COOCH2), 66.3 (C5), 76.6 (C4), 109.8 (C2), 173.6 (C=O); [α]D25 = 0.00 (c 0.1, CHCl3).
Compound 9. (Pathway B). 1.8 g of 8 (4.9 mmol) were dissolved in 40 mL of dry CH2Cl2 and 5.8 g of Amberlyst® 15 H+ resin were added. The suspension was stirred vigorously at r.t. until complete disappearing of the starting material was observed (4 h). The solution was filtered over a pad of Celite and the solvent evaporated. 1.6 g of 9 (>99.0%) were recovered as a yellowish oil. Rf (PE/EtOAc 1:1) 0.47. 1H NMR (300 MHz, CDCl3): δH = 4.23–4.09 (m, 2 H, C(1)H2), 3.92–3.86 (m, 1 H, C(2)H), 3.69 (dd, J = 11.5, 3.8 Hz, 1 H, C(3)Hb), 3.58 (dd, J = 11.5, 8.9 Hz, 1 H, C(3)Ha), 2.34 (t, J = 7.6 Hz, 2 H, CH2COOR), 1.69–1.52 (m, 2 H, CH2CH2COOR), 1.24, (s, 24 H, 12 × CH2), 0.87 (t, J = 6.7 Hz, 3 H, CH3). 13C NMR: δC = 14.1 (CH3), 22.7 (CH2), 24.9 (CH2), 25.4 (CH2), 29.1–29.7 (series of CH2), 31.9 (CH2), 34.2 (CH2), 63.1 (C1), 64.9 (C2), 76.6 (C3), 174.2 (C=O); [α]D25 = 0.00 (c 0.1, CHCl3).
Appendix A.3.2 Deprotection of compounds 6a–6d into 7a–7d: Pathways A and B
General method for deprotection of 6a–6d into 7a–7d obtained from routes A and B.
A mixture of CHCl
3–MeOH (100 mL, 1:1
v/v) containing 44 µL of concentrated HCl (12 N, 37%) was added dropwise to a CHCl
3–MeOH solution (100 mL, 1:1
v/v) of
8a–
d during 6 h at 0 °C. The obtained clear solution was left stirring at 4 °C overnight. A saturated solution of NaHCO
3 was then added slowly (15 min) and the resulting heterogeneous biphasic solution was stirred up to room temperature. The resulting solutions were extracted with CHCl
3 (3 × 250 mL), and the combined organic phases was washed with brine (3 × 100 mL) and dried over Na
2SO
4. Evaporation of the solvent followed by chromatography over freshly activated SiO
2 with CHCl
3 gave products
7a–
7d as pale–yellow oils. Yields are reported in
Table A1.
Table A1.
Data for the preparation of rac diacyl glycerols 6b–6d using 4 as common building block.
Table A1.
Data for the preparation of rac diacyl glycerols 6b–6d using 4 as common building block.
| Entry | Acylation | Scale 1 | Yield | Entry | Deprotection | Scale 1 | Yield |
|---|
| | | | | 4 | 6a→7a | 1.6 g | 45% |
| 1 | 4→6b | 2.2 g | 58% | 5 | 6b→7b | 700 mg | 50% |
| 2 | 4→6c | 1.0 g | 31% | 6 | 6c→7c | 450 mg | 89% |
| 3 | 4→6d | 1.0 g | – | 7 | 6d→7d | 910 mg 2 | 60% |
7a. Rf (9:1 PE/EtOAc) 0.15; 1H NMR (300 MHz, CDCl3): δH = 5.41–5.28 (m, 2 H, Z-CH=CH), 5.15–4.98 (m, 1 H, C(2)H), 4.32 (dd, J = 11.9, 4.5 Hz, 1 H, C(1)Hb), 4.22 (dd, J = 11.9, 5.7 Hz, 1 H, C(1)Ha), 3.78–3.56 (m, 2 H, C(3)H2), 2.32 (dd, J = 14.8, 7.3 Hz, 4 H, 2 × CH2COOR), 2.08–1.92 (m, 4 H, 2 × CH2-CH=CH-CH2), 1.70–1.55 (m, 4H, 2 CH2CH2COOR), 1.30, 1.26 (2 × br, 38 H, 19 × CH2), 0.96–0.76 (m, superimposition of 2 × t, apparent J = 6.8 Hz, 6 H, 2 × CH3). 13C NMR (75 MHz, CDCl3): δC = 14.3 (CH3), 22.9 (CH2), 25.1 (CH2), 27.3 (CH2), 27.4 (CH2), 29.3 (CH2), 29.5–29.9 (series of CH2), 30.0 (CH2), 32.1 (CH2), 34.3 (CH2), 61.7 (C3), 62.2 (C1), 72.3 (C2), 129.9 (Z-CH=CH), 130.2 (Z-CH=CH), 174.0 (C=O), 176.6 (C=O); [α]D25 = 0.00 (c 0.1, CHCl3).
7b. Rf (2:1 hexane/EtOAc) 0.30; 1H NMR (300 MHz, CDCl3): δH = 5.40–5.28 (m, 4 H, 2 × Z-CH=CH), 5.08 (quint, 1 H, J = 5.0 Hz, C(2)H), 4.31 (dd, J = 11.9, 4.6 Hz, 1 H, C(1)HHb), 4.24 (dd, J = 11.9, 4.6 Hz, 1 H, C(1)HaH), 3.74 (dd, J = 12.2, 4.7 Hz, 1 H, C(3)HHb), 3.72 (dd, J = 12.2, 4.7 Hz, 1 H, C(3)HaH), 2.39–2.37 (2 × t, J = 7.5 Hz, 4 H, 2 × CH2COOR), 2.08–1.90 (m, 8H, 2 × CH2– CH=CH-CH2), 1.67–1.56 (m, 4 H, 2 × CH2CH2COOR), 1.30, 1.26 (2 × br, 40 H, 20 × CH2), 0.87 (t, J = 6.8 Hz, 6 H, 2 × CH3). 13C NMR (75 MHz, CDCl3): δC = 14.1 (CH3), 22.1 (CH2), 24.8 (CH2), 24.9 (CH2), 25.6 (CH2), 27.0 (CH2), 27.1 (CH2), 27.2 (CH2), 29.0–29.2 (4 × CH2), 29.3 (CH2), 29.5 (CH2), 29.7 (CH2), 29.8 (CH2), 31.9 (CH2), 34.0 (CH2), 34.3 (CH2), 61.5 (C3), 62.0 (C1), 72.2 (C2), 129.8 (Z-CH=CH), 130.0 (Z-CH=CH), 173.5 (C=O), 173.9 (C=O); [α]D25 = 0.00 (c 0.1, CHCl3); ESI–MS m/z 643 as [M+Na]+; HRMS m/z: [M]+ calcd. for C39H72NO5: 634.5410, found C39H72NO5: 634.5272.
7c. Rf (PE/EtOAc 7:1) 0.30; 1H NMR (400 MHz, CDCl3): δH = 5.13–5.03 (m, 1H, C(2)H), 4.31 (dd, J = 11.9, 4.4 Hz, 1H, C(1)HHb), 4.22 (dd, J = 11.9, 5.8 Hz, 1H, C(1)HaH), 3.72 (d, J = 5.0 Hz, 1H, C(3)H2), 2.32 (dd, J = 9.0, 7.6 Hz, 4H, 2 CH2COOR), 1.61 (dd, J = 13.1, 6.8 Hz, 4H, 2 × CH2CH2COOR), 1.24 (s, 52H, 26 × CH2), 0.87 (t, J = 6.9 Hz, 6H, 2 × CH3). 13C NMR (75MHz, CDCl3): δC = 14.3 (CH3), 22.9 (CH2), 25.0 (CH2), 25.1 (CH2), 29.3–29.9 (series of CH2), 34.3 (CH2), 34.5 (CH2), 61.7 (C3), 62.2 (C1), 72.3 (C2), 173.7 (C=O), 174.0 (C=O); [α]D25 = 0.00 (c 0.1, CHCl3); ESI–MS m/z 591 as M+Na+;
7d. Rf (2:1 hexane/EtOAc) 0.40; 1H NMR (400 MHz, CDCl3): δH = 5.08 (quint, 1H, J = 8.2 Hz, C(2)H), 4.32 (dd, J = 11.9, 4.5 Hz, 1H, C(1)HHb), 4.23 (dd, J = 11.9, 5.7 Hz, 1H, C(1)HaH), 3.73 (d, J = 4.8 Hz, 1H, C(3)H2), 2.33 (dd, J = 15.9, 8.2 Hz, 4H, 2 × CH2COOR), 2.08 (s, 1H, –OH), 1.67–1–57 (m, 4H, 2 × CH2CH2COOR), 1.25 (s, 40H, 20 × CH2), 0.87 (t, J = 6.9 Hz, 6H, 2 × CH3). 13C NMR (75MHz, CDCl3): δC = 14.3 (CH3), 22.9 (CH2), 25.1 (CH2), 25.2 (CH2), 29.2 (CH2), 29.3 (CH2), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2), 29.8 (CH2), 29.9 (CH2), 30.0 (CH2), 32.1 (CH2), 34.3 (CH2), 34.5 (CH2), 61.7 (C3), 62.2 (C1), 72.4 (C2), 173.7 (C=O), 174.0 (C=O); [α]D25 = 0.00 (c 0.1, CHCl3);
Appendix A.3.3 Synthesis of Compounds 7a and (S)-(7a): Pathway C
Compound 8 (Pathway C). 3.7 g of α,β–isopropylidene–dl–glycerol (3b, 28.6 mmol) were dissolved in dry CH2Cl2 (75 mL) and palmitic acid (7.3 g, 28.6 mmol) was added. The solution was cooled to 0 °C using an ice bath, DMAP (1.0 g, 8.6 mmol) and EDC∙HCl (7.1 g, 37.2 mmol) were added together. The resulting solution was left under vigorous stirring at r.t. for 18 h. A quantity of 150 mL of saturated NaHCO3 was added to quench the reaction. The product was then extracted with CH2Cl2 (2 × 100 mL). The combined organic phases were dried over anhydrous MgSO4 and the crude material obtained after evaporation of the solvent was purified over SiO2 with PE:EtOAc (99:1 to 85:15, v/v) yielding 8 as a white powder (9.99 g, 94%). Rf (PE/EtOAc 9:1) 0.5. 1H NMR (300 MHz, CDCl3): δH = 4.33–4.27 (m, 1H, C(4)H), 4.19–4.05 (m, 3H, RCOOCHHb, RCOOCHaH, C(5)Hb), 3.74 (dd, J = 8.4, 6.2 Hz, 1 H, C(5)Ha), 2.34 (t, J = 7.6 Hz, 2 H, CH2COOR), 1.64–1.57 (m, 2 H, CH2CH2COOR), 1.43, 1.37 (2 × s, 6 H, (CH3)2C(2)), 1.25 (m, 24 H, 12 × CH2), 0.88 (t, J = 6.9 Hz, 3 H, CH3). 13C NMR: δC = 14.1 (CH3), 22.7 (CH2), 24.9 (CH2), 25.4 (CH2), 26.7 (CH2), 29.8–29.9 (series of CH2), 31.9 (CH2), 34.1 (CH2), 64.5 (COOCH2), 66.3 (C5), 76.6 (C4), 109.8 (C2), 173.6 (C=O); [α]D25 = 0.00 (c 0.1, CHCl3).
Compound 9 (Pathway C). A quantity of 9.99 g of 8 (26.9 mmol) was dissolved in a mixture of AcOH/H2O (100/25 mL, 4:1 v/v) and kept under vigorous stirring at 55 °C for 2 h. The solution was cooled to r.t. and 150 mL of saturated NaHCO3 were added dropwise until the solution was neutralized. The product was then extracted with ethyl acetate (2 × 100 mL). The combined organic phases were washed with brine (100 mL) and dried over anhydrous MgSO4. The resulting solution was evaporated yielding 9 as a white powder (8.90 g, 99%). 1H NMR (300 MHz, CDCl3): δH = 4.23–4.11 (m, 2 H, C(1)H2), 3.96–3.90 (m, 1 H, C(2)H), 3.70 (dd, J = 11.4, 3.8 Hz, 1 H, C(3)Hb), 3.60 (dd, J = 11.4, 5.7 Hz, 1 H, C(3)Ha), 2.80 (s, 2 H, 2 × OH), 2.35 (t, J = 7.4 Hz, 2 H, CH2COOR), 1.67–1.58 (m, 2 H, CH2CH2COOR), 1.26 (m, 24 H, 12 × CH2), 0.88 (t, J = 6.6 Hz, 3 H, CH3). 13C NMR: δC = 14.1 (CH3), 22.7 (CH2), 24.9 (CH2), 25.4 (CH2), 29.1–29.7 (series of CH2), 31.9 (CH2), 34.2 (CH2), 63.1 (C1), 64.9 (C2), 76.6 (C3), 174.2 (C=O); [α]D25 = 0.00 (c 0.1, CHCl3).
Compound 10. A quantity of 8.90 g of 9 (26.9 mmol) was dissolved in dry CH2Cl2 (250.0 mL) and imidazole (2.7 g, 40.4 mmol) was added. A solution of TBDMSCl (4.5 g, 29.6 mmol) in dry CH2Cl2 (50 mL) was added dropwise with an addition funnel and the resulting solution was left under vigorous stirring at r.t. for 18 h. The suspension was filtered over a pad of Celite and the solvent was evaporated. The crude material obtained was purified over SiO2 with PE:EtOAc (95:5 to 4:1, v/v) yielding 10 as a yellowish oil (8.11 g, 68%). Rf (PE/EtOAc 4:1) 0.8. 1H NMR (300 MHz, CDCl3): δH = 4.18–4.08 (m, 2 H, C(1)H2), 3.91–3.84 (m, 1 H, C(2)H), 3.68 (dd, J = 10.2, 5.6 Hz, 1 H, C(3)Hb), 3.60 (dd, J = 10.2, 4.6 Hz, 1 H, C(3)Ha), 2.34 (t, J = 7.6 Hz, 2 H, CH2COOR), 1.65–1.68 (m, 2 H, CH2CH2COOR), 1.25 (m, 24 H, 12 × CH2), 0.93–0.90 (m, 9H, t–Bu–Si), 0.88–0.86 (m, 3 H, CH3(CH2)n), 0.08 (s, 6 H, CH3Si). 13C NMR: 13C NMR: δC = –5.4 (2× CH3), 14.2 (CH3), 18.4 (Cq tBu), 22.8 (CH2), 25.8 (CH2), 25.9 (3× CH3), 29.3–29.8 (series of CH2), 32.0 (CH2), 34.3 (CH2), 63.8 (C1), 65.1 (C2), 70.1 (C3), 174.1 (C=O). [α]D25 = 0.00 (c 0.1, CHCl3).
Compound 11. A quantity of 5 g of 10 (11.2 mmol) was dissolved in dry CH2Cl2 (50.0 mL) and oleic acid (3.5 g, 12.3 mmol) was added. The solution was cooled to 0 °C using an ice bath before DMAP (0.4 g, 3.4 mmol) and EDC∙HCl (2.8 g, 14.6 mmol) were added together. The resulting solution was left under vigorous stirring at r.t. for 18 h. 75 mL of water were added to quench the reaction and the product was then extracted with CH2Cl2 (2 × 50 mL). The combined organic phases were dried over anhydrous MgSO4. The crude material obtained after evaporation of the solvent was purified over SiO2 with PE:EtOAc (99:1 to 9:1, v/v) giving 11 as a colorless oil (7.71 g, 97%). Rf (PE/EtOAc 16:1) 0.7. 1H NMR (300 MHz, CDCl3): δH = 5.36–5.32 (m, 2 H, Z-CH=CH), 5.09–5.05 (m, 1 H, CHOCOR), 4.33 (dd, J = 11.8, 3.7 Hz, 1 H, SiOCH2CHCHaHbOCOR), 4.16 (dd, J = 11.8, 6.2 Hz, 1 H, SiOCH2CHCHaHbOCOR), 3.71 (d, J = 5.3 Hz, 2 H, R(O)COCH2CHCHaHbOSi), 2.33–2.27 (m, 4 H, 2 × CH2COOR), 2.04–1.97 (m, 4 H, CH2-CH=CH-CH2), 1.64–1.58 (m, 4 H, 2 × CH2CH2COOR), 1.29 (m, 44 H, 22 × CH2), 0.91–0.85 (m, 15 H, t–Bu–Si, 2 × CH3), 0.05 (s, 6 H, CH3Si). 13C NMR: δC = –5.3 (2× CH3), 14.3 (CH3), 18.4 (Cq tBu), 22.8 (CH2), 25.1 (2× CH2), 25.9 (3× CH3), 27.3 (2× CH2), 29.2–29.9 (series of CH2), 32.0 (2× CH2), 34.3 (CH2), 34.4 (CH2), 61.6 (C1), 62.6 (C2), 71.8 (C3), 129.9 (Z-CH=CH), 130.2 (Z-CH=CH), 173.3 (C=O), 173.6 (C=O). [α]D25 = 0.00 (c 0.1, CHCl3).
Compound 7a. (Route C) A quantity of 7.71 g of 11 (10.9 mmol) was dissolved in a mixture of THF/MeCN (50/50 mL, 1:1 v/v) and Et3N·3HF (8.8 g, 54.3 mmol) was added slowly. The resulting solution was left under vigorous stirring at r.t. for 7 h. 150 mL of saturated NaHCO3 were added drop wise to quench the reaction and the product was then extracted with CH2Cl2 (2 × 100 mL). The combined organic phases were washed with water (100 mL), dried over anhydrous MgSO4 and concentrated, giving 7a as a colorless oil (6.36 g, 98%). 1H NMR (300 MHz, CDCl3): δH = 5.38–5.30 (m, 2 H, Z-CH=CH), 5.11–5.05 (m, 1 H, CHOCOR), 4.32 (dd, J = 11.8, 4.4 Hz, 1 H, C(1)Hb), 4.23 (dd, J = 11.8, 5.7 Hz, 1 H, C(1)Ha), 3.76–3.70 (m, 2 H, C(3)H2), 2.33 (dd, J = 14.7, 7.4 Hz, 4H, 2 × CH2COOR), 2.06–1.97 (m, 4 H, CH2-CH=CH-CH2), 1.68–1.56 (m, 4 H, 2 × CH2CH2COOR), 1.27 (m, 44 H, 22 × CH2), 0.91–0.85 (m, 6 H, 2 × CH3). 13C NMR: δC = 14.3 (CH3), 22.9 (CH2), 25.1 (CH2), 27.3 (CH2), 27.4 (CH2), 29.3 (CH2), 29.5–29.9 (series of CH2), 30.0 (CH2), 32.1 (CH2), 34.3 (CH2), 61.7 (C3), 62.2 (C1), 72.3 (C2), 129.9 (Z-CH=CH), 130.2 (Z-CH=CH), 174.0 (C=O), 176.6 (C=O); [α]D25 = 0.00 (c 0.1, CHCl3).
Compound (S)-8. A quantity of 3.7 g (R)-(−)-2,2-Dimethyl-1,3-dioxolane-4-methanol (S)-3b (15.1 mmol) was dissolved in dry CH2Cl2 (40 mL) and palmitic acid (3.8 g, 15.1 mmol) was added. The solution was cooled to 0 °C using an ice bath, before DMAP (0.5 g, 4.5 mmol) and EDC∙HCl (3.8 g, 19.6 mmol) were added together. The resulting solution was left under vigorous stirring at r.t. for 18 h. 75 mL of saturated NaHCO3 were added to quench the reaction and the product was then extracted with CH2Cl2 (2 × 50 mL). The combined organic phases were dried over anhydrous MgSO4. The crude material obtained after evaporation of the solvent was purified over SiO2 with PE:EtOAc (99:1 to 85:15, v/v) furnishing (S)-8 as a white powder (5.55 g, 98%). [α]D25 = 0.80 (c 0.05, CHCl3)
Compound (S)-9. A quantity of 5.55 g of (S)-8 (14.9 mmol) was dissolved in a mixture of AcOH/H2O (60/15 mL, 4:1 v/v) and left under vigorous stirring at 55 °C for 2 h. The solution was cooled to r.t. and 75 mL of saturated NaHCO3 were added dropwise until the solution was neutralized. The product was then extracted with ethyl acetate (2 × 50 mL). The combined organic phases were washed with brine (50 mL) and dried over anhydrous MgSO4. The solvent was evaporated furnishing (S)-9 as a white powder (4.61 g, 94%) [α]D25 = 0.40 (c 0.05, CHCl3)
Compound (R)-10. A quantity of 4.61 g of (S)-9 (13.94 mmol) was dissolved in dry CH2Cl2 (130.0 mL) and imidazole (1.42 g, 20.9 mmol) was added. A solution of TBDMSCl (2.3 g, 15.3 mmol) in dry CH2Cl2 (30 mL) was added dropwise via an addition funnel and the resulting solution was left under vigorous stirring at r.t. for 18 h. The suspension was filtered over a pad of Celite and solvent was evaporated. The crude material obtained was purified over SiO2 with PE:EtOAc (95:5 to 4:1, v/v) giving (R)-10 as a yellowish oil (4.77 g, 76%). [α]D25 = 1.73 (c 0.05, CHCl3)
Compound (R)-11. A quantity of 4.77 g of (R)-10 (10.7 mmol) was dissolved in dry CH2Cl2 (50 mL) and oleic acid (3.3 g, 11.7 mmol) was added. The solution was cooled to 0 °C using an ice bath before DMAP (0.4 g, 3.2 mmol) and EDC∙HCl (2.6 g, 13.9 mmol) were added. The resulting solution was left under vigorous stirring at r.t. for 18 h 75 mL of water were added to quench the reaction and the product was then extracted with CH2Cl2 (2 × 50 mL). The combined organic phases were dried over anhydrous MgSO4. The crude material obtained after evaporation of the solvent was purified over SiO2 with PE:EtOAc (99:1 to 9:1 v/v) giving (R)-11 as a colorless oil (7.35 g, 96%). (R)-11: [α]D25 = 2.31 (c 0.05, CHCl3)
Compound (S)-7a. A quantity of 7.35 g of (R)-11 (10.3 mmol) was dissolved in a mixture of THF/MeCN (50/50 mL, 1:1 v/v) and Et3N·3HF (8.3 g, 51.2 mmol) was added slowly. The resulting solution was left under vigorous stirring at r.t. for 7 h. 150 mL of saturated NaHCO3 were added dropwise to quench the reaction and the product was then extracted with CH2Cl2 (2 × 100 mL). The combined organic phases were washed with water (100 mL), dried over anhydrous MgSO4 and concentrated, giving (S)–7a as a colorless oil (6.08 g, 98%). [α]D25 = –0.60 (c 0.05, CHCl3)
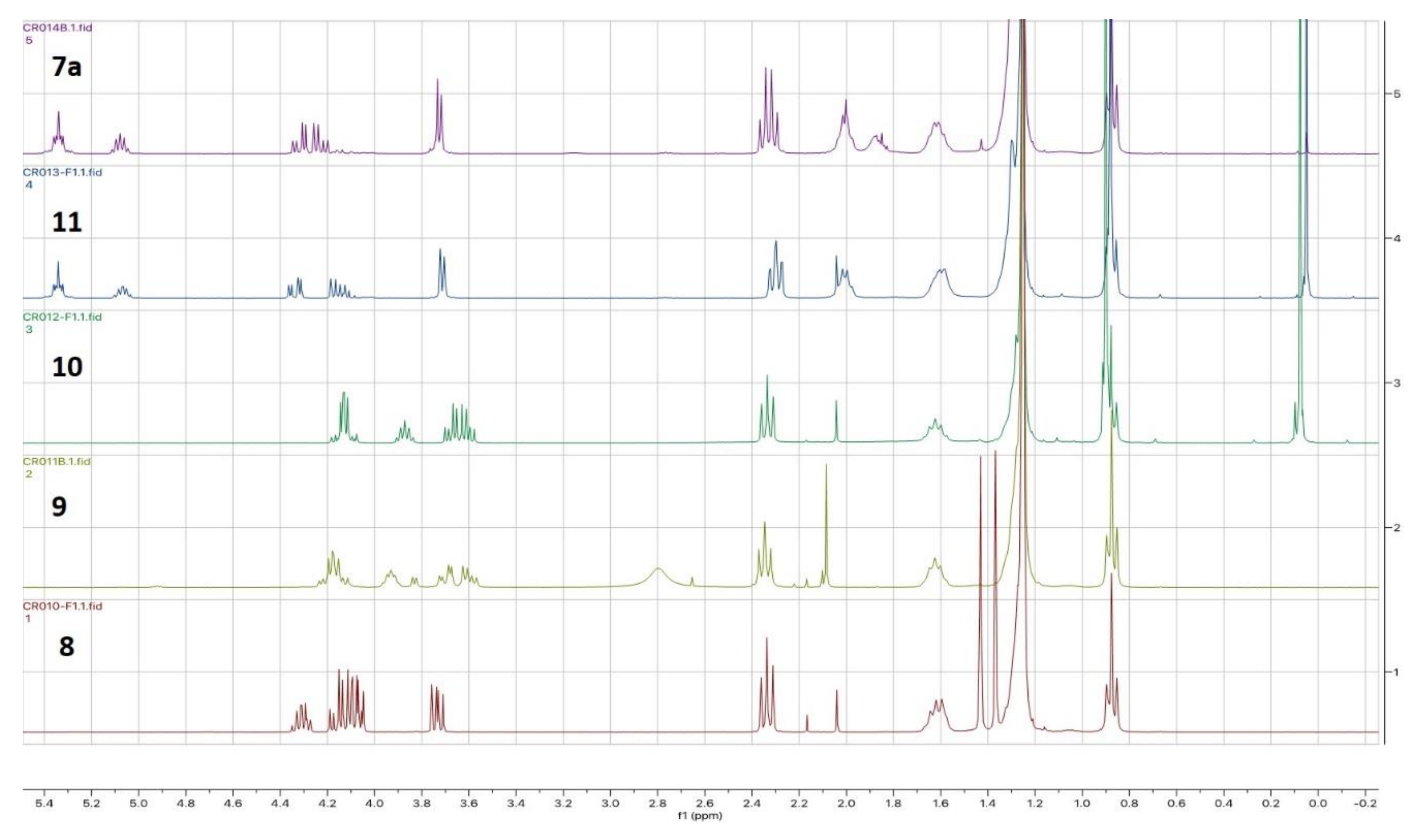
Figure A1.
1H NMR spectra (300 MHz, CDCl3) of the racemic compounds 8 to 7a obtained via Pathway C. The NMR spectra of compounds (S)-8 to (S)-7a are identical.
Figure A1.
1H NMR spectra (300 MHz, CDCl3) of the racemic compounds 8 to 7a obtained via Pathway C. The NMR spectra of compounds (S)-8 to (S)-7a are identical.
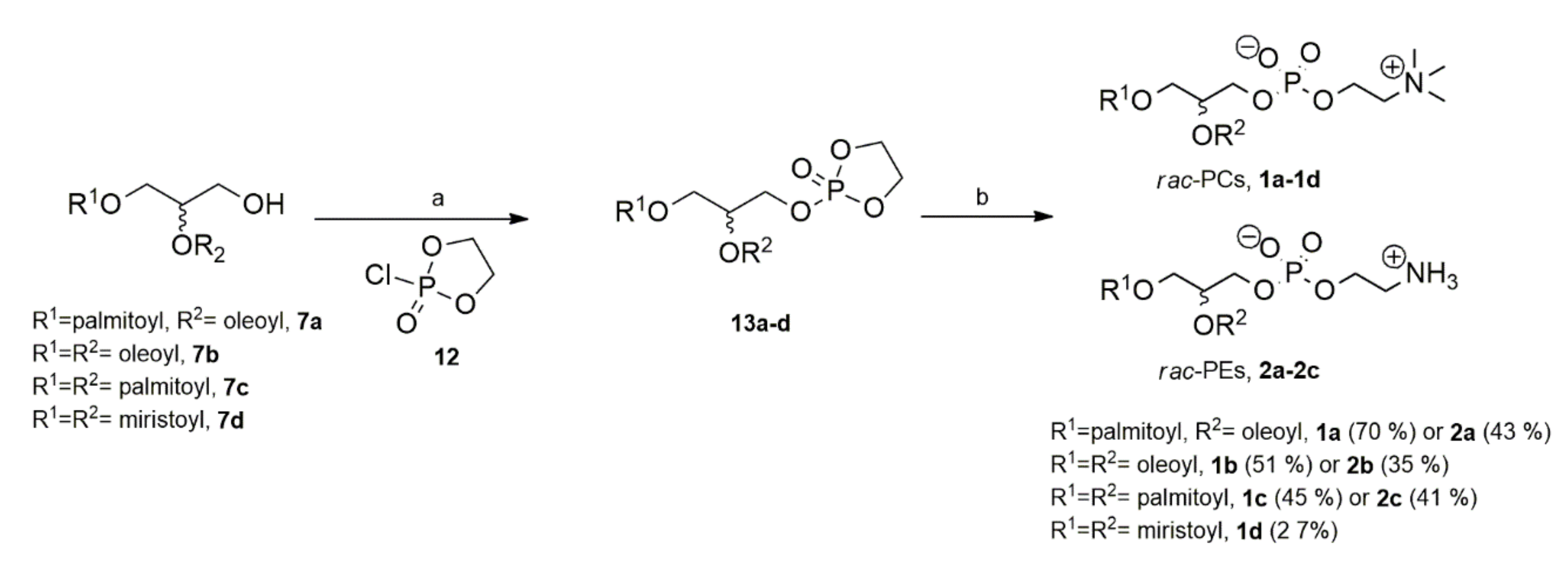
Appendix A.3.4 Synthesis of Compounds 1a–1d and 2a–2c
Synthesis of
13a–
13d and
(R)-
13a.
General method.
7a–
7d and
(S)-7a were dissolved in dry toluene (6 mL) with dry Et
3N (0.025 mL, 0.178 mmol) and cooled (0 °C). To this cold solution, a second solution prepared by dissolving 2-chloro-2-oxo-1,3,2-dioxaphospholane (
12, 1.1 equiv.) in dry toluene (4 mL) were slowly added and the resulting mixture was stirred at r.t. The white precipitate obtained after 16 h was filtered off over a Celite pad (2 cm thick, 4 cm Ø, filter porosity n°4) and the filtrate was evaporated as quickly as possible while keeping the temperature of the water bath below 20 °C.
1H NMR and
31P NMR (CDCl
3) were used to confirm the formation of
13a–
13d that were used without further purifications for the next steps. Yields are reported in
Table A2.
(R)-
13a was not used instead.
Table A2.
Data for the synthesis of 1a–1d and 2a–2c from 7a–7d.
Table A2.
Data for the synthesis of 1a–1d and 2a–2c from 7a–7d.
| Entry | Step | Scale 1 | Yield | Entry | Step | Scale 1 | Yield |
|---|
| 1 | 7a→13a→1a | 128 mg | 70% | 7 | 7a→13a→2a | 128 mg | 43% |
| 2 | 7a→13a→1a | 1.0 g | 65% | 8 | 7b→13b→2b | 56 mg | 35% |
| 3 | 7b→13b→1b | 56 mg | 51% | 9 | 7c→13c→2c | 82 mg | 41% |
| 4 | 7c→13c→1c | 82 mg | 27% | | | | |
| 5 | 7d→13d→1d | 100 mg | 45% | | | | |
| 6 | (S)–7a→(R)–13a | 50 mg | quant. | | | | |
13a. 1H NMR (300 MHz, CDCl3): δH = 5.39–5.30 (m, 2 H, Z-CH=CH), 5.28–5.21 (m, 1 H, C(2)H), 4.52–4.11 (m, 4 H, C(1)H2 and O-CH2-CH2-O), 3.89–3.76 (m, 2 H, C(1)H2), 2.34–2.30 (m, 4 H, 2 x CH2COOR), 2.01–1.94 (m, 4 H, 2 × CH2-CH=CH-CH2), 1.60–1.54 (m, 4 H, 2 x CH2CH2COOR), 1.30, 1.15 (2 x br, 38 H, 19 x CH2), 0.96–0.76 (m, superimposition of 2 x t, apparent J = 6.8 Hz, 6 H, 2 x CH3). 31P NMR (121.5 MHz, CDCl3): δP = 18.14.
13b. (The mixture contained unreacted 5b: ratio 5b/12a 1:4 by 1H NMR integration). 1H NMR (300 MHz, CDCl3): δH = 5.40–5.28 (m, 4 H, 2 x Z-CH=CH, 6b+7b), 5.27–5.20 (m, 1 H, C(2)H), 5.16–4.80 (m, 2 H, 1 x CH2NHR), 4.40–4.05 (m, 8 H, C(1)H2 and C(1)H2 5b+12b), 3.78–3.87 (m, 2 H, NHCH2CH2OR), 2.62–2.69 (m, 2 H, 1 x CH2-CH=CH-CH2), 2.40–2.25 (m, 4 H, CH2CH2COOR, 5b+12b), 2.11–1.96 (m, 6 H, 3 x CH2-CH=CH-CH2, 5b+12b), 1.60–1.55 (m, 4 H, 2 x CH2CH2COOR, 5b+12b), 1.39, 1.26 (2 x br, 40 H, 20 x CH2, 5b+12b), 0.96–0.76 (m, 6 H, 2 x CH3, 6b+7b). 31P NMR (121.5 MHz, CDCl3): δP = 17.63.
13c. 1H NMR (300 MHz, CDCl3, selected signals): δH = 5.30–5.16 (m, 1 H, C(2)H), 3.81–3.68 (m, 3H), 2.37–2.28 (m, 4 H, CH2CH2COOR), 1.70–1.53 (m, 4 H, 2 x CH2CH2COOR), 1.39, 1.25 (2 x br, 44 H, 22 x CH2), 0.87 (t, J = 6.9 Hz, 2 x CH3); 31P NMR (121.5 MHz, CDCl3): δP = 17.38.
13d. 1H NMR (300 MHz, CDCl3, selected signals): δH = 5.20–5.18 (m, 1 H, C(2)H), 2.40–2.23 (m, 4 H, CH2CH2COOR), 1.77–1.50 (m, 4 H, 2 x CH2CH2COOR), 1.22 (br, 40 H, 20 x CH2), 0.87 (t, J = 6.9 Hz, 2 x CH3); 31P NMR (121.5 MHz, CDCl3): δP = 17.68.
(R)-13a. 1H NMR (300 MHz, CDCl3): δH = 5.39–5.30 (m, 2 H, Z-CH=CH), 5.28–5.21 (m, 1 H, C(2)H), 4.52–4.11 (m, 4 H, C(1)H2 and O-CH2-CH2–O), 3.89–3.76 (m, 2 H, C(1)H2), 2.34–2.30 (m, 4 H, 2 x CH2COOR), 2.01–1.94 (m, 4 H, 2 x CH2-CH=CH-CH2), 1.60–1.54 (m, 4 H, 2 x CH2CH2COOR), 1.30, 1.15 (2 x br, 38 H, 19 x CH2), 0.96–0.76 (m, superimposition of 2 x t, apparent J = 6.8 Hz, 6 H, 2 x CH3). 31P NMR (121.5 MHz, CDCl3): δP = 18.14.
Synthesis of racemic phosphocholines 1a–
1d.
General method. Cyclic phosphotriesters
13a–13d were dissolved in dry MeCN (10 mL) and placed in a pressure tube (Sigma Aldrich) equipped with a rubber septum and connected to an Ar reservoir and the tube was placed in an ice bath (0 °C). Dry trimethylamine (0.5 mL, 0.49 mmol) was added, the tube sealed and the resulting white solution was kept stirring at 65 °C for 24 h. The solution was cooled to r.t. and the resulting mixture was purified by chromatography over freshly activated SiO
2 with CHCl
3/MeOH/H
2O 65:25:0.4
v/v/v furnishing products
1a–
1d as white waxes. Only the
1a reaction was scaled up to 1 g of
7a. Data were reported in
Table A2.
1a. Obtained 36 mg (70%), the scale–up reaction (1 gr) yielded 508 mg (65%); Rf (65: 25: 04, CHCl3: MeOH: H2O, v/v/v) 0.65. 1H NMR (300 MHz, CDCl3): δH = 5.41–5.27 (m, 2 H, Z-CH=CH), 5.23 (br, 1 H, C(2)H), 4.43–4.94 (m, 4 H, O-CH2-CH2-NMe3), 3.73 (br, 2 H, C(1)H2), 3.99 (br, 2 H, C(3)H2), 2.39–2.22 (m, 4 H, 2 × CH2COOR), 2.01–1.94 (m, 4 H, 2 x CH2-CH=CH-CH2), 1.66–1.50 (m, 4 H, 2 x CH2CH2COOR), 1.30, 1.15 (2 x br, 53 H, 19 x CH2 and N(CH3)3), 0.95–0.79 (m, superimposition of 2 x t, apparent J = 6.8 Hz), 6 H, 2 x CH3). 13C NMR (75 MHz, CDCl3): δC = (selected signals) 14.3 (CH3), 22.9 (CH2), 25.0 (CH2), 25.1 (CH2), 27.4 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2), 29.9–32.0 (series of CH2), 32.1 (CH2), 34.3 (CH2), 61.7 (C3), 62.9 (C1), 70.6 (C2, HSQC), 129.6 (Z-CH=CH), 130.2 (Z-CH=CH), 173.6 (C=O), 173.8 (C=O); 31P NMR (121.5 MHz, CDCl3): δP = −1.65; [α]D25 = 0.00 (c 0.1, CHCl3); [α]D25 = 0.00 (c 0.1, CHCl3); HRMS m/z: [M]+ calcd. for C42H82NNaO8P: 782.5675, found C42H82NNaO8P: 782.5670.
1b. Obtained 35.4 mg (51%); Rf (CHCl3: MeOH: H2O, 65: 25: 0.4 v/v/v) 0.64. 1H NMR (400 MHz, CDCl3): δH = 5.39–5.29 (m, 2 H, Z-CH=CH), 5.23 (br, 1 H, C(2)H), 4.51 (br, 2 H, OH), 4.16–4.11 (m, 2 H, C(1)H2); 4.11–3.96 (m, 6 H, O-CH2-CH2-NMe3 and C(1)H2), 2.37–2.26 (m, 4 H, 2 x CH2COOR), 2.06–1.93 (m, 4 H, 2 x CH2-CH=CH-CH2), 1.63–1.49 (m, 4 H, 2 x CH2CH2COOR), 1.38, 1.15 (2 x br, 53 H, 19 x CH2 and N(CH3)3), 0.95–0.79 (m, superimposition of 2 x t, apparent J = 6.8 Hz, 6H, 2 x CH3). 13C NMR (100 MHz, CDCl3): δC = (selected signals) 14.3 (CH3), 22.9 (CH2), 24.9 (CH2CH2COOR), 27.3 and 27.4 (CH2-CH=CH-CH2, HSQC), 29.4–29.5 (series of CH2), 29.8 (CH2), 29.9 (CH2), 30.0 (CH2), 32.1 (CH2), 34.1 (CH2COOR), 62.4 (C3, HSQC), 64.6 (C1, HSQC), 69.8 (C2, HSQC), 129.9 (Z-CH=CH), 130.2 (Z-CH=CH), 173.4 (C=O), 173.7 (C=O); 31P NMR (121.5 MHz, CDCl3): δP = −2.33; [α]D25 = 0.00 (c 0.1, CHCl3); HRMS m/z: [M]+ calcd. for C44H85NO8P: 786.6013, found C44H85NO8P: 786.6007.
1c. Obtained 33.0 mg (45%). Rf (CHCl3:MeOH :H2O, 65: 25: 0.4 v/v/v) 0.72. 1H NMR (400 MHz, CDCl3): δH = 5.17–5.22 (m, 1 H, C(2)H), 4.34–4.26 (m, 2 H, O-CH2-CH2-NH2), 4.12 (dd, J = 12.0, 2.8 Hz, 2 H, C(2)H2), 3.99–3.86 (m, 2 H, C(3)H2), 3.81–3.75 ( m, 2 H, CH2–NH2), 3.45 (s, 9 H, N(CH3)3), 2.28 (dd, 4 H, J = 15.4, 8 Hz, 4 H, 2 x CH2COOR), 1.62–1.51 (m, 4H, 2 x CH2CH2COOR), 1.30, 1.25 (2 x br, 48H, 24 x CH2), 0.87 (t, 6H, J = 6.9 Hz), 6 H, 2 x CH3). 13C NMR (100 MHz, CDCl3): δC = (selected signals) 14.3 (CH3), 25.1 (CH2), 25.2 (CH2), 29.2 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2), 29.8 (CH2), 29.9 (CH2), 30.0 (CH2), 32.0 (CH2), 34.4 (CH2), 34.5 (CH2), 63.2 (C3), 62.9 (C1), 70.6 (C2, HSQC), 173.4 (C=O), 173.8 (C=O); 31P NMR (121.5MHz, CDCl3): δP = −2.45; [α]D25 = 0.00 (c 0.1, CHCl3); HRMS m/z: [M]+ calcd. for C40H80NNaO8P: 756.5519, found C40H80NNaO8P: 756.5428.
1d. Obtained 28.7 mg (26%); Rf (CHCl3: MeOH: H2O, 65: 25: 04 v/v/v) 0.70. 1H NMR (300 MHz, CDCl3): δH = 5.24 (br, 1 H, C(2)H), 4.39–4.20 (m, 4 H, O-CH2-CH2-NMe3), 4.19–4.09 (m, 1 H, C(3)Hb), 4.04 (br, 1 H, C(1)H2), 3.76 (br, 2 H, C(3)Ha), 2.85 (br, 9 H, N(CH3), 2.37–2.30 (m, 4H, 2 x CH2COOR), 1.66–1.51 (m, 4H, 2 x CH2CH2COOR), 1.23 (br, 32H, 16 x CH2), 0.87 (t, 6H, J = 6.8 Hz, 2 x CH3). 13C NMR (75MHz, CDCl3): δC = (selected signals) 14.3 (CH3), 22.9 (CH2), 23.7 (CH2), 24.7 (CH2), 25.1 (CH2CH2COOR ), 27.4 (CH2), 29.5 (CH2), 29.8 (CH2), 29.6 (CH2), 32.1 (CH2), 34.4 (CH2COOR), 62.5 (C3, HSQC), 64.4 (C1, HSQC), 70.3 (C2, HSQC), 173.2 (C=O), 173.6 (C=O); 31P NMR (121.5MHz, CDCl3): δP = −2.05; [α]D25 = 0.00 (c 0.1, CHCl3); HRMS (m/z): [M]+ calcd. for C36H73NO8P: 678.5074, found C36H73NO8P: 678.5068.
Synthesis of racemic phosphoethanolamines 2a–2c.
General method.
13a–
14c were dissolved in dry CH
3CN (10 mL) in a pressure tube (Sigma Aldrich) equipped with a rubber septum and connected to an Ar reservoir and the tube was placed in an ice bath (0 °C). Dry NH
3 (3.5 bar, excess) was bubbled into the tube until the solution became white (2 min). The tube was sealed and the resulting white solution was kept under stirring at 65 °C for 24 h. The solution was cooled to r.t. and the resulting mixture was purified by chromatography over freshly activated SiO
2 with CHCl
3: MeOH: H
2O (65:25:0.4
v/v/v) yielding
2a–
2c as white waxes. Reactions carried out by using a 1.0 M solution of NH
3 in dry acetonitrile gave similar results. Data were reported in
Table A2 2a. Obtained 66.4 mg (43%); Rf (CHCl3: MeOH: H2O, 65: 25: 04 v/v/v) 0.67; 1H NMR (400 MHz, CDCl3): δH = 5.37–5.28 (m, 2 H, Z-CH=CH), 5.26–5.13 (m, 1 H, C(2)H), 4.41–4.33 (m, 1 H, C(1)Hb), 4.21–4.04 (m, 3 H, C(1)Ha and O-CH2-CH2–NH3+), 3.97–3.92 (m, 2 H, O-CH2-CH2–NH3+), 3.26–3.10 (m, 2 H, C(3)H2), 2.36–2.22 (m, 4 H, 2 x CH2COOR), 2.07 – 1.95 (m, 4 H, 2 x CH2-CH=CH-CH2), 1.66–1.51 (m, 4H, 2 x CH2CH2COOR), 1.25 (br, 44 H, 22 x CH2), 0.85 (t, J = 6.8 Hz, 6 H, 2 x CH3); 13C NMR (100 MHz, CDCl3): δC = (selected signals) 14.3 (CH3), 22.9 (CH2), 27.4 (series of CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2-CH=CH-CH2), 29.9–30.0 (series of CH2), 62.7 (C3), 64.2 (C1), 70.4 (C2), 129.9 (Z-CH=CH), 130.2 (Z-CH=CH), 173.2 (C=O), 173.6 (C=O); 31P NMR (121.5 MHz, CDCl3): δP = −0.17; [α]D25 = 0.00 (c 0.1, CHCl3); HRMS m/z: [M]+ calcd. for C39H77NO8P: 718.5387, found C39H77NO8P: 718.5381.
2b. Obtained 23.5 mg. Rf (CHCl3: MeOH: H2O, 65: 25: 04 v/v/v) 0.58; 1H NMR 400 MHz, CDCl3) δH = 8.49 (br s, 2.5 H, NH2 ⇌ NH3+), 5.38–5.28 (m, 4 H, 2 x Z-CH=CH), 5.24–5.18 (m, 1 H, C(1)H), 4.37 (dd, J = 12.0, 3.0 Hz, 1 H, C(2)Hb), 4.17–4.09 (m, 3H, C(1)Ha+C(3)H2), 3.95 (t, J = 6 Hz, 2 H, OCH2CH2NH2), 3.93 (br s, 2 H, OCH2CH2NH2), 2.29 (dd, J = 16.5, 8.3 Hz, 4 H, 2 x CH2CH2COOR), 2.01–1.93 (m, 8 H, 2 x CH2-CH=CH-CH2), 1.58 (br s, 4 H, 2 x CH2CH2COOR ), 1.39–1.22 (br s, 40 H, 20 x CH2), 0.88 (t, J = 10.0 Hz, 6 H, 2 x CH3); 13C NMR (100 MHz, CDCl3): δC = 14.3 (CH3), 22.9 (CH2), 25.1 (CH2), 25.2 (CH2), 27.4 (CH2), 29.4 (CH2), 29.5–29.8 (series of CH2), 30.0 (CH2), 32.1 (CH2), 34.3 (CH2), 34.3 (CH2),40.7 (CH2O, HSQC) 62.4 (C3), 63.7 (C1), 64.1 (CH2N, HSQC), 70.6 (C2, HSQC), 129.6 (Z-CH=CH), 130.2 (Z-CH=CH), 173.3 (C=O), 173.6 (C=O); 31P NMR (121.5MHz, CDCl3): δP = −2.24; [α]D25 = 0.00 (c 0.1, CHCl3); HRMS m/z: [M]+ calcd. for C41H78NNaO8P: 766.5339, found C39H77NO8P: 766.5357.
2c. Obtained 29.1 mg. Rf (CHCl3: MeOH: H2O, 65: 25: 04 v/v/v) 0.70; 1H NMR (400 MHz, CDCl3) selected signals: δH = 5.24–5.18 (m, 1 H, C(2)H), 4.0.2–3.99 (m, 2 H, C(2)H2), 4.33–4.45 (m, 2 H, C(3)H2), 4.27–4.19 (m, 2 H, O-CH2-CH2-NH2), 4.20–4.11 (m, 2 H, O-CH2-CH2-NH2), 2.37–2.18 (m, 4 H, 2 x CH2COOR), 1.68–1.50 (m, 4 H, 2 x CH2CH2COOR), 1.30, 1.26 (2 x br, 48H, 24 x CH2), 0.88 (t, 6H, J = 6.9 Hz), 6H, 2 x CH3). 13C NMR (100MHz, CDCl3): δC = (selected signals) 14.2 (CH3), 25.1 (CH2), 25.2 (CH2), 29.5 (CH2), 29.6 (CH2), 29.8–29.9 (series of CH2), 30.6 (CH2), 32.2 (CH2), 34.4 (CH2), 34.6 (CH2), 65.5 (C3), 65.6 (C1), 70.6 (C2, HSQC), 173.6 (C=O), 173.8 (C=O); 31P NMR (121.5 MHz, CDCl3): δP = −2.45; [α]D25 = 0.00 (c 0.1, CHCl3); HRMS m/z: [M]+ calcd. for C37H75NO8P: 692.5230, found C37H75NO8P: 692.5225.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





