
Two Faces of Water in the Formation and Stabilization of Multicomponent Crystals of Zwitterionic Drug-Like Compounds

 ,
,
Abstract
:1. Introduction
- (i).
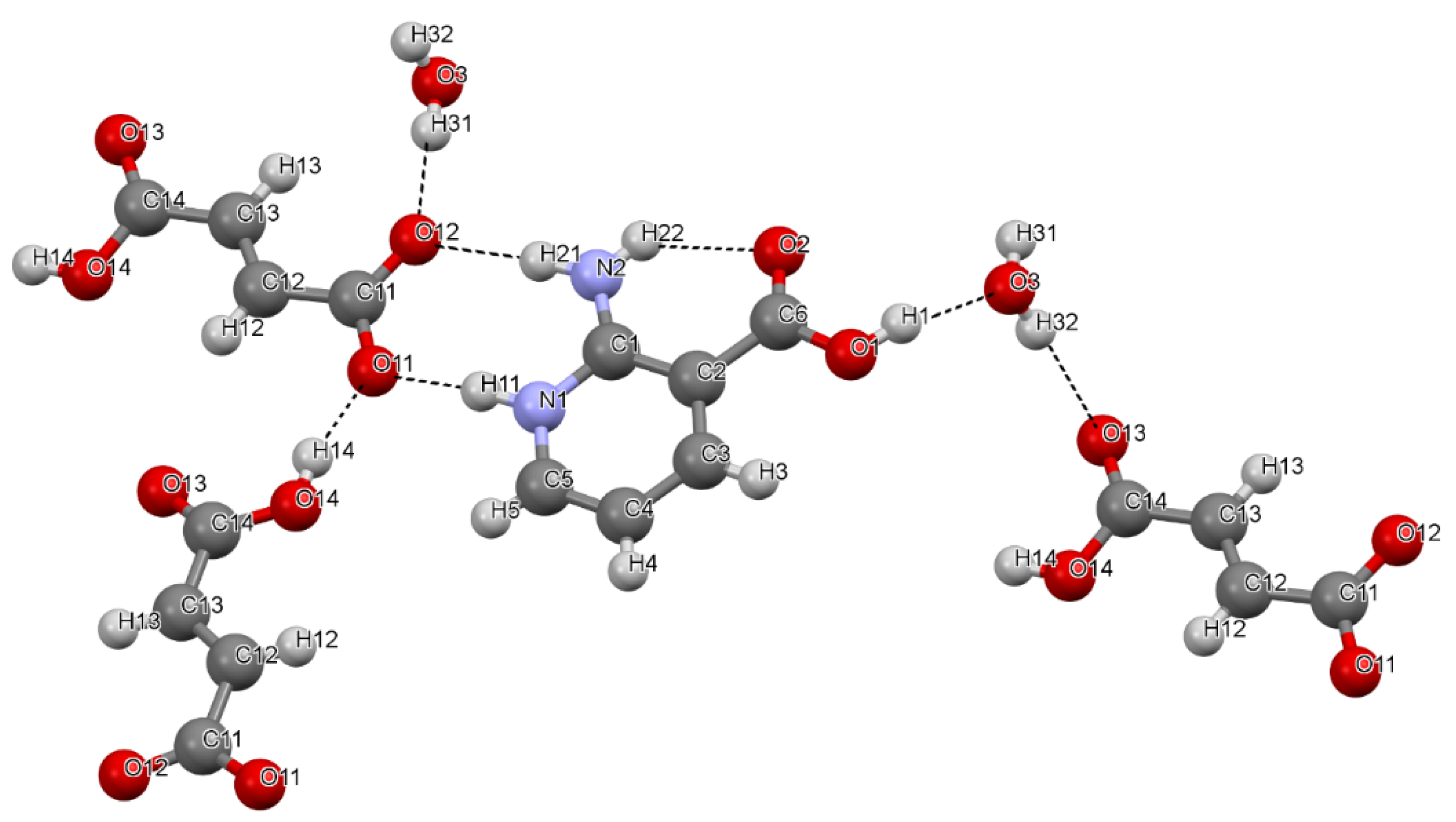
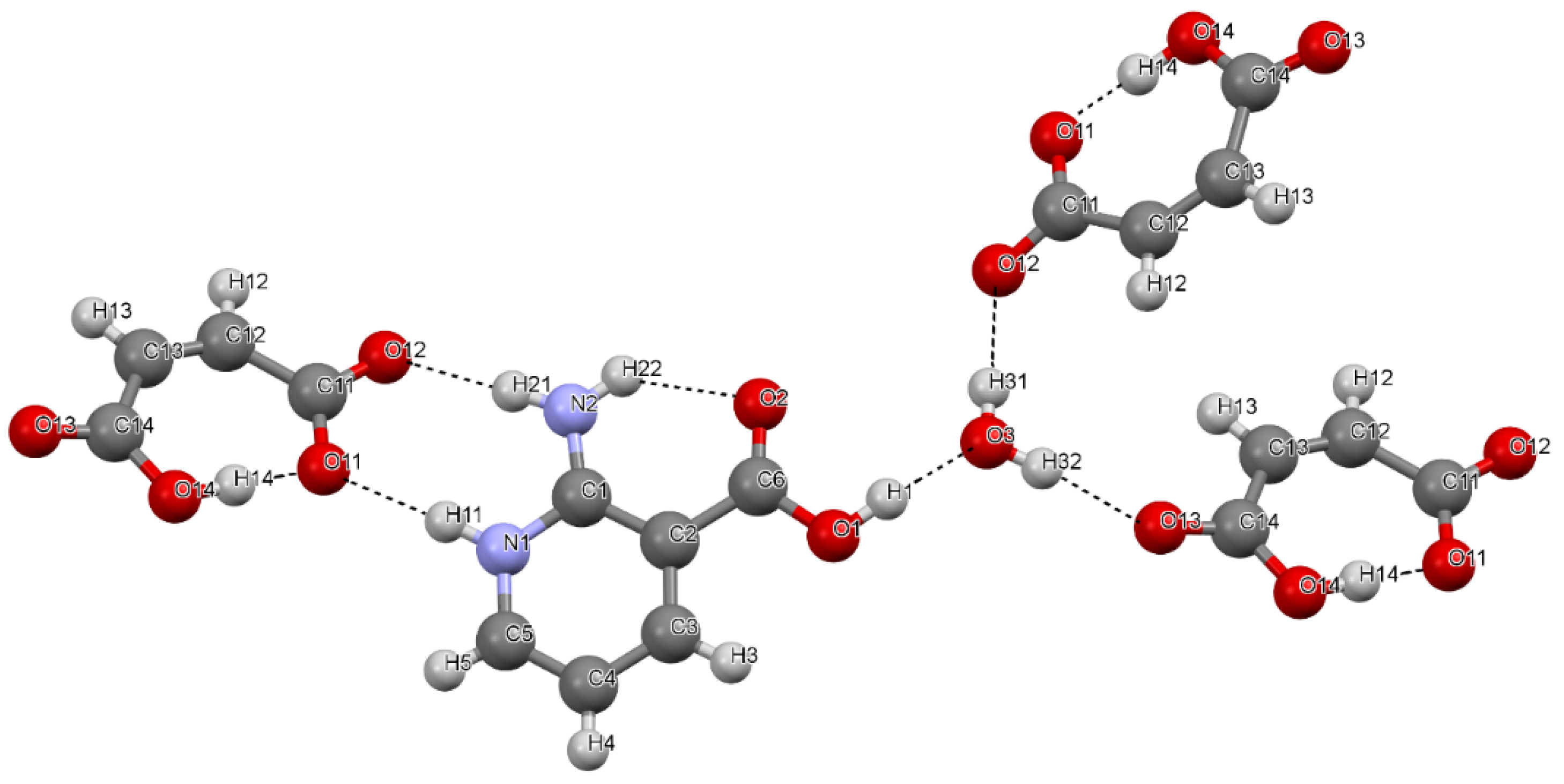
- To characterize the structure and hydrogen bond (H-bond) network in two multicomponent crystals—[2AmNic+Mle+H2O] (1:1:1) and [2AmNic+Fum+H2O] (1:1:1)—by X-ray analysis, terahertz Raman spectroscopy, and periodic density functional theory (DFT) calculations. 2AmNic denotes 2-amino-nicotinic acid, while Mle and Fum stand for maleic and fumaric acids, respectively.
- (ii).
- To reveal the structure-directing role of the water molecule in the considered crystals.
- (iii).
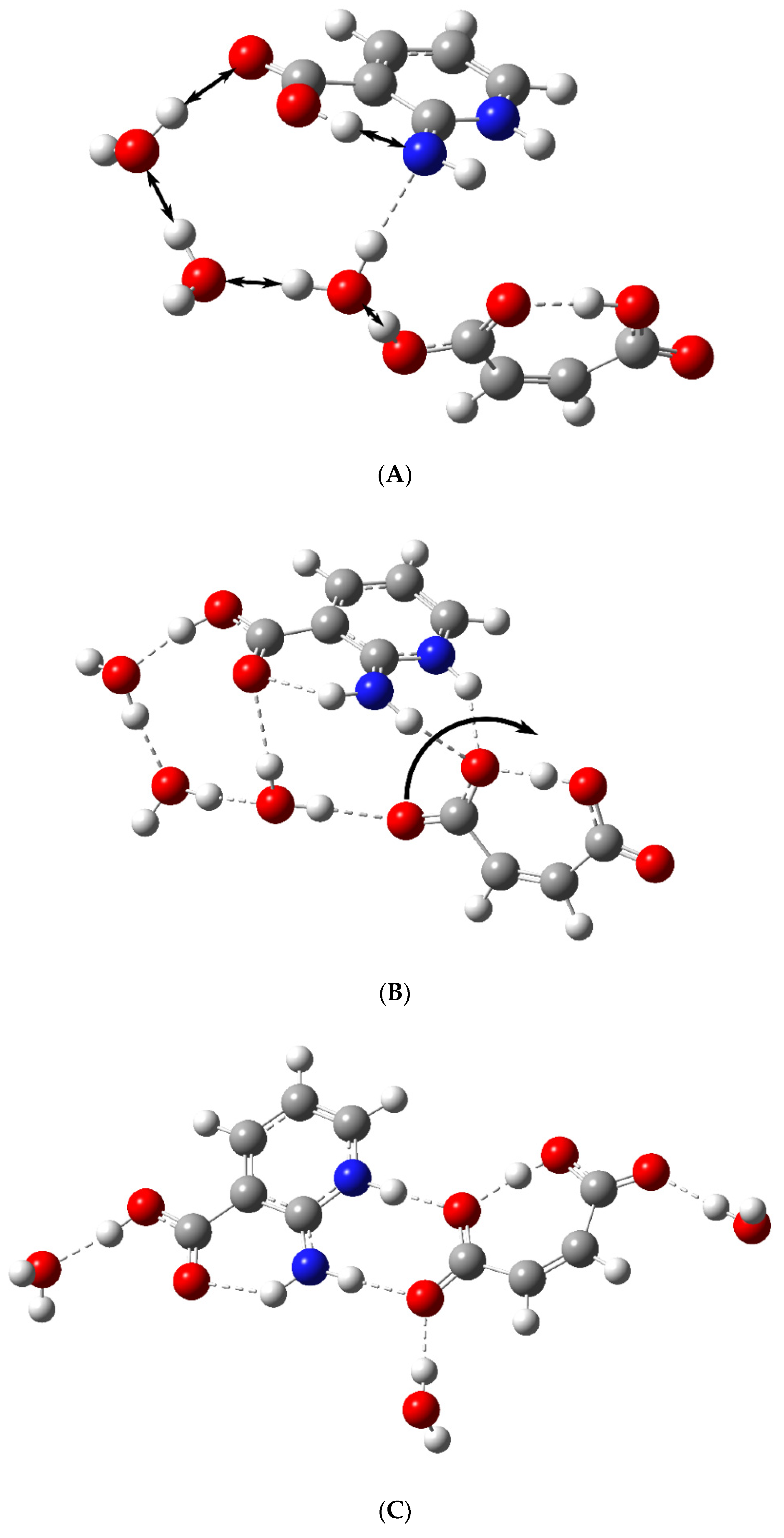
- To theoretically substantiate the scheme of proton transfer from the dicarboxylic acid to the zwitterion by means of water wires.
2. Materials and Methods
2.1. Compounds and Solvents
2.2. Preparation Procedures
2.3. Thermal Analysis
2.3.1. Differential Scanning Calorimetry (DSC)
2.3.2. Thermogravimetric Analysis (TGA)
2.4. Single Crystal and Powder X-ray Diffraction (XRD) Experiments
2.5. Raman Spectroscopy
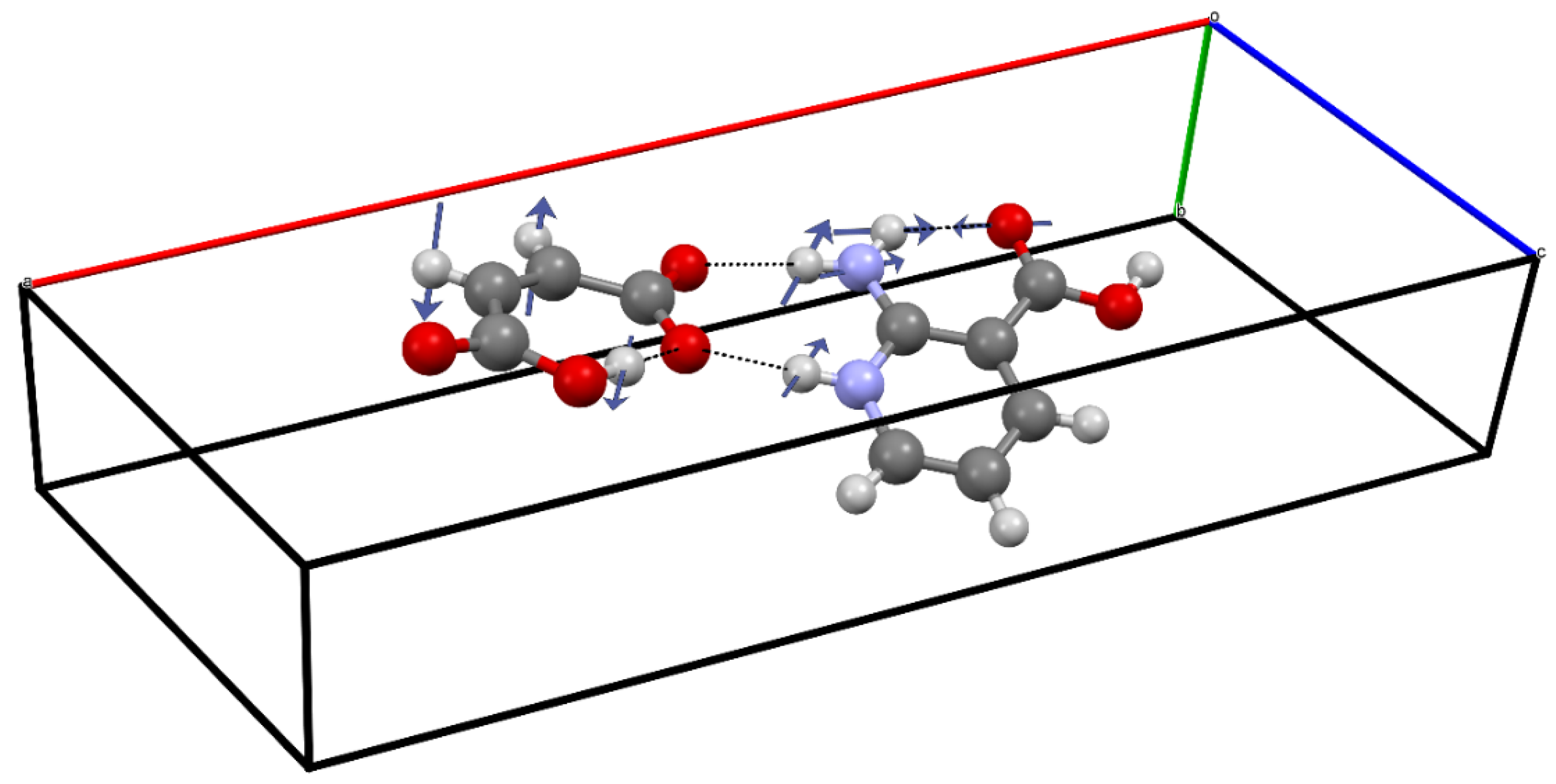
2.6. Periodic (Solid-State) DFT Computations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fragment 1 | Experiment | PBE-D3/6-31G** | B3LYP/6-31G** |
|---|---|---|---|
| O12…H21-N2 | 2.877 (175.7) | 2.848 (176.4) | 2.867 (176.0) |
| O11…H11-N1 | 2.662 (175.3) | 2.668 (173.5) | 2.681 (173.7) |
| O12…H31-O3 | 2.702 (174.4) | 2.684 (177.9) | 2.697 (178.0) |
| O13…H32-O3 | 2.859 (167.8) | 2.801 (172.2) | 2.828 (171.2) |
| O11…H14-O14 | 2.559 (164.0) | 2.540 (160.6) | 2.547 (161.9) |
| O3…H1-O1 | 2.582 (165.9) | 2.545 (165.4) | 2.571 (165.9 |

| Fragment 1 | Experiment | PBE-D3/6-31G** | B3LYP/6-31G** |
|---|---|---|---|
| O12…H21-N2 | 2.816 (176.4) | 2.804 (170.4) | 2.811 (171.7) |
| O11…H11-N1 | 2.816 (170.1) | 2.816 (173.7) | 2.818 (172.8) |
| O12…H31-O3 | 2.717 (177.2) | 2.701 (176.0) | 2.706 (176.4) |
| O13…H32-O3 | 2.771 (177.5) | 2.701 (173.4) | 2.706 (177.6) |
| O3…H1-O1 | 2.567 (173.0) | 2.536 (173.6) | 2.550 (174.4) |
| O11…H14-O14 (intra) | 2.460 (174.9) | 2.462 (176.1) | 2.460 (174.9) |

3. Results
3.1. Crystal Structure and H-Bond Network
3.2. The Structure-Directing Role of the Water Molecule
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwoerer, M.; Wolf, H.C. Organic Molecular Solids; Wiley: Weinheim, Germany, 2008. [Google Scholar]
- Qiu, Y.; Chen, Y.; Zhang, G.G.Z.; Yu, L.; Mantri, R.V. Developing Solid Oral Dosage Forms: Pharmaceutical Theory and Practice; Elsevier Science: London, UK, 2016. [Google Scholar]
- Sathisaran, I.; Dalvi, S.V. Engineering Cocrystals of Poorly Water-Soluble Drugs to Enhance Dissolution in Aqueous Medium. Pharmaceutics 2018, 10, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wang, Y.; Yang, F.; Zhang, X.; Hu, W. Cocrystal Engineering: A Collaborative Strategy toward Functional Materials. Adv. Mater. 2019, 31, 1902328. [Google Scholar] [CrossRef]
- Kavanagh, O.N.; Croker, D.M.; Walker, G.M.; Zaworotko, M.J. Pharmaceutical cocrystals: From serendipity to design to application. Drug Discov. Today 2019, 24, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Duggirala, N.K.; Perry, M.L.; Almarsson, Ö.; Zaworotko, M.J. Pharmaceutical cocrystals: Along the path to improved medicines. Chem. Commun. 2016, 52, 640–655. [Google Scholar] [CrossRef] [PubMed]
- Bolla, G.; Nangia, A. Pharmaceutical cocrystals: Walking the talk. Chem. Commun. 2016, 52, 8342–8360. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.J.; Steed, J.W. Pharmaceutical cocrystals, salts and multicomponent systems; intermolecular interactions and property based design. Adv. Drug Del. Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aakeröy, C.B.; Hussain, I.; Desper, J. 2-Acetaminopyridine: A Highly Effective Cocrystallizing Agent. Cryst. Growth Des. 2006, 6, 474–480. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Forbes, S.; Desper, J. Altering physical properties of pharmaceutical co-crystals in a systematic manner. CrystEngComm 2014, 16, 5870–5877. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.D.B.; Bradner, M.W.; Fleischman, S.; Morales, L.A.; Moulton, B.; Rodríguez-Hornedo, N.; Zaworotko, M.J. Crystal engineering of the composition of pharmaceutical phases. Chem. Commun. 2003, 2, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Corpinot, M.K.; Bučar, D.-K. A Practical Guide to the Design of Molecular Crystals. Cryst. Growth Des. 2019, 19, 1426–1453. [Google Scholar] [CrossRef] [Green Version]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Lemmerer, A.; Bernstein, J.; Kahlenberg, V. One-pot covalent and supramolecular synthesis of pharmaceutical co-crystals using the API isoniazid: A potential supramolecular reagent. CrystEngComm 2010, 12, 2856–2864. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Crystal Engineering: A Holistic View. Angew. Chem. Int. Ed. 2007, 46, 8342–8356. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.O.; Voronin, A.P.; Vener, M.V.; Churakov, A.V.; Perlovich, G.L. Specific features of supramolecular organisation and hydrogen bonding in proline cocrystals: A case study of fenamates and diclofenac. CrystEngComm 2018, 20, 6970–6981. [Google Scholar] [CrossRef]
- Bolla, G.; Nangia, A. Novel pharmaceutical salts of albendazole. CrystEngComm 2018, 20, 6394–6405. [Google Scholar] [CrossRef] [Green Version]
- Voronin, A.P.; Surov, A.O.; Churakov, A.V.; Parashchuk, O.D.; Rykounov, A.A.; Vener, M.V. Combined X-ray Crystallographic, IR/Raman Spectroscopic, and Periodic DFT Investigations of New Multicomponent Crystalline Forms of Anthelmintic Drugs: A Case Study of Carbendazim Maleate. Molecules 2020, 25, 2386. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Sadeghi, F.; Molčanov, K.; Zarȩba, J.K.; Gomila, R.M.; Frontera, A. Recurrent Supramolecular Motifs in a Series of Acid–Base Adducts Based on Pyridine-2,5-Dicarboxylic Acid N-Oxide and Organic Bases: Inter- and Intramolecular Hydrogen Bonding. Cryst. Growth Des. 2020, 20, 1738–1751. [Google Scholar] [CrossRef]
- Yadav, B.; Balasubramanian, S.; Chavan, R.B.; Thipparaboina, R.; Naidu, V.G.M.; Shastri, N.R. Hepatoprotective Cocrystals and Salts of Riluzole: Prediction, Synthesis, Solid State Characterization, and Evaluation. Cryst. Growth Des. 2018, 18, 1047–1061. [Google Scholar] [CrossRef]
- Thomas, S.P.; Kumar, V.; Alhameedi, K.; Guru Row, T.N. Non-Classical Synthons: Supramolecular Recognition by S…O Chalcogen Bonding in Molecular Complexes of Riluzole. Chem. Eur. J. 2019, 25, 3591–3597. [Google Scholar] [CrossRef] [PubMed]
- Babu, N.J.; Reddy, L.S.; Nangia, A. Amide−N-Oxide Heterosynthon and Amide Dimer Homosynthon in Cocrystals of Carboxamide Drugs and Pyridine N-Oxides. Mol. Pharm. 2007, 4, 417–434. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D.; Gavezzotti, A. Supramolecular Synthons: Validation and Ranking of Intermolecular Interaction Energies. Cryst. Growth Des. 2012, 12, 5873–5877. [Google Scholar] [CrossRef]
- Vener, M.V.; Levina, E.O.; Koloskov, O.A.; Rykounov, A.A.; Voronin, A.P.; Tsirelson, V.G. Evaluation of the lattice energy of the two-component molecular crystals using solid-state density functional theory. Cryst. Growth Des. 2014, 14, 4997–5003. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Voth, G.A. Infrared Spectrum of the Hydrated Proton in Water. J. Phys. Chem. Lett. 2011, 2, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Kaila, V.R.I.; Hummer, G. Energetics and dynamics of proton transfer reactions along short water wires. PCCP 2011, 13, 13207–13215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zatula, A.S.; Ryding, M.J.; Uggerud, E. Concerted proton migration along short hydrogen bonded water bridges in bipyridine–water clusters. PCCP 2012, 14, 13907–13909. [Google Scholar] [CrossRef] [PubMed]
- Freier, E.; Wolf, S.; Gerwert, K. Proton transfer via a transient linear water-molecule chain in a membrane protein. Proc. Natl. Acad. Sci. USA 2011, 108, 11435–11439. [Google Scholar] [CrossRef] [Green Version]
- Di Donato, M.; van Wilderen, L.J.G.W.; Van Stokkum, I.H.M.; Stuart, T.C.; Kennis, J.T.M.; Hellingwerf, K.J.; van Grondelle, R.; Groot, M.L. Proton transfer events in GFP. PCCP 2011, 13, 16295–16305. [Google Scholar] [CrossRef] [PubMed]
- Grigorenko, B.; Polyakov, I.; Nemukhin, A. Mechanisms of ATP to cAMP Conversion Catalyzed by the Mammalian Adenylyl Cyclase: A Role of Magnesium Coordination Shells and Proton Wires. J. Phys. Chem. B 2020, 124, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. SADABS, Program for Scaling and Correction of Area Detector Data; University of Göttingen: Lower Saxony, Germany, 1997. [Google Scholar]
- Sheldrick, G. A short history of SHELX. Acta Cryst. Sect. A Found. Cryst. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. WIREs Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Bifurcated Hydrogen Bonds: Three-Centered Interactions. J. Phys. Chem. A 1998, 102, 9925–9932. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Bodensteiner, M.; Tolstoy, P.M.; Denisov, G.S.; Shenderovich, I.G. P═O Moiety as an Ambidextrous Hydrogen Bond Acceptor. J. Phys. Chem. C 2018, 122, 1711–1720. [Google Scholar] [CrossRef]
- Vener, M.V. Model study of the primary H/D isotope effects on the NMR chemical shift in strong hydrogen-bonded systems. Chem. Phys. 1992, 166, 311–316. [Google Scholar] [CrossRef]
- Jóźwiak, K.; Jezierska, A.; Panek, J.J.; Goremychkin, E.A.; Tolstoy, P.M.; Shenderovich, I.G.; Filarowski, A. Inter- vs. Intramolecular Hydrogen Bond Patterns and Proton Dynamics in Nitrophthalic Acid Associates. Molecules 2020, 25, 4720. [Google Scholar] [CrossRef]
- Müller-Dethlefs, K.; Hobza, P. Noncovalent Interactions: A Challenge for Experiment and Theory. Chem. Rev. 2000, 100, 143–168. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Evans, T.A.; Seddon, K.R.; Pálinkó, I. The C–H…Cl hydrogen bond: Does it exist? New J. Chem. 1999, 23, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Nishio, M. CH/π hydrogen bonds in crystals. CrystEngComm 2004, 6, 130–158. [Google Scholar] [CrossRef]
- Martinez, C.R.; Iverson, B.L. Rethinking the term “pi-stacking”. Chem. Sci. 2012, 3, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Bartashevich, E.V.; Tsirelson, V.G. Interplay between non-covalent interactions in complexes and crystals with halogen bonds. Russ. Chem. Rev. 2014, 83, 1181–1203. [Google Scholar] [CrossRef]
- Melikova, S.M.; Voronin, A.P.; Panek, J.; Frolov, N.E.; Shishkina, A.V.; Rykounov, A.A.; Tretyakov, P.Y.; Vener, M.V. Interplay of π-stacking and inter-stacking interactions in two-component crystals of neutral closed-shell aromatic compounds: Periodic DFT study. RSC Adv. 2020, 10, 27899–27910. [Google Scholar] [CrossRef]
- Mattei, A.; Li, T. Intermolecular Interactions and Computational Modeling. In Pharmaceutical Crystals; John Wiley & Sons: Hoboken, USA, 2018; pp. 123–167. [Google Scholar] [CrossRef]
- Burrows, A.D. Crystal Engineering Using Multiple Hydrogen Bonds. In Supramolecular Assembly via Hydrogen Bonds I; Mingos, D.M.P., Ed.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 55–96. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Seddon, K.R. The hydrogen bond and crystal engineering. Chem. Soc. Rev. 1993, 22, 397–407. [Google Scholar] [CrossRef]
- Subramanian, S.; Zaworotko, M.J. Exploitation of the hydrogen bond: Recent developments in the context of crystal engineering. Coord. Chem. Rev. 1994, 137, 357–401. [Google Scholar] [CrossRef]
- Desiraju, G.R. Designer crystals: Intermolecular interactions, network structures and supramolecular synthons. Chem. Commun. 1997, 16, 1475–1482. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Peraka, K.S.; Zaworotko, M.J. Chapter 2 The Role of Hydrogen Bonding in Co-crystals. In Co-Crystals: Preparation, Characterization and Applications; The Royal Society of Chemistry: Croydon, UK, 2018; pp. 33–79. [Google Scholar] [CrossRef]
- Medvedev, A.G.; Churakov, A.V.; Prikhodchenko, P.V.; Lev, O.; Vener, M.V. Crystalline Peroxosolvates: Nature of the Coformer, Hydrogen-Bonded Networks and Clusters, Intermolecular Interactions. Molecules 2021, 26, 26. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Salmon, D.J. Building co-crystals with molecular sense and supramolecular sensibility. CrystEngComm 2005, 7, 439–448. [Google Scholar] [CrossRef]
- Elder, D.P.; Holm, R.; Diego, H.L.d. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. Int. J. Pharm. 2013, 453, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Manin, A.N.; Voronin, A.P.; Shishkina, A.V.; Vener, M.V.; Churakov, A.V.; Perlovich, G.L. Influence of Secondary Interactions on the Structure, Sublimation Thermodynamics, and Solubility of Salicylate:4-Hydroxybenzamide Cocrystals. Combined Experimental and Theoretical Study. J. Phys. Chem. B 2015, 119, 10466–10477. [Google Scholar] [CrossRef] [PubMed]
- Voronin, A.P.; Perlovich, G.L.; Vener, M.V. Effects of the crystal structure and thermodynamic stability on solubility of bioactive compounds: DFT study of isoniazid cocrystals. Comput. Theor. Chem. 2016, 1092, 1–11. [Google Scholar] [CrossRef]
- Landeros-Rivera, B.; Moreno-Esparza, R.; Hernández-Trujillo, J. Theoretical study of intermolecular interactions in crystalline arene–perhaloarene adducts in terms of the electron density. RSC Adv. 2016, 6, 77301–77309. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Hoser, A.A.; Varughese, S.; Kamiński, R.; Malinska, M.; Stachowicz, M.; Pedireddi, V.R.; Woźniak, K. Structural and Energetic Analysis of Molecular Assemblies in a Series of Nicotinamide and Pyrazinamide Cocrystals with Dihydroxybenzoic Acids. Cryst. Growth Des. 2017, 17, 4918–4931. [Google Scholar] [CrossRef]
- Tao, Q.; Hao, Q.-Q.; Voronin, A.P.; Dai, X.-L.; Huang, Y.; Perlovich, G.L.; Lu, T.-B.; Chen, J.-M. Polymorphic Forms of a Molecular Salt of Phenazopyridine with 3,5-Dihydroxybenzoic Acid: Crystal Structures, Theoretical Calculations, Thermodynamic Stability, and Solubility Aspects. Cryst. Growth Des. 2019, 19, 5636–5647. [Google Scholar] [CrossRef]
- Manin, A.N.; Voronin, A.P.; Drozd, K.V.; Churakov, A.V.; Perlovich, G.L. Pharmaceutical salts of emoxypine with dicarboxylic acids. Acta Crystallogr. Sect. C 2018, 74, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.O.; Churakov, A.V.; Perlovich, G.L. Three Polymorphic Forms of Ciprofloxacin Maleate: Formation Pathways, Crystal Structures, Calculations, and Thermodynamic Stability Aspects. Cryst. Growth Des. 2016, 16, 6556–6567. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Simagina, A.A.; Churakov, A.V.; Skachilova, S.Y.; Perlovich, G.L. Saccharin salts of biologically active hydrazone derivatives. New J. Chem. 2015, 39, 8614–8622. [Google Scholar] [CrossRef]
- Chan, H.C.S.; Kendrick, J.; Neumann, M.A.; Leusen, F.J.J. Towards ab initio screening of co-crystal formation through lattice energy calculations and crystal structure prediction of nicotinamide, isonicotinamide, picolinamide and paracetamol multi-component crystals. CrystEngComm 2013, 15, 3799–3807. [Google Scholar] [CrossRef] [Green Version]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between interaction energy, intermolecular distance and electron density properties in hydrogen bonded complexes under external electric fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Kuznetsov, M.L. Can halogen bond energy be reliably estimated from electron density properties at bond critical point? The case of the (A)nZ—Y…X− (X, Y = F, Cl, Br) interactions. Int. J. Quantum Chem. 2019, 119, e25869. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
- Korlyukov, A.A.; Nelyubina, Y.V. Quantum chemical methods in charge density studies from X-ray diffraction data. Russ. Chem. Rev. 2019, 88, 677–716. [Google Scholar] [CrossRef]
- Dem’yanov, P.; Polestshuk, P. A Bond Path and an Attractive Ehrenfest Force Do Not Necessarily Indicate Bonding Interactions: Case Study on M2X2 (M = Li, Na, K; X = H, OH, F, Cl). Chem. Eur. J. 2012, 18, 4982–4993. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, S. Why Bond Critical Points Are Not “Bond” Critical Points. Chem. Eur. J. 2018, 24, 5401–5405. [Google Scholar] [CrossRef] [PubMed]
- Iogansen, A.V. Direct proportionality of the hydrogen bonding energy and the intensification of the stretching ν(XH) vibration in infrared spectra. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1999, 55, 1585–1612. [Google Scholar] [CrossRef]
- Rozenberg, M.; Loewenschuss, A.; Marcus, Y. An empirical correlation between stretching vibration redshift and hydrogen bond length. PCCP 2000, 2, 2699–2702. [Google Scholar] [CrossRef]
- Musin, R.N.; Mariam, Y.H. An integrated approach to the study of intramolecular hydrogen bonds in malonaldehyde enol derivatives and naphthazarin: Trend in energetic versus geometrical consequences. J. Phys. Org. Chem. 2006, 19, 425–444. [Google Scholar] [CrossRef]
- Filarowski, A.; Koll, A.; Sobczyk, L. Intramolecular Hydrogen Bonding in o-hydroxy Aryl Schiff Bases. Curr. Org. Chem. 2009, 13, 172–193. [Google Scholar] [CrossRef]
- Medvedev, A.G.; Mikhaylov, A.A.; Chernyshov, I.Y.; Vener, M.V.; Lev, O.; Prikhodchenko, P.V. Effect of aluminum vacancies on the H2O2 or H2O interaction with a gamma-AlOOH surface. A solid-state DFT study. Int. J. Quantum Chem. 2019, 119, e25920. [Google Scholar] [CrossRef]
- Musso, F.; Casassa, S.; Corno, M.; Ugliengo, P. How strong are H-bonds at the fully hydroxylated silica surfaces? Insights from the B3LYP electron density topological analysis. Struct. Chem. 2017, 28, 1009–1015. [Google Scholar] [CrossRef]
- Vener, M.V.; Manaev, A.V.; Egorova, A.N.; Tsirelson, V.G. QTAIM Study of Strong H-Bonds with the O−H…A Fragment (A=O, N) in Three-Dimensional Periodical Crystals. J. Phys. Chem. A 2007, 111, 1155–1162. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Mazák, K.; Noszál, B. Physicochemical Properties of Zwitterionic Drugs in Therapy. ChemMedChem 2020, 15, 1102–1110. [Google Scholar] [CrossRef]
- Yang, Z.; Li, Q.; Yang, G. Zwitterionic structures: From physicochemical properties toward computer-aided drug designs. Future Med. Chem. 2016, 8, 2245–2262. [Google Scholar] [CrossRef] [PubMed]
- Mesallati, H.; Mugheirbi, N.A.; Tajber, L. Two Faces of Ciprofloxacin: Investigation of Proton Transfer in Solid State Transformations. Cryst. Growth Des. 2016, 16, 6574–6585. [Google Scholar] [CrossRef] [Green Version]
- Mazzenga, G.C.; Berner, B. The transdermal delivery of zwitterionic drugs I: The solubility of zwitterion salts. J. Control. Release 1991, 16, 77–88. [Google Scholar] [CrossRef]
- Gunnam, A.; Suresh, K.; Ganduri, R.; Nangia, A. Crystal engineering of a zwitterionic drug to neutral cocrystals: A general solution for floxacins. Chem. Commun. 2016, 52, 12610–12613. [Google Scholar] [CrossRef] [PubMed]
- Manallack, D.T.; Prankerd, R.J.; Yuriev, E.; Oprea, T.I.; Chalmers, D.K. The significance of acid/base properties in drug discovery. Chem. Soc. Rev. 2013, 42, 485–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, P.H.; Wermuth, C.G.; Pure, I.U.o.; Chemistry, A. Handbook of Pharmaceutical Salts Properties, Selection, and Use; Wiley: Weinheim, Germany, 2008. [Google Scholar]
- Han, H.-K.; Choi, H.-K. Improved absorption of meloxicam via salt formation with ethanolamines. Eur. J. Pharm. Biopharm. 2007, 65, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Gwak, H.-S.; Choi, J.-S.; Choi, H.-K. Enhanced bioavailability of piroxicam via salt formation with ethanolamines. Int. J. Pharm. 2005, 297, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Tunon, I.; Rinaldi, D.; Ruiz-Lopez, M.F.; Rivail, J.L. Hydroxide Ion in Liquid Water: Structure, Energetics, and Proton Transfer Using a Mixed Discrete-Continuum ab Initio Model. J. Phys. Chem. 1995, 99, 3798–3805. [Google Scholar] [CrossRef]
- Nemukhin, A.V.; Topol, I.A.; Grigorenko, B.L.; Burt, S.K. On the Origin of Potential Barrier for the Reaction OH- + CO2 → HCO3- in Water: Studies by Using Continuum and Cluster Solvation Methods. J. Phys. Chem. B 2002, 106, 1734–1740. [Google Scholar] [CrossRef]
- da Silva, E.F.; Svendsen, H.F.; Merz, K.M. Explicitly Representing the Solvation Shell in Continuum Solvent Calculations. J. Phys. Chem. A 2009, 113, 6404–6409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vener, M.V.; Shenderovich, I.G.; Rykounov, A.A. A qualitative study of the effect of a counterion and polar environment on the structure and spectroscopic signatures of a hydrated hydroxyl anion. Theor. Chem. Acc. 2013, 132, 1361. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Sun, J.; Bousquet, D.; Forbert, H.; Marx, D. Glycine in aqueous solution: Solvation shells, interfacial water, and vibrational spectroscopy from ab initio molecular dynamics. J. Chem. Phys. 2010, 133, 114508. [Google Scholar] [CrossRef]


| Fragment 1 | R(O∙∙∙H), Å | −ΔHHB 2, kJ/mol | −ΔHHB 3, kJ/mol | EHB 4, kJ/mol |
|---|---|---|---|---|
| [2AmNic+Fum+H2O] (1:1:1) | ||||
| O12…H21-N2 | 1.840 | 23.4 | - | 26.8 |
| O11…H11-N1 | 1.628 | 34.0 | - | 45.7 |
| O12…H31-O3 | 1.709 | 29.3 | 27.8 | 35.8 |
| O13…H32-O3 | 2.034 | 17.2 | 18.3 | 24.3 |
| O11…H14-O14 | 1.563 | 38.5 | 39.7 | 52.9 |
| O3…H1-O1 | 1.571 | 37.9 | 40.7 | 52.1 |
| ∑(–ΔHHB/EHB) | - | 180.3 (84.4) | (86.8) | 237.6 (112.1) |
| [2AmNic+Mle+H2O] (1:1:1) | ||||
| O12…H21-N2 | 1.792 | 25.4 | - | 30.5 |
| O11…H11-N1 | 1.785 | 25.7 | - | 30.2 |
| O12…H31-O3 | 1.725 | 28.5 | 24.8 | 34.9 |
| O13…H32-O3 | 1.797 | 25.2 | 17.6 | 28.5 |
| O3…H1-O1 | 1.525 | 41.5 | 43.5 | 58.2 |
| ∑(–ΔHHB/EHB) | - | 146.3 (95.2) | (85.9) | 182.3 (121.6) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Surov, A.O.; Vasilev, N.A.; Churakov, A.V.; Parashchuk, O.D.; Artobolevskii, S.V.; Alatortsev, O.A.; Makhrov, D.E.; Vener, M.V. Two Faces of Water in the Formation and Stabilization of Multicomponent Crystals of Zwitterionic Drug-Like Compounds. Symmetry 2021, 13, 425. https://0-doi-org.brum.beds.ac.uk/10.3390/sym13030425
Surov AO, Vasilev NA, Churakov AV, Parashchuk OD, Artobolevskii SV, Alatortsev OA, Makhrov DE, Vener MV. Two Faces of Water in the Formation and Stabilization of Multicomponent Crystals of Zwitterionic Drug-Like Compounds. Symmetry. 2021; 13(3):425. https://0-doi-org.brum.beds.ac.uk/10.3390/sym13030425
Chicago/Turabian StyleSurov, Artem O., Nikita A. Vasilev, Andrei V. Churakov, Olga D. Parashchuk, Sergei V. Artobolevskii, Oleg A. Alatortsev, Denis E. Makhrov, and Mikhail V. Vener. 2021. "Two Faces of Water in the Formation and Stabilization of Multicomponent Crystals of Zwitterionic Drug-Like Compounds" Symmetry 13, no. 3: 425. https://0-doi-org.brum.beds.ac.uk/10.3390/sym13030425