Chiroptical Properties of Amino Acids: A Density Functional Theory Study
by
 and
and
Martine Adrian-Scotto
1,*, 
Serge Antonczak
1,
Jan Hendrik Bredehöft
2,
Søren V. Hoffmann
3 and
Uwe J. Meierhenrich
1 1
Laboratoire de Chimie des Molécules Bioactives et des Arômes UMR 6001 CNRS-UNSA, Université de Nice-Sophia Antipolis, 28 Avenue Valrose, 06108 Nice, France
2
Institute for Applied and Physical Chemistry, University of Bremen, Leobener Str. NW2, 28359 Bremen, Germany
3
Institute for Storage Ring Facilities, Aarhus University, Ny Munkegade 120, DK-8000 Aarhus C, Denmark
*
Author to whom correspondence should be addressed.
Symmetry 2010, 2(2), 935-949; https://0-doi-org.brum.beds.ac.uk/10.3390/sym2020934
Submission received: 6 March 2010
/
Revised: 6 April 2010
/
Accepted: 14 April 2010
/
Published: 19 April 2010
(This article belongs to the Special Issue Symmetry of Life and Homochirality)
Abstract
:Amino acids are involved in many scientific theories elucidating possible origins of life on Earth. One of the challenges when discussing the evolutionary origin of biopolymers such as proteins and oligonucleotides in living organisms is the phenomenon that these polymers implement monomers of exclusively one handedness, a feature called biomolecular homochirality. Many attempts have been made to understand this process of racemic symmetry breaking. Assuming an extraterrestrial origin of the molecular building blocks of living organisms, their susceptibility to asymmetric photolysis by the absorption of circularly polarized electromagnetic radiation in interstellar space was proposed. In order to predict whether the interaction of circularly polarized light with various racemic amino acids can induce an enantiomeric excess, we investigated the electronic and chiroptical properties of the amino acids valine and isovaline by a molecular modelling approach based on quantum chemistry (Density Functional Theory). The average spectra of both L-valine and L-isovaline have been produced on the basis of Boltzmann population analysis using computed spectra for the various conformations of each amino acid.
1. Introduction
Amino acids have been known and studied for a long time, for the crucial role they play in biological organisms: 22 of them, called the proteinogenic amino acids, are found in contemporary proteins. The composition of proteins has evolved over time [1], and amino acids are thought to be deeply involved in prebiotic molecular processes triggering the origin of life on Earth [2]. The way these so important small molecules appeared at the primitive Earth’s surface gives rise to various assumptions. Among the different hypotheses on the prebiotic formation of amino acids, one seriously considers the contribution of carbonaceous chondrite meteorites [3]. The most famous example is the Murchison meteorite found in 1969, in which a wide variety of organic compounds was detected, not only hydrocarbons and amino acids [4] but also diamino acids [5] and even nucleobases [6]. Moreover, recent studies have confirmed that amino acids are also present in comets, with the detection of glycine in samples returned to Earth by NASA’s Stardust spacecraft [7]. This is not surprising since some meteorites could originate from comets. Today, it is seriously considered that a huge quantity of cometary [8] and meteoritic [9] matter was delivered to the early Earth and that the basic building blocks of modern biochemistry were already present in the early solar system, as the result of an extraterrestrial synthesis, made in the dense clouds of the interstellar medium (ISM).
Laboratory simulation experiments have shown that cometary ice chemistry may lead to the formation of amino acids [10,11,12]. A recent study on the evolution of the interstellar ice analogues has shown how the glycine molecule could be produced in the protostellar environments before its introduction into comets [13].
Even though by now we have reached a fair degree of understanding how life‘s molecules are produced abiotically, the origin of homochirality still remains a major unanswered question. Except for glycine, all proteinogenic amino acids have a chiral center with their α-carbon atom. Yet only the l-configuration is used in the construction of proteins. The sugars that make up the backbone material of DNA and RNA show the same phenomenon, they are chiral, but only their d-enantiomer is used in the construction of biopolymers. However when starting from achiral precursors, a chiral process would be needed to form products enantioselectively. Life itself is such a chiral process, but how the first breaking of symmetry occurred is still not known. Furthermore, enantioselective analyses of carbonaceous meteorite samples have revealed enantiomeric excesses within amino acid series: the l-configuration is favoured [14,15]. This observation means that amino acids delivered by extraterrestrial objects could have seeded the primitive Earth with left-handed molecules before the origin of life.
Where does racemic symmetry breaking in extraterrestrial amino acids come from? The presence of asymmetric light sources in space could explain this phenomenon: the interaction of circularly polarized radiation with organic matter is likely to produce a slight enantiomeric enhancement. Asymmetric photolysis allows the preferential destruction of one enantiomer during the photodegradation of a racemic mixture by circularly polarized light (CPL) [16,17,18,19]. Then, even a slight enantiomeric excess may under space conditions be amplified to a much more important gap between l- and d- configurations amounts.
Detection of circularly polarized electromagnetic radiation in interstellar environments was first reported by Bailey in 1998 [20]. Asymmetric photolysis in space gave rise to many further discussions, articles and books of which we can only mention a few [21,22,23,24,25]. Asymmetric syntheses by circularly polarized light were performed too [26,27], leading to a small enantiomeric excess. In situ determination of enantiomeric excesses in extraterrestrial objects has now become part of space research programs. The European Space Agency ESA has designed the ambitious ROSETTA mission. A rendez-vous with the Comet 67P/Churyumov-Gerasimenko is planned for 2014. The aim of the project is to identify chiral organic molecules in situ on the surface of a comet nucleus and to detect possible enantiomeric excesses in them. During the envisaged four years of the mission, the Rosetta probe will orbit the comet and deploy a small landing module called Philae, for ground-based observations. On board this landing unit, a multi-column enantio-selective gas chromatograph (GC) coupled to a linear reflectron time-of-flight mass-spectrometer is installed. This instrument, the COmetary Sampling and ACquisition experiment COSAC, will allow separation of chiral molecules, among which aminoacids [2,28,29].
Two enantiomers of a chiral molecule have the same intrinsic properties, except for their circular dichroism: they exhibit distinct responses to left- and right-circularly polarized light. Several studies, generally at room temperature, were performed in order to measure the vacuum-ultraviolet circular dichroism (VUVCD) spectra of amino and also diamino acids in aqueous solution, using synchrotron-radiation CD spectrophotometers [18,19,30,31,32]. Such instruments are capable of reaching wavelengths under 190 nm, down to 130 nm. The aim of these experiments was to study the susceptibility of the considered molecules to asymmetric photolysis, by highlighting at which wavelengths the effect was significant and also what sign it would have.
In this context, molecular modelling can be a useful tool to access the knowledge of photon absorption by amino acids. Electronic transitions can be estimated using quantum mechanical calculations applied to the considered structures. We present here our results obtained for the l- form of valine and isovaline.
2. Procedures of Quantum Computations
All quantum computations were performed by using the Gaussian03 [33] program, on an IBM cluster/P1600Power4+ machine located at the French National Computer Center for Higher Education (CINES). Full geometry optimisations in vaccum were carried out in the framework of density functional theory (DFT), using the Becke 3-Lee-Yang-Parr (B3LYP) exchange-correlation functional at the 6-31G(d,p) basis set level. Solvent effects, in terms of continuum methodology for instance, have not been taken into account since the representation of molecules in solution does not match with space conditions. Moreover, the zwitterionic form adopted by amino acids in aqueous solution was observed to be unstable in the gas phase (without any water molecules around), and could not be reproduced by DFT calculations. It has been checked that obtained structures were true minima of the Potential Energy Surface (PES) by verifying they do not exhibit vibrational imaginary frequencies. The ab initio time-dependent density functional theory (TDDFT) technique has been applied to the calculation of the electronic circular dichroism, i.e. energies and rotational strengths of electronic excitations. The functional and basis set were B3LYP and 6-31G(d,p), respectively. Hybrid functionals and the B3LYP functional in particular have previously been shown to be capable of accurate prediction of Electronic Circular Dichroism (ECD) characteristics [34,35,36,37]. The graphical interface GaussView4 [38] for Windows was used for preparing input for submission to Gaussian and also for visualizing the outputs, in particular all ECD spectra.
Since valine and isovaline are relatively flexible molecules, a complete conformational analysis was required to characterize the multiple possible conformations. Thus multiple scan calculations have been performed by modifying dihedral angles around Cα-Cβ, Cα-N and Cα-C bonds by semiempirical methods, in order to characterize each of the stationary points depending on these three main dihedral angles. Each of the stationary points found was then optimised by our DFT method. Thermodynamic corrections, calculated at 298 K and 1 atm by using B3LYP/6-31G(d,p) frequencies, have been added to the energies of each optimized structure leading to an estimation of the free energies for each conformation. The free energy values have then been used to obtain the Boltzmann population of conformers at 298,15 K. Boltzmann populations have been estimated according to:
where:
with Z being the partition function and Gi the absolute free energy of the considered conformation.
Such a procedure could also be applied to very low temperatures as those encoutered in interstellar regions (almost 2-3 K). In this article, only the spectra produced using free energies computed at 298 K are shown since the goal of this work is to propose a protocol that should be useful for comparisons with data produced in laboratory conditions. However, we have simulated such spectra at very low temperature and only slight modifications of the overall shape for each average spectra are noticed.
Circular dichroism is induced by the interaction between electric and magnetic dipole transition moments of chromophores [39,40,41]. It is related to the rotational strength, R, which is theoretically defined by:
where: R n → j is the rotational strength of the electric transition from the “n” to “j” states,
R n → j = Im < n | μ | j > ● < j | m | n >
- μ and m are the electric and magnetic moments, respectively, « ● » is the dot product
- and Im means the imaginary part of a complex number.
The rotational strength R is thus equal to the imaginary part of the scalar product of electric and magnetic transition moments.
The ECD spectra are automatically generated from electronic excitation energies and velocity rotational strengths by the Gaussview program (by overlapping the Gaussian function for each transition [41]). The rotational strength is expressed in cgs units.
3. Results and Discussion
3.1. Conformational Analysis of l-valine and l-isovaline
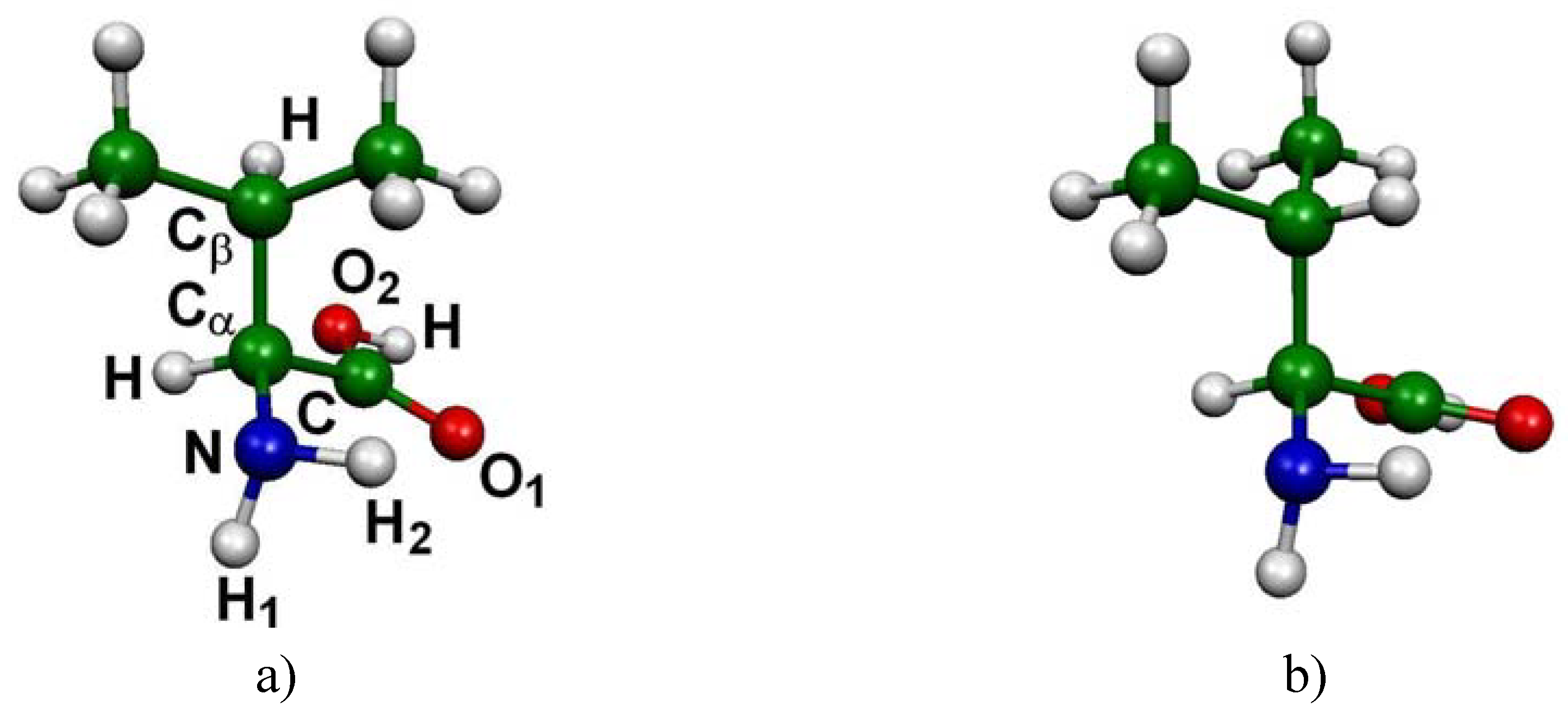
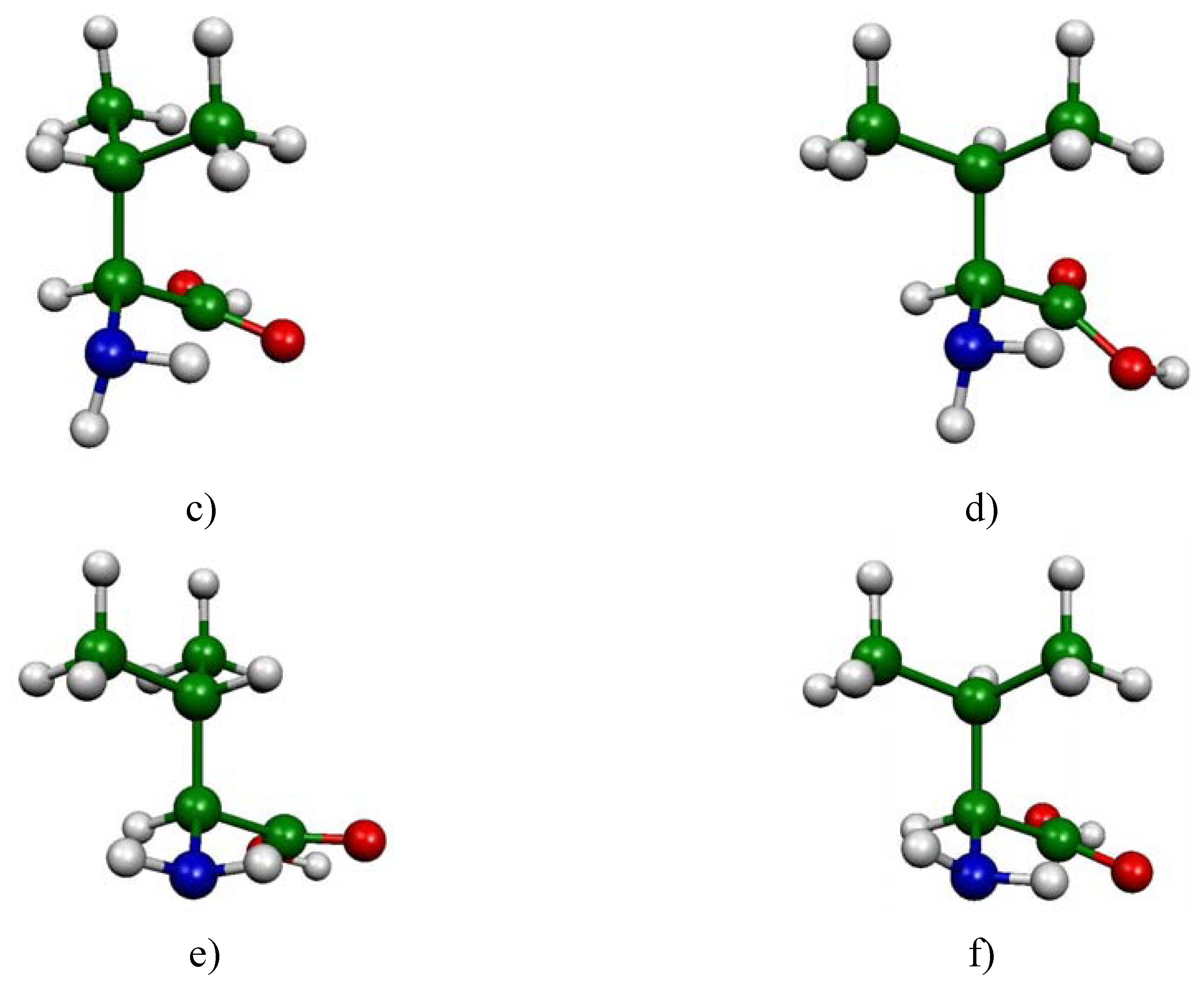
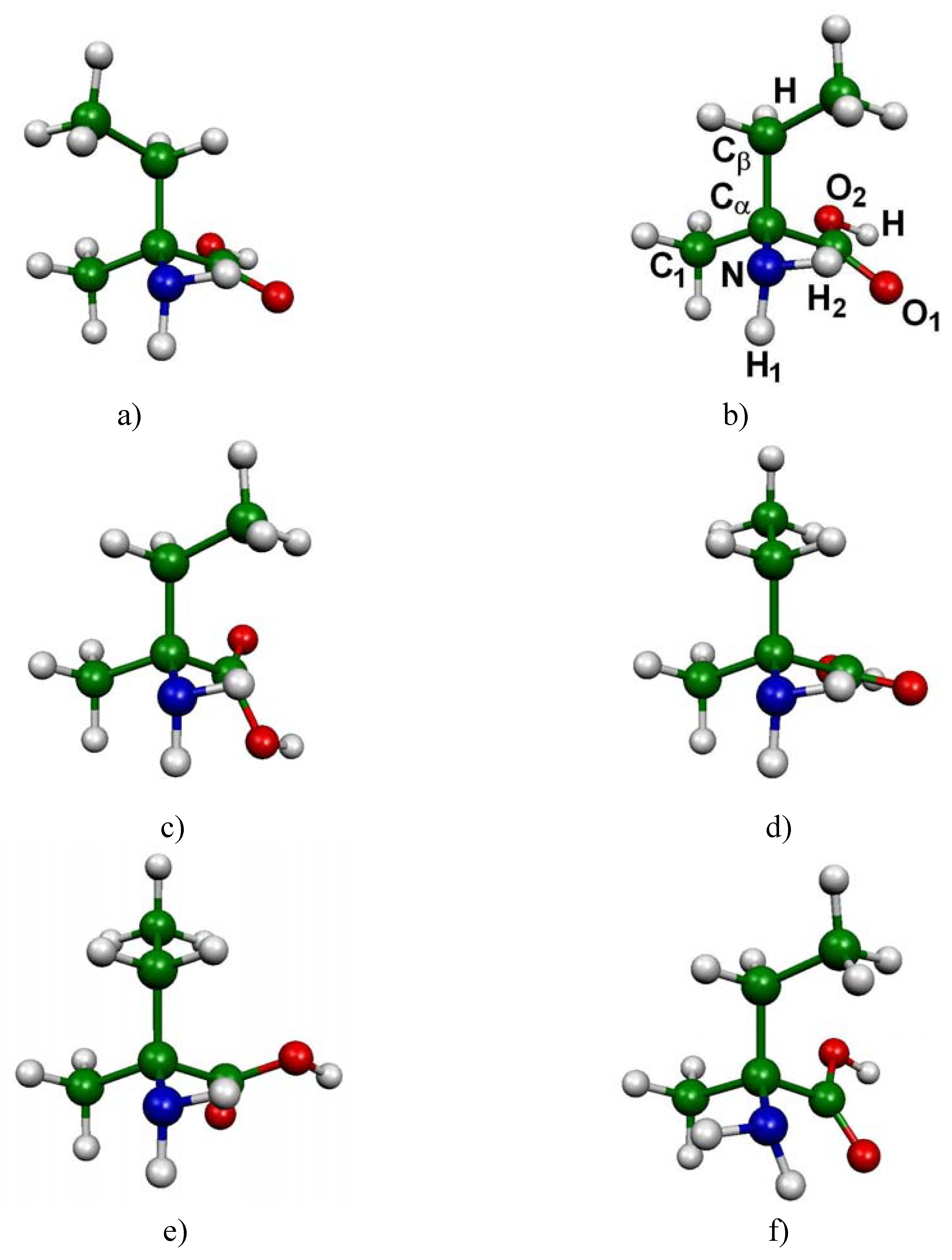
In the case of l-valine, 14 low-lying energy conformers were identified. The main structural differences between these conformers can be expressed by means of three dihedral angles a = H-Cα-Cβ-H, b = O1-C-Cα-Cβ and c = H1-N-Cα-Cβ (see Figure 1a for atom numbering). The corresponding absolute free energies are listed in Table 1 (in Hartree), as well as the free energy differences with respect to the CC1-CO1-CN1 most stable conformation (ΔG in kJ/mol). In the last column the populations obtained from Boltzmann analysis are reported.
The computations have revealed six main minima, which possess a different structural arrangement. In figure 1 are reported these six conformations corresponding to the most significantly populated neutral molecular structures of l-valine (their cumulative proportions reach 90% of the total population). The three most stable conformers only differ by the orientation of the -CHCH3CH3 group (Figure 1 a,b,c). The difference between the fourth conformer and the first one stems from the rotation of the carboxyl group (Figure 1 a,d). Conformers 5 and 6 (Figure 1 e,f) have to be compared to conformers 1 and 2 respectively (Figure 1 a,b) since the difference here lies in the orientation of the amino group.
Analogous characterizations were performed for the case of l-isovaline: 18 conformations have been identified. We present in Table 2 the results related to these 18 conformers. Here again, three dihedral angles are used to differentiate the general structure of each conformer: a′ = C1-Cα-Cβ-Η, b′ = O1-C-Cα-Cβ and c′ = H1-N-Cα-Cβ (see Figure 2a for atom numbering).
Figure 2 gives the geometrical arrangements of the six conformers of l-isovaline presenting the most important weights in Boltzmann analysis, obtained from DFT B3LYP/6-31G(d,p) optimizations.
Three conformers differ mainly by the rotation of the -CH2CH3 group (Figure 2 a,b,d), and present positions for the amino and carboxyl groups that are approximately the same. The rotation of the carboxyl group leads to two other conformations (Figure 2 c,e). The last conformer (Figure 2 f) is the only one for which a change in the position of the amino group is detected (among the six most stable conformations).
3.2. Computed ECD Spectra of l-valine and l-isovaline
The ECD intensities directly reflect individual electron transitions. Here we have computed excitation energies and rotational strengths for each located minimum of interest.
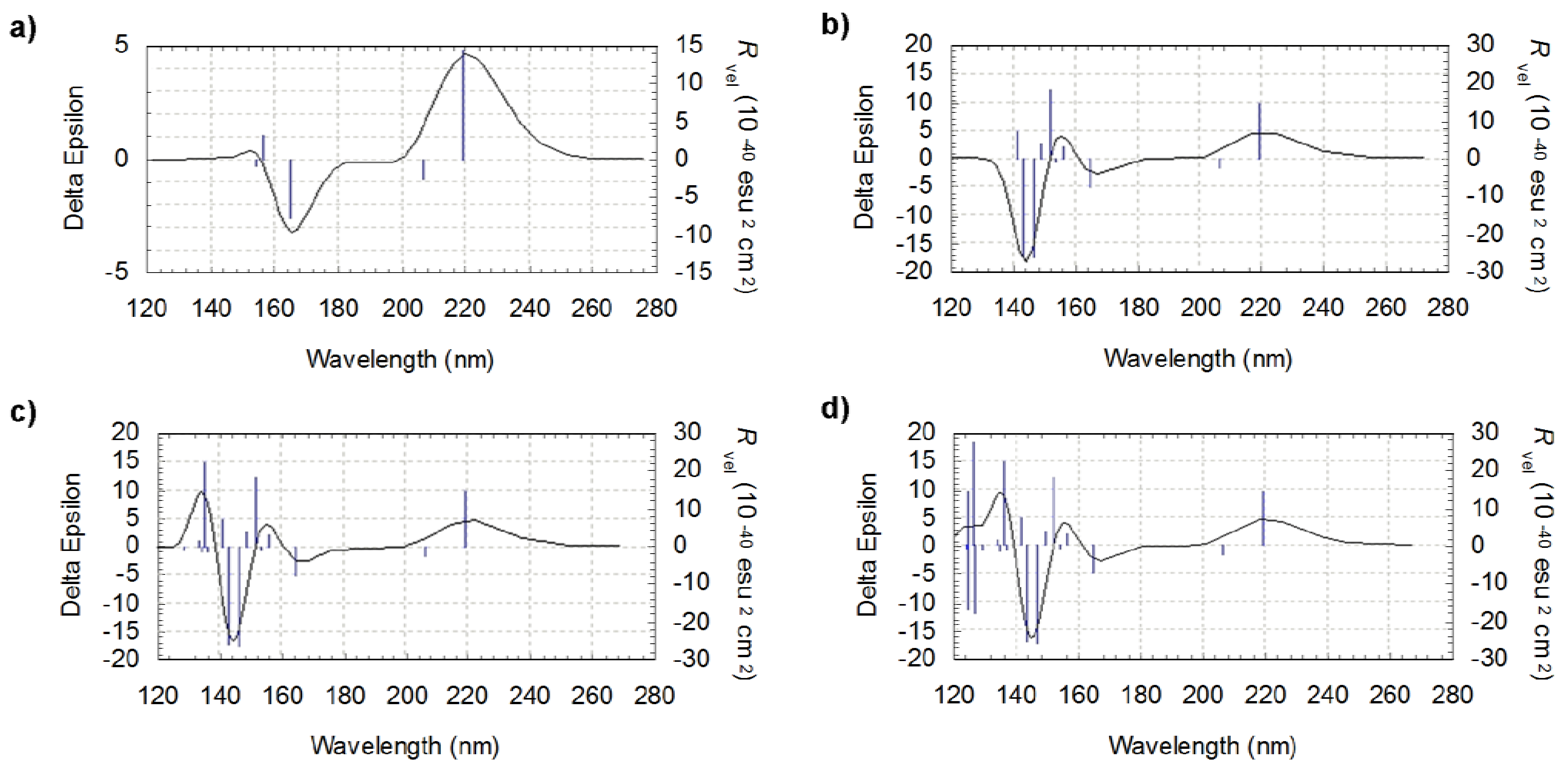
The number of considered excited states determines the extent of the spectrum: increasing this number results in adding peaks on the lower wavelengths side. This is illustrated in figure 3, where the ECD spectra for the lowest energy l-valine conformer (CC1-CO1-CN1) are presented. They have been produced taking into account respectively 5, 10, 15 and 20 excited states. Excitation energy is given in nm, rotational strength (R) in 10-40 esu2 cm2 cgs unit.
The wavelengths related to the first peak in each of the spectra in Figure 3 are: 154.1 nm, 141.5 nm, 129.1 nm and 124.2 nm, respectively. Since the spectral band of interest for us starts at approximately 130 nm, the most appropriate choice corresponds in this case to a number of states of 20. The same applies for the other conformers of l-valine and also to the l-isovaline ones.
We intend to compare in subsequent studies theoretically obtained ECD spectra of amino acids with experimental measurements by UV-CPL, with wavelengths between 140 and 280 nm. The choice of a 120–270 nm spectral band therefore seems appropriate.
We present now in Figure 4 and Figure 5 the results of electronic circular dichroism calculations obtained for the most significantly populated neutral molecular structures of the two studied amino acids, l-valine and l-isovaline respectively. It has to be noted that for each selected conformation, the TDDFT calculations lead to a good agreement between Rlength and Rvelocity values, which confirms the choice of the basis set [36].
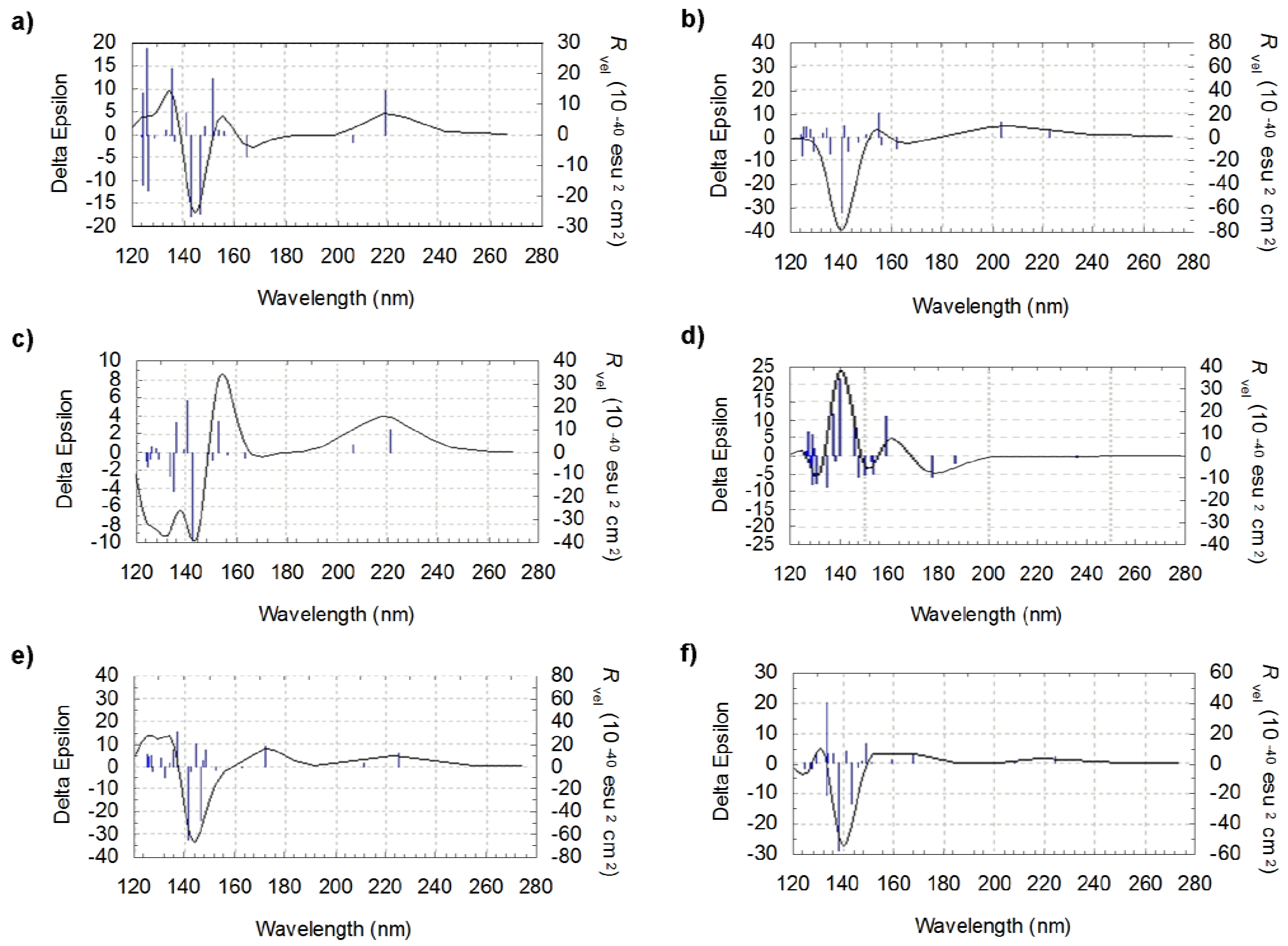
It can be noted that the rotational strengths of the various individual conformers drastically differ in sign and/or magnitude leading to large differences not only in the shape of the spectra but also in the relative intensities of the peaks. The region around 140 nm seems to be particularly interesting since, at this wavelength, the rotational strength reaches intense values. In this zone, the rotational strength is found to be negative for five conformers out of six and positive in one case only (Figure 4d). When comparing the various geometries (Figure 1), it appears that the position of the carboxyl group should play an important role in fixing the sign of the peaks
The differences that appear according to the position of the amino group are more difficult to point out precisely. Direct comparisons, considering the a and e structures in Figure 1 on one side and the b and f structures on the other (for wich the only structural difference is related to a rotation of the amino group), suggest that this group may play a role around 120–130 nm. However, analyzing the role of individual functional groups remains a challenging task since, as pointed out by our results, small differences in structure and thus in the position of each functional group, may lead to important modifications of the peak intensities for a given wavelength.
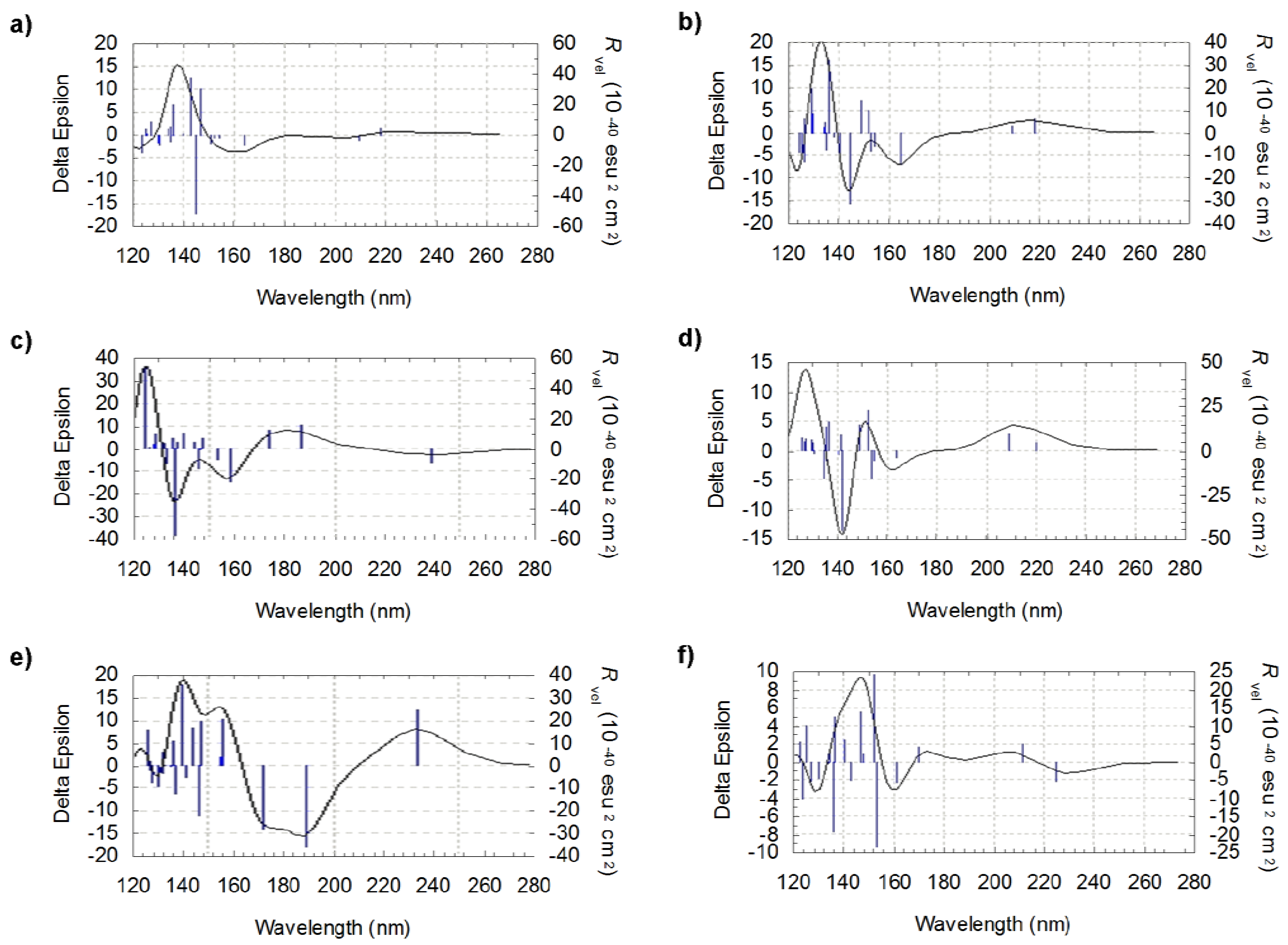
Inspecting the structures of l-isovaline in Figures 2a and c and their respective spectra in Figure 5, for which the main structural difference arises from the position of the carboxyl group, one notes the appearance of negative peaks around 140 nm. But the comparison between a and b strucures leads to the same remark though only the orientation of the –CH2CH3 group has been modified. Here again, these results confirm the fact that it remains relatively difficult to analyse the shape of the spectra by taking into account individually the orientations of the substituent. Moreover, in the case of l-isovaline the increase in complexity of the spectra may stem from the presence of an additional functional group on the asymmetric carbon of l-isovaline.
Generally speaking, the CD response of both amino acids is found to depend clearly on the molecular geometry. An experimentally detected spectrum can be considered as the averaged response of a collection of numerous individual molecules. In order to simulate accurately this averaged spectra by theoretical methods, it appears important to follow a procedure ensuring that the weight of each conformation in the resulting plot is relevant. One solution lies in considering the relative population of each conformer in term of percentage following Boltzmann statistics, based on relative free energies. These distributions are presented in Table 1 and Table 2 for l-valine and l-isovaline, respectively. Relative weigths have been used to produce averaged spectra for both amino acids. Thus the “real” computed spectra can be defined as a sum of individual responses (i.e. individual spectra as presented above), each of them being weighted by its occurence (here presented as a percentage). The two “real” computed spectra for both l-valine and l-isovaline are presented in Figure 6.
These theoretical curves have been obtained at the TDDFT/B3LYP/6-31G(d,p) level as a Boltzmann statistics analysis upon all the conformers of each amino acid, i.e. for every low-lying stationary point, taking into account the 20 lowest energy transitions.
Since the wavelengths corresponding to the various excited states are different from one conformer to another, it was essential to generate, from the Gaussian results, a set of continuous values of wavelengths and corresponding rotational strengths for every ECD spectrum. The curves presented in figure 6 were obtained from these sets.
A deconvolution of these spectra is necessary to obtain discrete values corresponding to excitation peaks. However, obtaining such a general shape, that corresponds to a consistent distribution between all the relevant structures for each amino-acid, must avoid using a single non-representative structure which could have led to a non-representative simulated spectrum. Here, note the high weight of the most stable conformers in each distribution (57.1% and 45.8% for l-valine and l-isovaline respectively). In the present case, using just these two structures (Figure 1a, Figure 2a and respective spectra) could lead to a good idea of the general shape of the spectrum for each molecule, but a complete analysis is necessary for describing precisely each ECD response. For instance, in the case of l-valine, taking into account the participation of all conformers to produce the averaged spectra leads to an increase of the rotational strength around 155 nm and a decrease around 130 nm with respect to the spectrum obtained for the most stable structure. Concerning l-isovaline, the combination of all spectra has highlighted additional peaks around 125 nm which are not present in the spectrum of the most stable conformer.
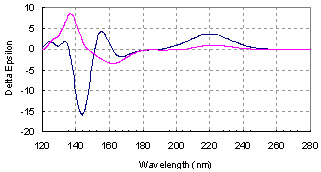
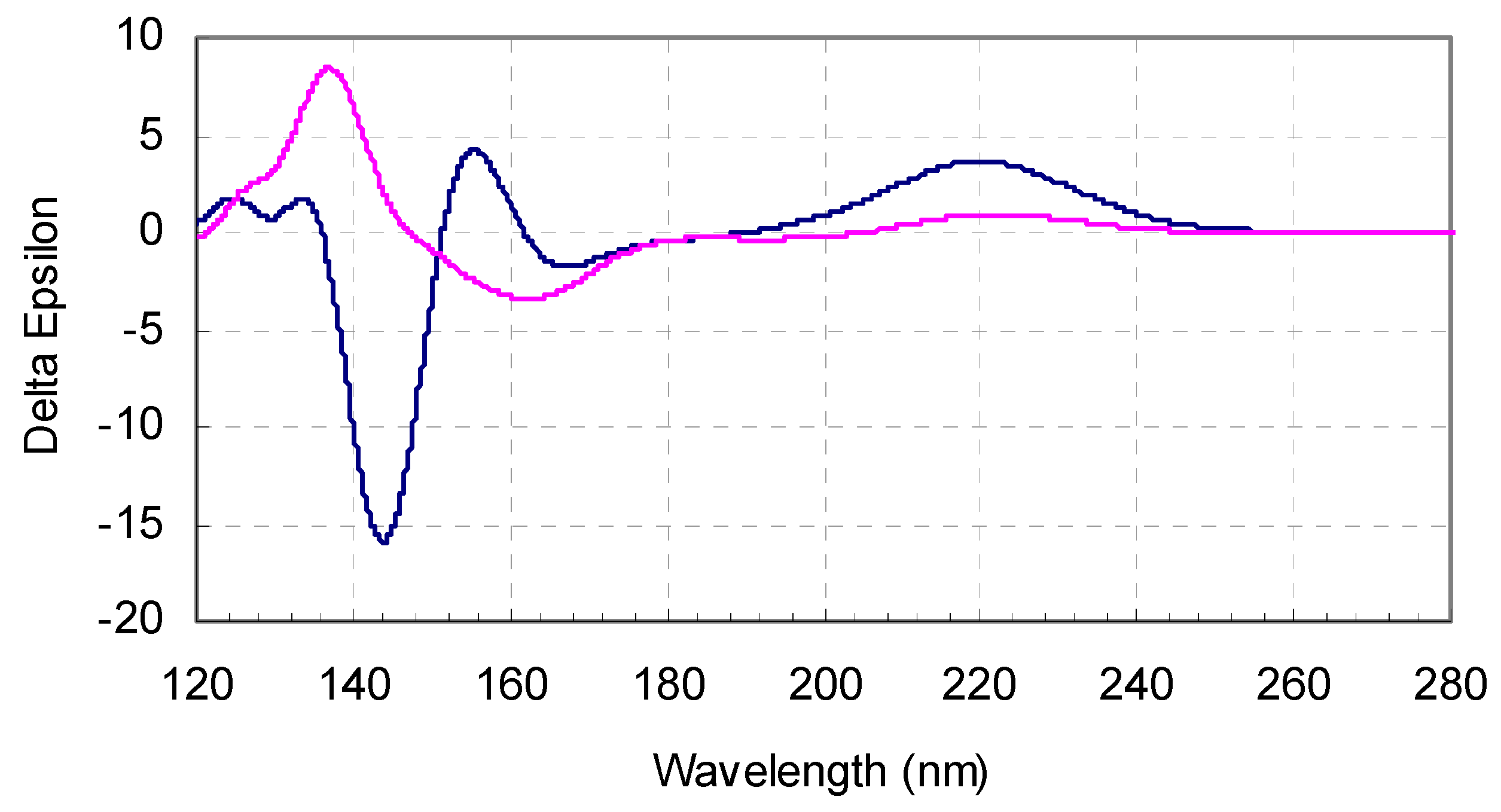
Direct comparison of the spectra for the two amino-acids also leads to interesting remarks: the magnitude of the ECD response is far larger around 140 nm than between 200 and 300 nm, and the shape of the spectra is different in this region for the two amino acids, even opposite in sign. Between 121 and 136 nm, the responses are of the same sign. Then the spectrum of l-isovaline presents a maximum at 137 nm, while that of the l-valine stays negative from 136 to 151 nm, with a minimum at 144 nm. l-valine reaches its maximum intensity of ECD signal at 155 nm, while l-isovaline presents a minimum at 162 nm.
An earlier attempt has been conducted [32] to obtain an average spectrum that takes into account different possible conformations of an amino acid. In this study, only the two most probable conformations of L-valine, obtained by molecular modelling protocols were considered. Though these conformations were not optimized at the DFT method, the proposed procedure was clearly a step forward the production of realistic spectroscopic responses since it took into account the interaction of this amino-acid with a surface. Most of the experiments in the present field of research are based on the detection of spectroscopic properties of molecules condensed on crystals transparent over a wide range of wavelengths. Thus, the combination of a Boltzmann population analysis, as proposed in this work, coupled with the characterization of the most probable conformations of the molecule of interest interacting with a surface would then be a breakthrough for the prediction of electronic CD spectra.
4. Conclusions
Generally, when computing ECD characteristics of a chiral molecule, only the results relative to the most stable structure are reported. Here we have taken into account the possible conformations of each amino acid in their neutral form as occurring in gas phase at room temperature and have weighted their respective contributions according to a Boltzmann statistical method. The theoretically obtained results of ab initio calculations for l-valine and l-isovaline were thus averaged and led to plausible ECD spectra. This way we hope to avoid quantitative differences between computed and experimental data, due to the problem of conformational flexibility. The next step will be to generate in the same way ECD spectra of other molecules of exobiological interest and to compare the results with those experimentally obtained by our team. According to remarks made in previous studies [37], the computed ECD spectra by the use of the TDDFT/B3LYP method with the 6-31G* basis set should correctly reflect the signs and shapes of the experimental ones, provided a certain red-shift is applied that further studies will allow to quantify.
Acknowledgements
Distant computational support was provided by the CINES (Centre Informatique National de l’Enseignement Supérieur) in Montpellier, France. This work was funded by the Groupe de Recherche en Exobiologie (GDR Exobio) from CNRS and ANR-07-BLAN-0293.
References
- Jordan, I.K.; Kondrashov, F.A.; Adzhubei, I.A.; Wolf, Y.I.; Koonin, E.V.; Kondrashov, A.S.; Sunyaev, S. A universal trend of amino acid gain and loss in protein evolution. Nature 2005, 433, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Meierhenrich, U.J. Amino Acids and the Asymmetry of Life; Springer: Heidelberg, Germany/New York, NY, USA, 2008. [Google Scholar]
- Cronin, J.R.; Pizzarello, S. Amino acids in meteorites. Adv. Space Res. 1983, 3, 5–18. [Google Scholar] [CrossRef]
- Kvenvolden, K; Lawless, J.; Pering, K.; Peterson, E.; Flores, J.; Ponnamperuma, C.; Kaplan, I.R.; Moore, C. Evidence for extraterrestrial amino-acids and hydrocarbons in the Murchison meteorite. Nature 1970, 228, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Meierhenrich, U.J.; Muñoz Caro, G.M.; Bredehöft, J.H.; Jessberger, E.K.; Thiemann, W. Identification of diamino acids in the Murchison meteorite. Proc. Natl. Acad. Sci. USA 2004, 101, 9182–9186. [Google Scholar] [CrossRef] [PubMed]
- Martins, Z.; Botta, O.; Fogel, M.L.; Sephton, M.A.; Glavin, D.P.; Watson, J.S.; Dworkin, J.P.; Schwartz, A.W.; Ehrenfreund, P. Extraterrestrial nucleobases in the Murchison meteorite. Earth Planet. Sci. Lett. 2008, 270, 130–136. [Google Scholar] [CrossRef]
- Elsila, J.E.; Glavin, D.P.; Dworkin, J.P. Cometary glycine detected in samples returned by Stardust. Meteorit. Planet. Sci. 2009, 44, 1323–1330. [Google Scholar] [CrossRef]
- Oró, J. Comets and the formation of biochemical compounds on the primitive Earth. Nature 1961, 190, 389–390. [Google Scholar] [CrossRef]
- Cronin, J.R.; Chang, S. Organic matter in meteorites: Molecular and isotopic analyses of the Murchison meteorite. In The chemistry of life’s origins; Greenberg, J.M., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1993; pp. 209–258. [Google Scholar]
- Kobayashi, K.; Kasamatsu, T.; Kaneko, T.; Koike, J.; Oshima, T.; Saito, T.; Yamamoto, T.; Yanagawa, H. Formation of amino acid precursors in cometary ice environments by cosmic radiation. Adv. Space Res. 1995, 16, 21–26. [Google Scholar] [CrossRef]
- Muñoz Caro, G.M.; Meierhenrich, U.J.; Schutte, W.A.; Barbier, B.; Arcones Segovia, A.; Rosenbauer, H.; Thiemann, W.; Brack, A.; Greenberg, J.M. Amino acids from ultraviolet irradiation of interstellar ice analogues. Nature 2002, 416, 403–406. [Google Scholar] [CrossRef]
- Bernstein, M.P.; Dworkin, J.P.; Sandford, S.A.; Cooper, G.W.; Allamandola, L.J. Racemic amino acids from the ultraviolet photolysis of interstellar ice analogues. Nature 2002, 416, 401–403. [Google Scholar] [CrossRef]
- Bossa, J.-B.; Duvernay, F.; Theulé, P.; Borget, F.; d’Hendecourt, L.; Chiavassa, T. Methylammonium methylcarbamate thermal formation in interstellar ice analogs: A glycine salt precursor in protostellar environments. Astron. Astrophys. 2009, 506, 601–608. [Google Scholar] [CrossRef]
- Pizzarello, S. The Chemistry of Life’s Origin: A Carbonaceous Meteorite Perspective. Acc. Chem. Res. 2006, 39, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Glavin, D.P.; Dworkin, J.P. Enrichment of the amino acid L-isovaline by aqueous alteration on CI and CM meteorite parent bodies. Proc. Natl. Acad. Sci. USA 2009, 106, 5487–5492. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, W.; Braun, E. Photochemische Erzeugung optisch aktiver Stoffe. Die Naturwissenschaften 1929, 17, 227–228. [Google Scholar] [CrossRef]
- Kuhn, W; Knopf, E. Photochemische Erzeugung optisch aktiver Stoffe. Naturwissenschaften 1930, 18, 183. [Google Scholar] [CrossRef]
- Meierhenrich, U.J.; Nahon, L.; Alcaraz, C.; Bredehöft, J.H.; Hoffmann, S.V.; Barbier, B.; Brack, A. Asymmetric Vacuum UV photolysis of the Amino Acid Leucine in the Solid State. Angew. Chem. Int. Ed. 2005, 44, 5630–5634. [Google Scholar] [CrossRef] [PubMed]
- Meierhenrich, U.J.; Filippi, J.-J.; Meinert, C.; Hoffmann, S.V.; Bredehöft, J.H.; Nahon, L. Photolysis of rac-Leucine with Circularly Polarized Synchrotron Radiation. Chem. Biodiv. 2010, in press. [Google Scholar] [CrossRef]
- Bailey, J.; Chrysostomou, A.; Hough, J.H.; Gledhill, T.M.; McCall, A.; Clark, S.; Ménard, F.; Tamura, M. Circular Polarization in Star-Formation Regions: Implications for Biomolecular Homochirality. Science 1998, 281, 672–674. [Google Scholar] [CrossRef]
- Meierhenrich, U.J.; Thiemann, W.; Rosenbauer, H. Molecular parity violation via comets? Chirality 1999, 11, 575–582. [Google Scholar] [CrossRef]
- Cerf, C.; Jorissen, A. Is amino-acid homochirality due to asymmetric photolysis in space ? Space Sci. Rev. 2000, 92, 603–612. [Google Scholar] [CrossRef]
- Jorissen, A.; Cerf, C. Asymmetric photoreactions as the origin of biomolecular homochirality: a critical review. Orig. Life Evol. Biosph. 2002, 32, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Meierhenrich, U.J.; Thiemann, W. Photochemical concepts on the origin of biomolecular asymmetry. Orig. Life Evol. Biosph. 2004, 34, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Takahashi, J-I.; Kaneko, T.; Marumo, K.; Kobayashi, K. Asymmetric synthesis of amino acid precursors in interstellar complex organics by circularly polarized light. Earth Planet. Sci. Lett. 2007, 254, 106–114. [Google Scholar] [CrossRef]
- Thiemann, W.; Rosenbauer, H.; Meierhenrich, U.J. Conception of the ‘chirality-experiment’ on esa's mission rosetta to comet 46P/wirtanen. Adv. Space Res. 2001, 27, 323–328. [Google Scholar] [CrossRef]
- Nuevo, M.; Meierhenrich, U.J.; Muñoz Caro, G.M.; Dartois, E.; d’Hendecourt, L.; Deboffle, D.; Auger, G.; Blanot, D.; Bredehöft, J.H.; Nahon, L. The effects of circularly polarized light on amino acid enantiomers produced by the UV irradiation of interstellar ice analogs. Astron. Astrophys. 2006, 457, 741–751. [Google Scholar] [CrossRef]
- Thiemann, W.; Meierhenrich, U.J. ESA Mission ROSETTA Will Probe for Chirality of Cometary Amino Acids. Orig. Life Evol. Biosph. 2001, 31, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Goesmann, F.; Rosenbauer, H.; Roll, R.; Szopa, C.; Raulin, F.; Sternberg, R.; Israel, G.; Meierhenrich, U.J.; Thiemann, W.; Munoz-Caro, G. Cosac, The Cometary Sampling and Composition Experiment on Philae. Space Sci. Rev. 2007, 128, 257–280. [Google Scholar] [CrossRef]
- Gekko, K.; Matsuo, K. Vacuum-Ultraviolet Circular Dichroism Analysis of Biomolecules. Chirality 2006, 18, 329–334. [Google Scholar] [CrossRef]
- Bredehöft, J.H.; Breme, K.; Meierhenrich, U.J.; Hoffmann, S.V.; Thiemann, W. Chiroptical Properties of Diamino Carboxylic Acids. Chirality 2007, 19, 570–573. [Google Scholar] [CrossRef]
- Kaneko, F.; Yagi-Watanabe, K.; Tanaka, M.; Nakagawa, K. Natural Circular Dichroism Spectra of Alanine and Valine Films in Vaccum Ultraviolet Region. J. Phys. Soc. Jpn. 2009, 78, 013001:1–013001:4. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A., Jr.; Stratmann, R.E.; Burant, J.C.; Dapprich, S.; Millam, J.M.; Daniels, A.D.; Kudin, K.N.; Strain, M.C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G.A.; Ayala, P.Y.; Cui, Q.; Morokuma, K.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Cioslowski, J.; Ortiz, J.V.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M.W.; Johnson, B.G.; Chen, W.; Wong, M.W.; Andres, J.L.; Head-Gordon, M.; Replogle, E.S.; Pople, J.A. Gaussian03; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Bour, P. Simulation of Electronic Circular Dichroism with Rigid Kohn-Sham Orbitals: A Computational Experiment. J. Phys. Chem. A 1999, 103, 5099–5104. [Google Scholar] [CrossRef]
- Pecul, M.; Ruud, K.; Helgaker, T. Density functional theory calculation of electronic circular dichroism using London orbitals. Chem. Phys. Lett. 2004, 38, 110–119. [Google Scholar] [CrossRef]
- Stephens, P.J.; McCann, D.M.; Devlin, F.J.; Cheeseman, J.R.; Frisch, M.J. Determination of the Absolute Configuration of [32](1,4)Barrelenophanedicarbonitrile Using Concerted Time-Dependent Density Functional Theory Calculations of Optical Rotation and Electronic Circular Dichroism. J. Am. Chem. Soc. 2004, 126, 7514–7521. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, E.; Tanaka, K; Verotta, L.; Nakanishi, K.; Berova, N.; Rosini, C. Determination of the absolute configurations of flexible molecules: Synthesis and theoretical simulation of electronic circular dichroism/optical rotation of some pyrrolo[2,3-b]indoline alkaloids - A case study. Chirality 2007, 19, 434–445. [Google Scholar] [PubMed]
- GaussView4; Gaussian, Inc.: Pittsburgh, PA, USA, 2006.
- Berova, N.; Nakanishi, K.; Woody, R.W. Circular dichroism - Principles and Applications, 2nd ed.; Wiley-VCH: New York, NY, USA, 2000; pp. 1–36. [Google Scholar]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Harada, N. ECD Cotton Effect Approximated by the Gaussian Curve and Other Methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Conformations of l-valine: the six most significant conformers according to Boltzmann statistics analysis based on DFT B3LYP/6-31G(d,p) computations at 298 K (a) CC1-CO1-CN1 (b) CC3-CO5-CN1 (c) CC3-CO3-CN3 (d) CC1-CO3-CN1 (e) CC1-CO5-CN3 (f) CC3-CO5-CN3.
Figure 1.
Conformations of l-valine: the six most significant conformers according to Boltzmann statistics analysis based on DFT B3LYP/6-31G(d,p) computations at 298 K (a) CC1-CO1-CN1 (b) CC3-CO5-CN1 (c) CC3-CO3-CN3 (d) CC1-CO3-CN1 (e) CC1-CO5-CN3 (f) CC3-CO5-CN3.


Figure 2.
Conformations of l-isovaline: the six most significant conformers according to a Boltzmann statistics analysis based on DFT B3LYP/6-31G(d,p) computations at 298 K (a) CC3-CO5-CN1 (b) CC2-CO5-CN1 (c) CC3-CO6-CN1 (d) CC1-CO3-CN1 (e) CC1-CO1-CN1 (f) CC3-CO7-CN3.
Figure 2.
Conformations of l-isovaline: the six most significant conformers according to a Boltzmann statistics analysis based on DFT B3LYP/6-31G(d,p) computations at 298 K (a) CC3-CO5-CN1 (b) CC2-CO5-CN1 (c) CC3-CO6-CN1 (d) CC1-CO3-CN1 (e) CC1-CO1-CN1 (f) CC3-CO7-CN3.

Figure 3.
Calculated ECD spectra of lowest energy l-valine conformer (CC1-CO1-CN1) at B3LYP/6-31G(d,p) (a) with 5 excited states, (b) with 10 excited states, (c) with 15 excited states and (d) with 20 excited states.
Figure 3.
Calculated ECD spectra of lowest energy l-valine conformer (CC1-CO1-CN1) at B3LYP/6-31G(d,p) (a) with 5 excited states, (b) with 10 excited states, (c) with 15 excited states and (d) with 20 excited states.

Figure 4.
Theoretical CD curves for l-valine: comparison for the six most significant conformers (a) CC1-CO1-CN1 (b) CC3-CO5-CN1 (c) CC3-CO3-CN3 (d) CC1-CO3-CN1 (e) CC1-CO5-CN3 (f) CC3-CO5-CN3.
Figure 4.
Theoretical CD curves for l-valine: comparison for the six most significant conformers (a) CC1-CO1-CN1 (b) CC3-CO5-CN1 (c) CC3-CO3-CN3 (d) CC1-CO3-CN1 (e) CC1-CO5-CN3 (f) CC3-CO5-CN3.

Figure 5.
Theoretical CD curves for l-isovaline: comparison for the six most significant conformers (a) CC3-CO5-CN1 (b) CC2-CO5-CN1 (c) CC3-CO6-CN1 (d) CC1-CO3-CN1 (e) CC1-CO1-CN1 (f) CC3-CO7-CN3
Figure 5.
Theoretical CD curves for l-isovaline: comparison for the six most significant conformers (a) CC3-CO5-CN1 (b) CC2-CO5-CN1 (c) CC3-CO6-CN1 (d) CC1-CO3-CN1 (e) CC1-CO1-CN1 (f) CC3-CO7-CN3

Figure 6.
Boltzmann population weighted theoretical CD curves for l-valine (blue line) and l-isovaline (red line).
Figure 6.
Boltzmann population weighted theoretical CD curves for l-valine (blue line) and l-isovaline (red line).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
l-valine conformers from a DFT B3LYP/6-31G(d,p) calculation at T = 298 K.
| Conformers of l-valine | a (°) | b (°) | c (°) | G (H) | ΔG (kJ/mol) | Ni (%) |
|---|---|---|---|---|---|---|
| CC1-CO1-CN1 | - 65.09 | 107.81 | 161.20 | - 402.259301 | 0 | 57.11 |
| CC3-CO5-CN1 | 176.19 | 81.36 | 169.74 | - 402.258267 | 2.72 | 19.09 |
| CC3-CO3-CN3 | 63.77 | 93.75 | 164.41 | - 402.257400 | 4.99 | 7.62 |
| CC1-CO3-CN1 | - 64.83 | - 61.41 | 169.05 | - 402.257202 | 5.51 | 6.18 |
| CC1-CO5-CN3 | - 61.83 | 100.36 | - 78.17 | - 402.256512 | 7.32 | 2.97 |
| CC3-CO5-CN3 | - 177.86 | 74.45 | - 63.29 | - 402.255997 | 8.68 | 1.72 |
| CC2-CO5-CN3 | 65.86 | 90.48 | - 78.67 | - 402.255919 | 8.88 | 1.59 |
| CC2-CO3-CN1 | 66.46 | - 80.25 | 170.29 | - 402.255659 | 9.56 | 1.20 |
| CC2-CO5-CN1 | - 177.29 | - 103.76 | - 55.53 | - 402.255367 | 10.33 | 0.88 |
| CC1-CO3-CN3 | - 62.34 | - 73.94 | - 65.18 | - 402.254890 | 11.58 | 0.53 |
| CC1-CO4-CN2 | - 59.19 | - 27.97 | 71.21 | - 402.254699 | 12.08 | 0.44 |
| CC2-CO3-CN3 | 66.45 | - 83.21 | - 67.26 | - 402.254449 | 12.74 | 0.33 |
| CC2-CO4-CN2 | 71.40 | - 19.53 | 64.27 | - 402.254102 | 13.65 | 0.23 |
| CC3-CO4-CN2 | - 165.38 | 1.57 | 68.87 | - 402.253352 | 15.62 | 0.10 |
ΔG represents the difference between G and the most stable conformer’s (CC1-CO1-CN1) free energy
Table 2.
l-isovaline conformers from a DFT B3LYP/6-31G(d,p) calculation.
| Conformers of l-isovaline | a′ (°) | b′ (°) | c′ (°) | G (H) | ΔG (kJ/mol) | Ni (%) |
|---|---|---|---|---|---|---|
| CC3-CO5-CN1 | - 58.65 | 116.70 | 173.69 | - 402.260832 | 0 | 45.75 |
| CC2-CO5-CN1 | - 178.95 | 109.38 | 175.79 | - 402.259992 | 2.21 | 18.79 |
| CC3-CO6-CN1 | - 60.04 | - 35.03 | 178.53 | - 402.259440 | 3.66 | 10.47 |
| CC1-CO3-CN1 | 63.79 | 87.52 | 176.02 | - 402.259037 | 4.71 | 6.83 |
| CC1-CO1-CN1 | 58.37 | - 128.60 | 177.65 | - 402.258809 | 5.31 | 5.36 |
| CC3-CO7-CN3 | - 51.81 | 139.77 | 83.15 | - 402.258525 | 6.06 | 3.97 |
| CC3-CO3-CN2 | - 58.36 | 100.69 | - 78.12 | - 402.258061 | 7.28 | 2.43 |
| CC1-CO1-CN2 | 63.11 | - 104.99 | - 55.65 | - 402.257413 | 8.98 | 1.22 |
| CC2-CO4-CN3 | - 56.49 | - 26.85 | 66.56 | - 402.257375 | 9.08 | 1.17 |
| CC3-CO1-CN2 | - 58.52 | - 77.94 | - 63.91 | - 402.256869 | 10.41 | 0.69 |
| CC2-CO2-CN3 | - 170.84 | 136.57 | 91.62 | - 402.256855 | 10.44 | 0.68 |
| CC2-CO3-CN2 | - 176.70 | 93.45 | - 68.44 | - 402.256749 | 10.72 | 0.60 |
| CC2-CO6-CN1 | - 178.54 | - 58.33 | - 179.59 | - 402.256739 | 10.75 | 0.60 |
| CC1-CO3-CN2 | 62.35 | 80.52 | - 68.14 | - 402.256619 | 11.06 | 0.53 |
| CC2-CO1-CN2 | - 175.29 | - 103.38 | - 53.81 | - 402.256494 | 11.39 | 0.46 |
| CC2-CO4-CN3 | - 172.29 | - 17.68 | 70.12 | - 402.255819 | 13.16 | 0.23 |
| CC1-CO4-CN3 | 70.08 | - 4.62 | 61.99 | - 402,255366 | 14.35 | 0.14 |
| CC1-CO2-CN3 | 65.73 | - 169.89 | 60.42 | - 402,255011 | 15.28 | 0.10 |
ΔG represents the difference between G and the most stable conformer’s (CC3-C05-CN1) free energy
© 2010 by the authors; licensee MDPI, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Adrian-Scotto, M.; Antonczak, S.; Bredehöft, J.H.; Hoffmann, S.V.; Meierhenrich, U.J. Chiroptical Properties of Amino Acids: A Density Functional Theory Study. Symmetry 2010, 2, 935-949. https://0-doi-org.brum.beds.ac.uk/10.3390/sym2020934
AMA Style
Adrian-Scotto M, Antonczak S, Bredehöft JH, Hoffmann SV, Meierhenrich UJ. Chiroptical Properties of Amino Acids: A Density Functional Theory Study. Symmetry. 2010; 2(2):935-949. https://0-doi-org.brum.beds.ac.uk/10.3390/sym2020934
Chicago/Turabian StyleAdrian-Scotto, Martine, Serge Antonczak, Jan Hendrik Bredehöft, Søren V. Hoffmann, and Uwe J. Meierhenrich. 2010. "Chiroptical Properties of Amino Acids: A Density Functional Theory Study" Symmetry 2, no. 2: 935-949. https://0-doi-org.brum.beds.ac.uk/10.3390/sym2020934