1. Introduction
Calcite, aragonite, and vaterite are the major polymorphs of calcium carbonate. Among them, calcite is the most stable and abundant form at ambient conditions. In some marine and terrestrial systems, where biotic or abiotic kinetically controlled processes control the formation of aragonite, it may be present as the major polymorph [
1,
2]. The presence of aragonite in soil is, however, attributed to formation at a high temperature and/or sufficiently high Mg concentration [
3,
4]. The last form, vaterite, is a metastable mineral and rarely found in natural environments [
1]. Hence, vaterite and aragonite are considered to be precursors for calcite formation via Ostwald ripening [
5,
6]. Ostwald ripening leads to the dissolution of metastable polymorphs of calcium carbonate (vaterite) and the subsequent crystallization of stable polymorphs [
6] (calcite and/or aragonite, depending on solution conditions).
Due to the high solubility of carbonate minerals and daily variations in soil water content, the dissolution and precipitation of carbonate minerals frequently occur in calcareous soils and sediments. The calcium concentration varies as these processes occur. Ions other than Ca may also be sequestered by coprecipitation. The extent of ion sequestration depends on ion concentrations and may vary from trace (like heavy metals) to major components (like magnesium (Mg)). Typically, Mg, strontium (Sr), and manganese (Mn) are major impurities in natural calcites [
1]. Natural calcite with a considerable amount of Mg in its structure is usually called Mg-calcite [
1,
2]. In marine environments, where the Mg concentration is relatively high, ~50 mmol/L ≅ 5 × Ca concentration [
7,
8], the formation of Mg-calcites is common [
1]. Mg coprecipitation with calcite has been extensively studied in the context of marine environments [
9]. These results are not directly transferable to terrestrial systems, where low supersaturation conditions are omnipresent in the soil solution of calcareous soils.
Groundwater is the major source of irrigation water in arid and semi-arid regions [
10]. The quality of groundwater thus dominantly affects the salt accumulation or migration in the soil profiles [
11,
12]. Recent investigations have shown an important shift in the ionic composition of irrigation water towards relatively high concentrations of Mg, such that the Mg to Ca ratio exceeds unity [
13,
14]. Using Mg-enriched irrigation water gradually changes the ionic composition of the soil solution, as well as the composition of exchangeable ions in the solid phase (i.e., clays) [
15]. At the same time, this process may affect the composition of precipitated calcium carbonate, leading to the formation of Mg-calcite and/or other polymorphs. These reactions affect the solubility of minerals and, in turn, the equilibrium concentration of ions. Therefore, the solution speciation predicted with thermodynamically valid solubility constants for pure phases may not correctly simulate real solution speciation.
The interaction of Mg with calcite has been studied using microscopic and spectroscopic techniques, as well as with kinetic models [
8,
16,
17]. Typically, a partition coefficient is used to quantify the amount of foreign ions (e.g., Mg) in the calcite structure [
9,
18,
19]. The high variability in Mg uptake that has been previously reported can be attributed to differences in experimental conditions such as supersaturation conditions and methods of preparing Mg-calcite. Synthesizing Mg-calcite at a relatively high supersaturation (Ω ≈ 50, Ω is defined in Equation (2)) usually incorporates low amounts of Mg into the calcite structure (≈0–1 mole %) compared to very high supersaturation (Ω > 200), which leads to the incorporation of significant amounts of Mg (≈14 mole %) [
20,
21]. The equilibrium pH may also affect the incorporation rate. Keeping the pH constant even at very low supersaturation (Ω = 5–11) induces Mg incorporation (7–10 mol%) [
9]. Moreover, the incorporation rate can be controlled by changes in the solution chemistry, precipitation rate, and temperature [
19]. For instance, an increase in the Mg to Ca ratio in a solution leads to a decrease in partition coefficients [
9] and could reduce the calcite growth rates [
1,
9,
18] by inhibiting calcite nucleation via Mg adsorption on growth sites, distortion of the calcite lattice, changes in the surface charge, and cation dehydration [
22]. As a result, in the presence of Mg ions, calcite morphology may change, and new faces may develop on edges and corners [
19].
Prolonged drought and the unbalanced use of groundwater as irrigation water may lead to changes in the ionic composition of groundwater. In some arid and semi-arid regions, with abundant calcareous soils, such changes often imply a relative increase in the aqueous Mg ion concentration and the aqueous Mg to Ca ratio. Subsequently, such processes are expected to have a long-term effect on the carbonate minerals in the respective soils. The aim of this study was, therefore, to investigate the impact of the aqueous Mg to Ca ratio on newly forming calcium carbonate minerals and to quantify the resulting Mg-uptake at conditions relevant for calcareous soils in arid- and semi-arid regions.
To facilitate the experimental procedure, the recrystallization of metastable vaterite to calcite (or aragonite) (according to the Ostwald ripening process) is employed in order to enable calcite (or aragonite) growth in simple batch-type experiments at five Mg to Ca ratios, three different pH conditions, and in the absence and presence of kaolinite and bentonite. This experimental method simulates low supersaturation (Ω = 3–4). Vaterite is not regarded as a component of interest in the investigated chemical system. Rather, it is the source that provides a constant supply of calcium (Ca) and carbonate ions at constant supersaturation with respect to calcite (and aragonite), as dictated by the differences in the solubility products of the respective polymorphs, and thus enables constant crystal growth at low supersaturation in very simple batch-type experiments. The Mg uptake is quantified via both solution chemistry and the digestion of Mg-doped calcium carbonate. In addition, the Mg-doped precipitates are characterized by X-ray diffraction and Rietveld refinement, as well as by scanning electron microscopy (SEM-EDX). On the basis of the experimental results, a regular mixing model for the dilute (Mg,Ca)CO3 solid solution is suggested.
2. Materials and Methods
2.1. Synthesis of Vaterite
Vaterite was synthesized by a rapid mixing of 10-mL 0.33-M CaCl
2 (CaCl
2·2H
2O, p.a., from Merck KGaA, Darmstadt, Germany) and 10-mL 0.33-M Na
2CO
3, (Na
2CO
3, anhydrous, p.a., from Merck KGaA, Darmstadt, Germany) in a water/ethylene glycol (80/20%,
v/
v) mixture [
23] (ethylene glycol, p.a., Merck KGaA, Darmstadt, Germany). After less than one minute of mixing, the precipitated solid phase was removed from the solution by filtration using a 10-µm Millipore
® filter paper (Merck KGaA, Darmstadt, Germany) and a suction pump. The solid phase was washed with a water and ethanol (50/50%,
v/
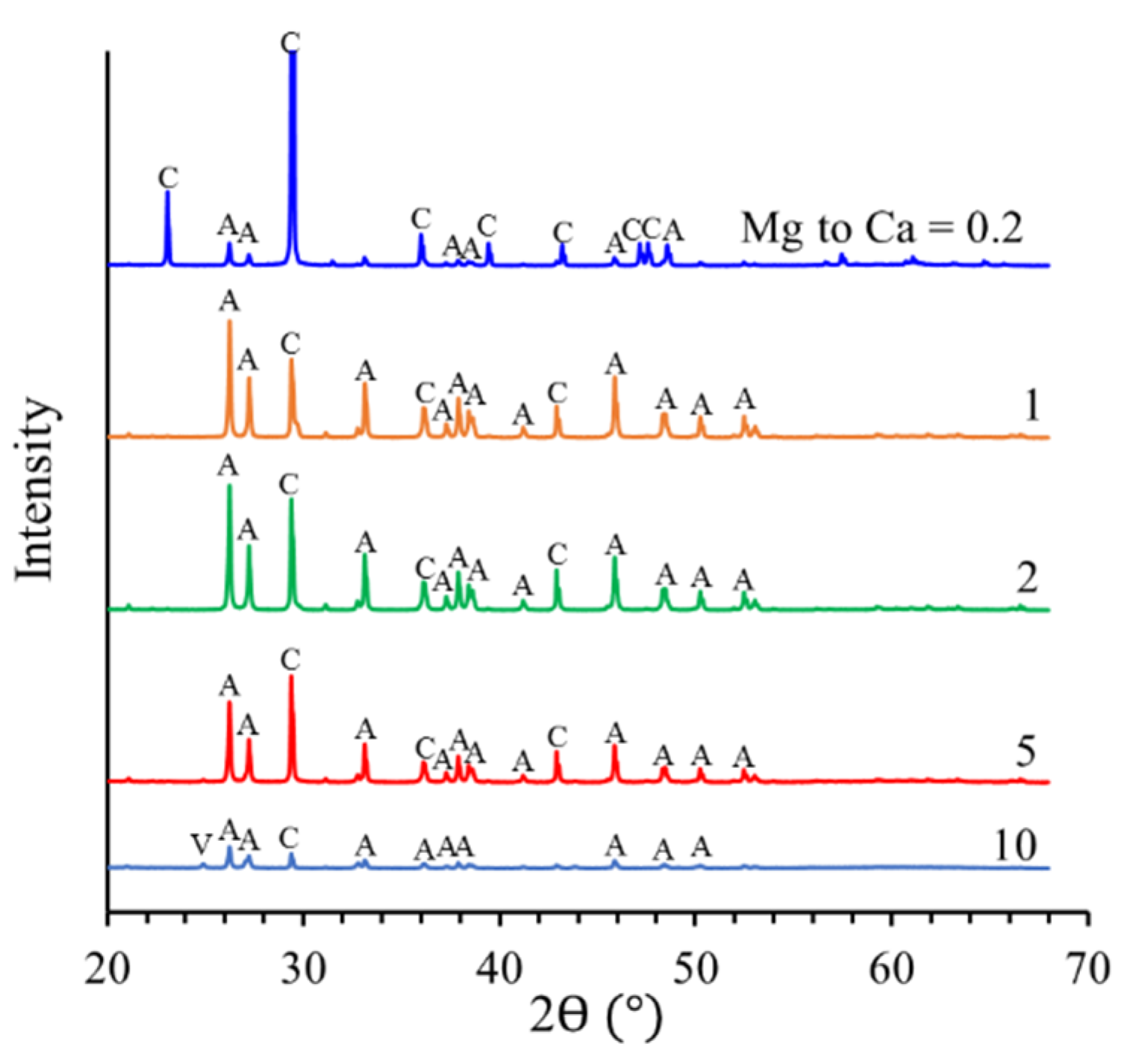
v) mixture (ethanol absolute, Merck KGaA, Darmstadt, Germany) and dried at 60 °C. X-ray diffraction of the originally synthesized solid phase (
Figure S1), shows that about 98% of the solid phase is vaterite. MilliQ water (18.2 MΩ∙cm, <4 ppb DOC) was used for the preparation of the solutions. The experiments were carried out at room temperature (20 ± 1 °C).
2.2. Mg-Calcite Coprecipitation Experiments
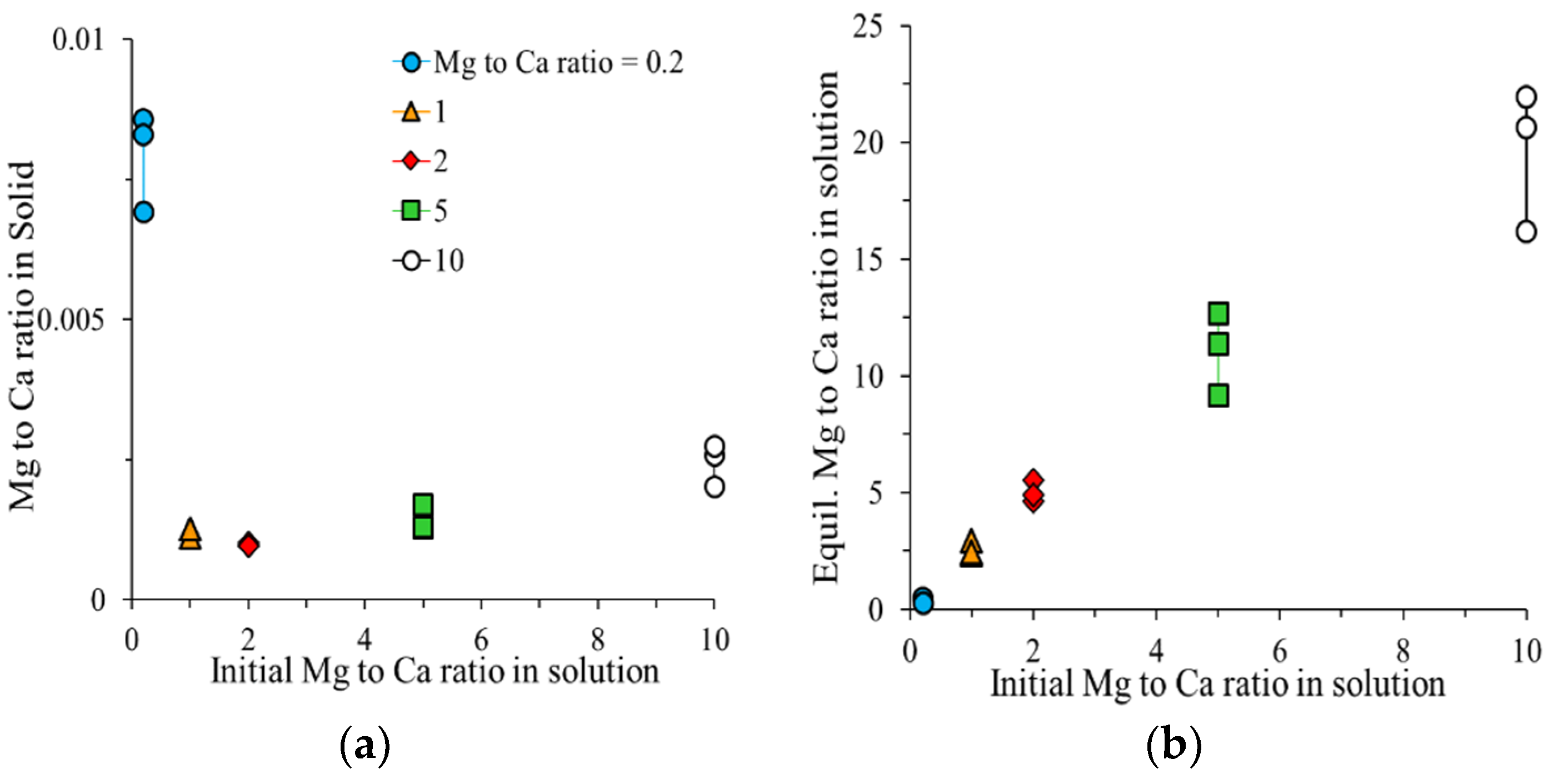
To evaluate the effect of a Mg to Ca ratio on Mg incorporation into calcite, solutions with five initial Mg to Ca ratios (0.2, 1, 2, 5, and 10) were reacted with vaterite (1 g/L) to simulate the relevant conditions for the precipitation of solid phases in soils, irrigated with Mg-enriched water. In these experiments, Ca
2+ is provided by vaterite dissolution (log
10 K
sp = −7.91). The Ca concentrations provided by vaterite dissolution were calculated by PHREEQC (Version 2, US Geological Survey, Reston, VA, USA [
24]) and the PSI/Nagra chemical thermodynamic database [
25]. The calculated Ca concentrations at pH 7.5, 8, and 9 were 5, 2.5, and 0.8 mmol/L, respectively (assuming equilibrium with atmospheric CO
2). The pH of the suspensions was adjusted to 7.5, 8, and 9 by adding precalculated volumes of HCl to the centrifuge tubes (50 mL) (dilutions from HCl, 1M, Titrisol, Merck, KGaA, Darmstadt, Germany). The amount of added Mg
2+ (MgCl
2·6H
2O, p.a., Merck, KGaA, Darmstadt, Germany) at each Mg to Ca ratio was chosen based on the expected (calculated) Ca
2+ concentration. After solution preparation, a mass of 0.04 g of vaterite was added to the centrifuge tube to obtain a 1-g/L suspension. In the experiments, a low supersaturation with respect to calcite (Ω
3 to 4) was established due to vaterite dissolution. All the experiments were carried out at a 0.025-mol/L NaCl background electrolyte concentration.
The saturation indices (
SI) of the solutions (Equation (1)) were calculated using PHREEQC.
The saturation state (Ω) of the solutions with respect to calcite (Equation (2)) was subsequently obtained as
where
IAP is the activity product of Ca
2+ and CO
32− ions, as calculated by PHREEQC, and
is the solubility product of calcite (
.
The suspensions were shaken at 20 rpm for 7 days, and then centrifuged. The supernatants were collected to measure the equilibrium pH, total dissolved concentrations of Ca2+ and Mg2+, and total inorganic carbon (TIC). The concentrations of Ca and Mg in the supernatants were measured by ICP-OES (Perkin Elmer, optima 8300). The total inorganic carbon was measured by expulsion with phosphoric acid.
2.3. Mineralogy and Morphology of the Precipitated Solid Phase
Solids were separated from the solution by centrifugation. To eliminate soluble salts from the precipitated solid phase, a volume of 5 mL of ethanol was added to the solid after centrifugation and decantation. The suspensions were rapidly shaken and centrifuged again. The supernatants were discarded, and the solid was air-dried. An aliquot of the solid (10 mg) was transferred to centrifuge tubes and digested with 10-mL 0.1-M HCl in order to determine the contents of Ca and Mg in the solid by ICP-OES and ICP-MS (Thermo Element XR), respectively. The remaining solids were characterized by X-ray powder diffraction (XRD, BRUKER D8-advance, Karlsruhe, Germany), scanning electron microscopy (SEM-EDX, FEI Quanta 650 FEG environmental SEM, Eindhoven, The Netherlands). SEM-EDX spectra of selected areas were acquired by use of a Thermo Scientific UltraDryTM silicon drift X-ray detector (Peltier cooled) and analyzed by Thermo Fisher Scientific Pathfinder software, version 2.8. Rietveld refinement (using the TOPAS 6.0 software, BRUKER, Karlsruhe, Germany) was used to quantify the solid-phase composition (i.e., vaterite/aragonite/calcite ratio) and to characterize the resulting calcite.
In this study, the incorporation of Mg into calcite is expressed as (Equation (3)):
where
is the mole fraction of MgCO
3 in the precipitated solid.
2.4. The Partition Coefficient of Mg in the Calcite Structure
The homogeneous Henderson–Kracek partition coefficient (
D) (Equation (4)) for Mg between calcite and the solution phase was used to quantify Mg incorporation into calcite.
is the mole fraction of Ca in the solid (calcite), and and , are the molar equilibrium concentrations of Mg and Ca ions in the solution, respectively.
is calculated from the known mole fraction of Mg in the solid (calcite):
In this study, was obtained by two methods:
A: by calculating the magnesium percentage in calcite from digestion and the subsequent solution analysis (
X1). The percent mole fraction of Mg incorporated into the solid is then given by Equation (6):
where [Ca] and [Mg] are the concentrations of Ca and Mg ions in the solution after digestion as measured by ICP-OES and ICP-MS (iCAP
TM TQ ICP-MS, Thermo Fisher Scientific, Waltham, MA, USA), respectively. Mg-calcite% is the percentage of Mg-calcite in the solid phase, as obtained from Rietveld refinement of the XRD data. Method A assumes (as supported by SEM-EDX) that Mg is incorporated only into calcite and not into vaterite or aragonite.
B: by using SEM-EDX results to calculate
and (
X2) using Equation (7):
where Mg and Ca atom % are the fractions of Mg and Ca measured at various points on Mg-calcite crystals by SEM-EDX.
2.5. Modeling
Experimental partition coefficients were further used to calculate the dimensionless Guggenheim parameter,
a0 (Equation (8)), which characterizes the non-ideality of the Mg-calcite solid solution (as described, e.g., in Heberling et al. [
26]).
where
and
are the solubility products of calcite and magnesite (
KSP(magnesite) = 10
−8.29), respectively [
27].
The free energy of mixing, d
GM (Equation (9)), of the precipitated Mg-calcite is described relative to the free energy of a hypothetical mechanical mixture of equal composition without any molecular interactions between the components [
28]. It was assumed that the dilute Mg-calcite is a mixture between magnesite and calcite.
The quantities dG0 (magnesite) and dG0 (calcite) are standard free energies of formation at 298 K for magnesite (−1030.6 kJ/mol) and calcite (−1128.8 kJ/mol), respectively.
d
GM can be decomposed into an ideal free energy of mixing (
and an excess free energy of mixing (d
GE).
For the dilute Mg-calcite, we assume random mixing. Then, the entropy contribution to
can be calculated as:
where
R and
T are the universal gas constant (
R = 8.3145 J/mol/K) and the absolute temperature (K), respectively.
For nonideal solid solutions, Guggenheim [
29] proposed to describe d
GE by:
This relates the free energy calculations to the Guggenheim parameter, a0, which we determined from experimental partition coefficients (Equation (8)). Higher order parameters were not considered (a1 = a2 … = 0). Combining Equations (4)–(8) with Equations (9)–(12) allows calculating the free energy of the Mg-calcite solid solution on the basis of the experimental results.
2.6. Incorporation of Mg2+ in Calcite in the Presence of Clay Minerals
To evaluate the effect of clay minerals in soils on the incorporation of Mg2+ into calcite, four solutions with varying Mg to Ca ratios (0.2, 1, 2, and 5) were equilibrated with vaterite in the presence of either bentonite or kaolinite. Georgia kaolinite (KGa-1) was obtained from the Source Clay Repository of the Clay Minerals Society. Its specific surface area determined by BET (N2 adsorption, Quantachrome Autosorb Gas Sorption System, Quantachrome, Odelzhausen, Germany) was found to be 15.6 m2/g. The cation exchange capacity (CEC) of KGa-1 was 0.015 molc/kg. Bentonite (Na-montmorillonite) was obtained from LEM (Laboratoire Environnement et Mineralurgie, Nancy, France). The specific surface area and CEC for this bentonite were 75.4 m2/g and 0.9 molc/kg, respectively.
To investigate the effects of kaolinite and bentonite clays, the procedure was as described in
Section 2.2. The pH of the suspension was adjusted to 8 by adding HCl. Known amounts of bentonite or kaolinite were added to the centrifuge tubes (50 mL) to obtain 0.2- and 1-g/L suspensions for each clay. After 24 h of equilibration time, an amount yielding 1 g/L of vaterite was added. Suspensions were shaken at low speed for 7 days, then centrifuged. The supernatants were collected to measure the final pH and the concentrations of Ca, Mg, and TIC. The solid phase was washed with 5-mL ethanol to remove the soluble salts and then air-dried. Finally, a mass of 10 mg of the solid phases was introduced into 10-mL 0.1-M HCl to measure the concentrations of the Ca and Mg ions incorporated in the carbonate phases, and the solid was characterized by XRD and SEM-EDX.
4. Discussion
In contrast to previous studies, which involved slightly higher supersaturation [
9], our experiments aimed at simulating the effect of Mg to Ca ratios on the formation of calcite polymorphs in groundwater like media and were carried out at relatively low supersaturation (Ω = 3 to 4). Such low supersaturations were realized by using vaterite as a precursor of calcite formation [
6].
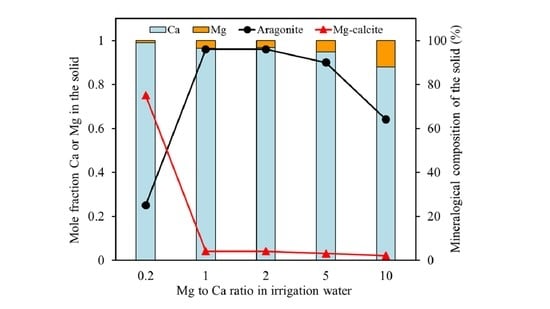
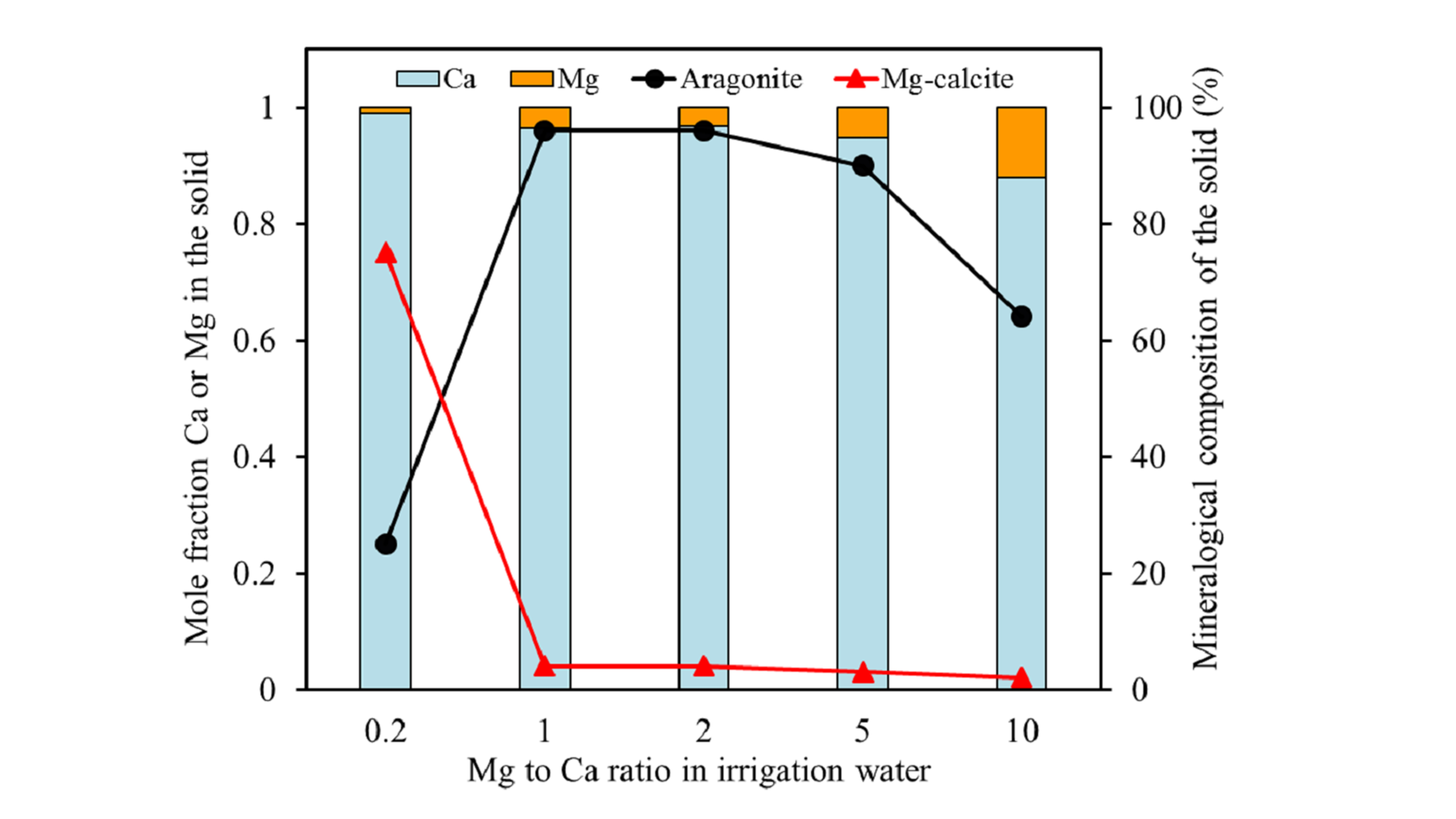
The results obtained for the Mg coprecipitation experiments with calcite yielded a white precipitate, which consisted of Mg-calcite, aragonite, and vaterite, depending on the Mg to Ca ratio of the solution phase. Only calcite incorporated significant amounts of Mg
2+ ions. The amount of Mg incorporation was controlled by the Mg to Ca concentration ratio in the solution. As such, the mole fraction of Mg in Mg-calcite increased with the increasing Mg to Ca ratios. The same trend has been reported in earlier studies at higher supersaturations [
2,
9,
19]. For example, Mucci and Morse [
9] reported that the amount of Mg incorporated in the calcite structure increased from 2.7 to 11.2% when increasing the solution Mg to Ca ratio from 1 to 10.3.
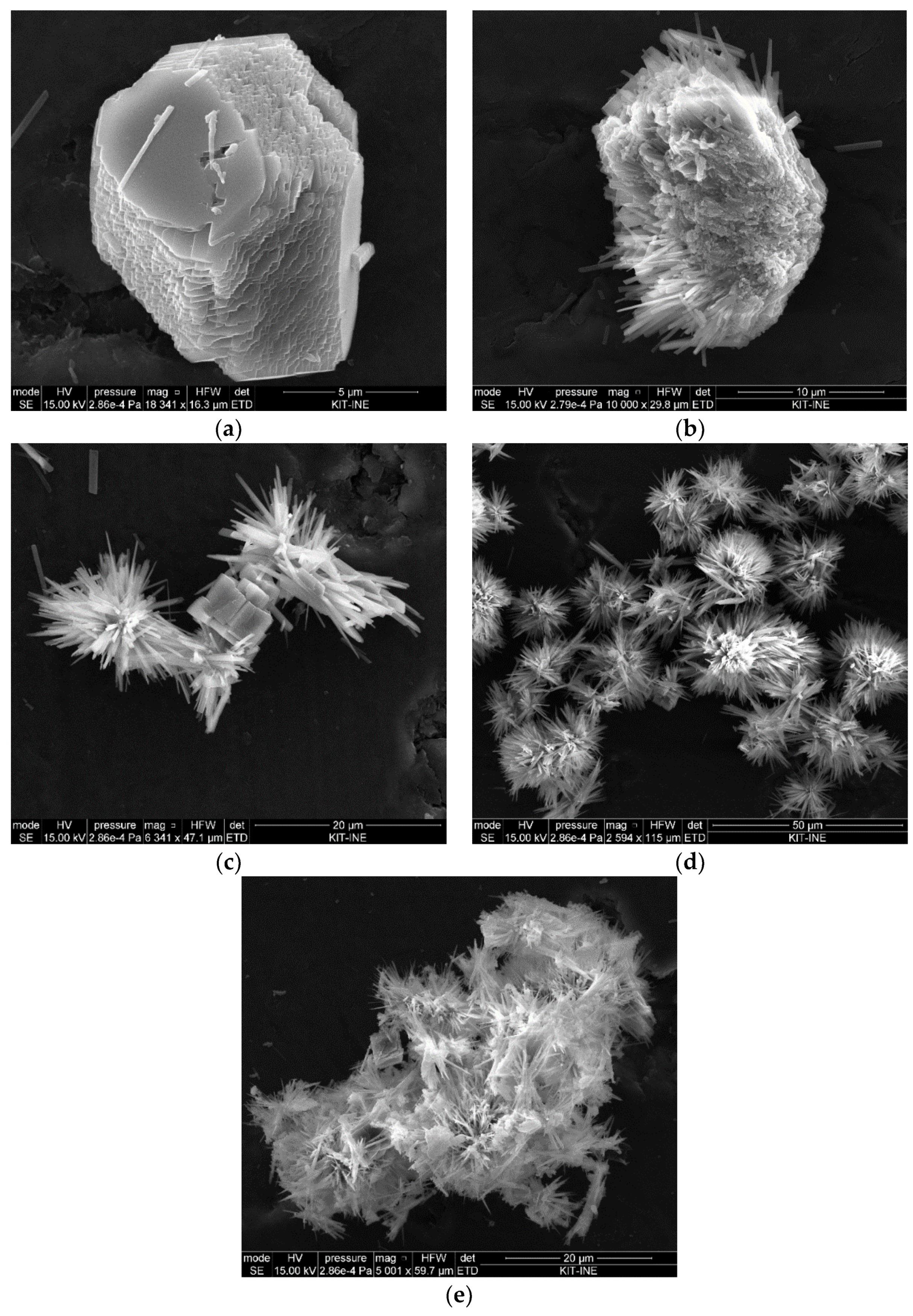
The Mg to Ca ratio in solution affected the morphology of the CaCO
3 polymorphs. At the lowest Mg to Ca ratio (i.e., 0.2), the morphologies of the precipitated solid differ from that of pure calcite (rhombohedral morphology). At this ratio, the modification involves new faces on the edges and corners as stepped and kinked surfaces. Similar morphological changes in the presence of low Mg concentration were observed by Zhang and Dawe [
19]. The formation of stepped and kinked surfaces indicates a higher affinity of some edges and corner sites for Mg incorporation, as suggested earlier [
18,
19]. As the Mg concentration increases (i.e., at a Mg to Ca ratio larger than 0.2), aragonite becomes the dominant CaCO
3 polymorph. Previous studies based on the same concept of calcite crystallization from a vaterite precursor in the absence of Mg have showed that the vaterite-calcite transformation in aqueous solution at room temperature was completed in less than two days, and aragonite was not observed at any stage of the transformation [
31]. Therefore, it can be confirmed that the presence of Mg inhibits calcite formation, while aragonite formation is favored. The SEM-EDX analyses of precipitated solids at Mg to Ca ratios above 0.2 revealed that Mg ions are not incorporated into aragonite. Aragonite precipitation is, thus, not much affected by Mg ions and can outcompete calcite precipitation. The formation thermodynamics of (Mg,Ca)CO
3 (see below) indicate that, with increasing the Mg incorporation into calcite, the forming Mg-calcite becomes less stable than aragonite, which offers a likely explanation for the experimental observations. The results indicated that, at about 1 mole % Mg content, Mg-calcite becomes more soluble than aragonite, but they also showed that low amounts of Mg-calcite with up to 12 mole % may form via kinetic controls.
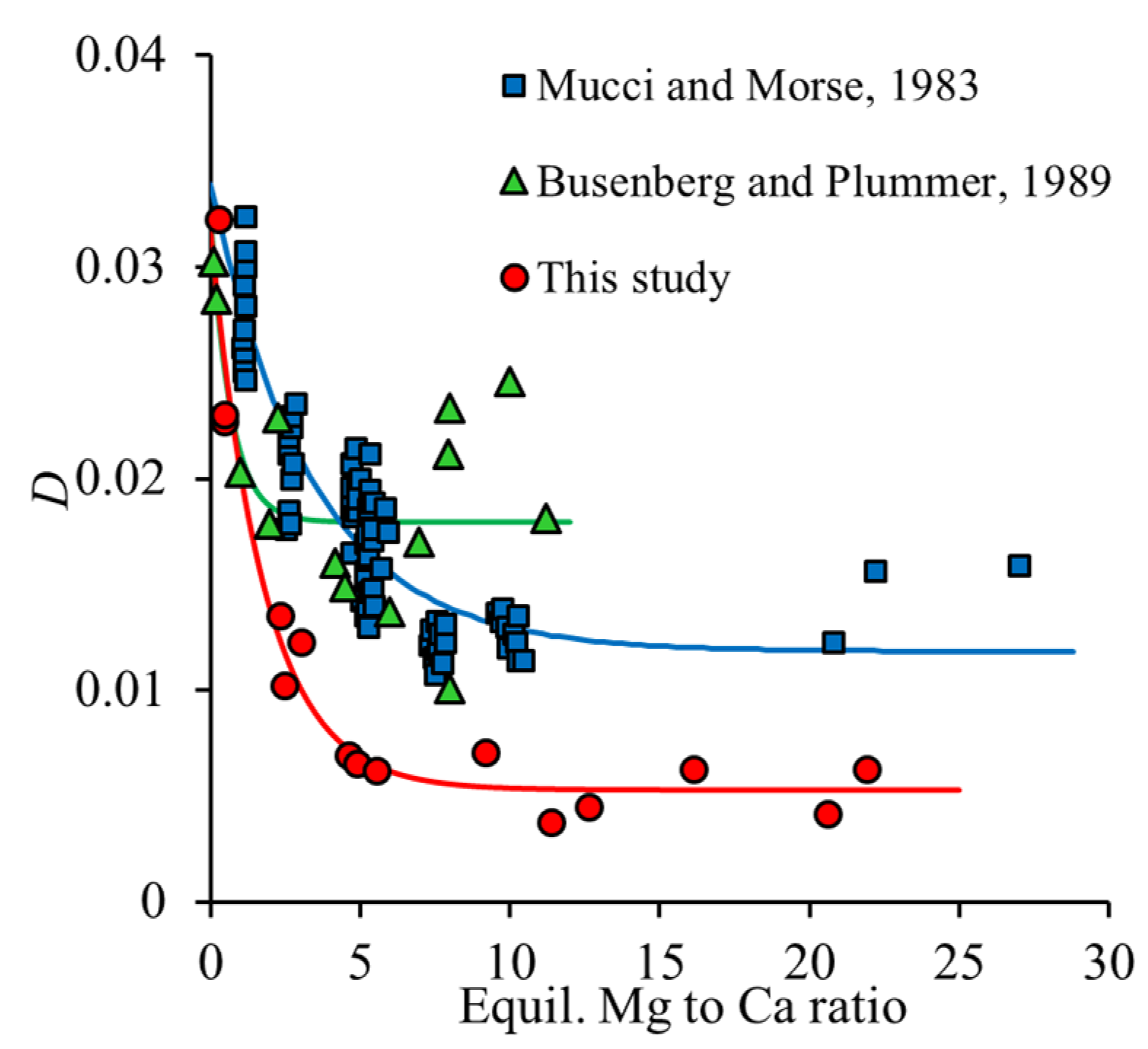
As shown in
Figure 4, a decrease in the calculated
D values was observed as a function of the Mg to Ca ratio in the solution. For comparison, the experimental data of Mucci and Morse [
9], obtained in seawater at a slightly higher supersaturation (Ω
3–17), are also presented in
Figure 4. The same trend and comparable partition coefficients were observed by Busenberg and Plummer [
27]. The derived partition coefficients of our study were systematically lower than those reported by Mucci and Morse [
9] or Busenberg and Plummer [
27]. The averages were 1.1 × 10
−2, 1.79 × 10
−2, and 2.0 × 10
−2, respectively. We suggest that this difference mainly reflects the expected kinetic effect that higher supersaturation leads to higher partition coefficients during the incorporation of ions, for which incorporation is unfavorable, i.e.,
D < 1 [
32].
All D values are considerably lower than what would be expected for an ideal solid solution. With log(Ksp(magnesite)) = −8.29 and log(Ksp(calcite)) = −8.48, Dideal is 1.55.
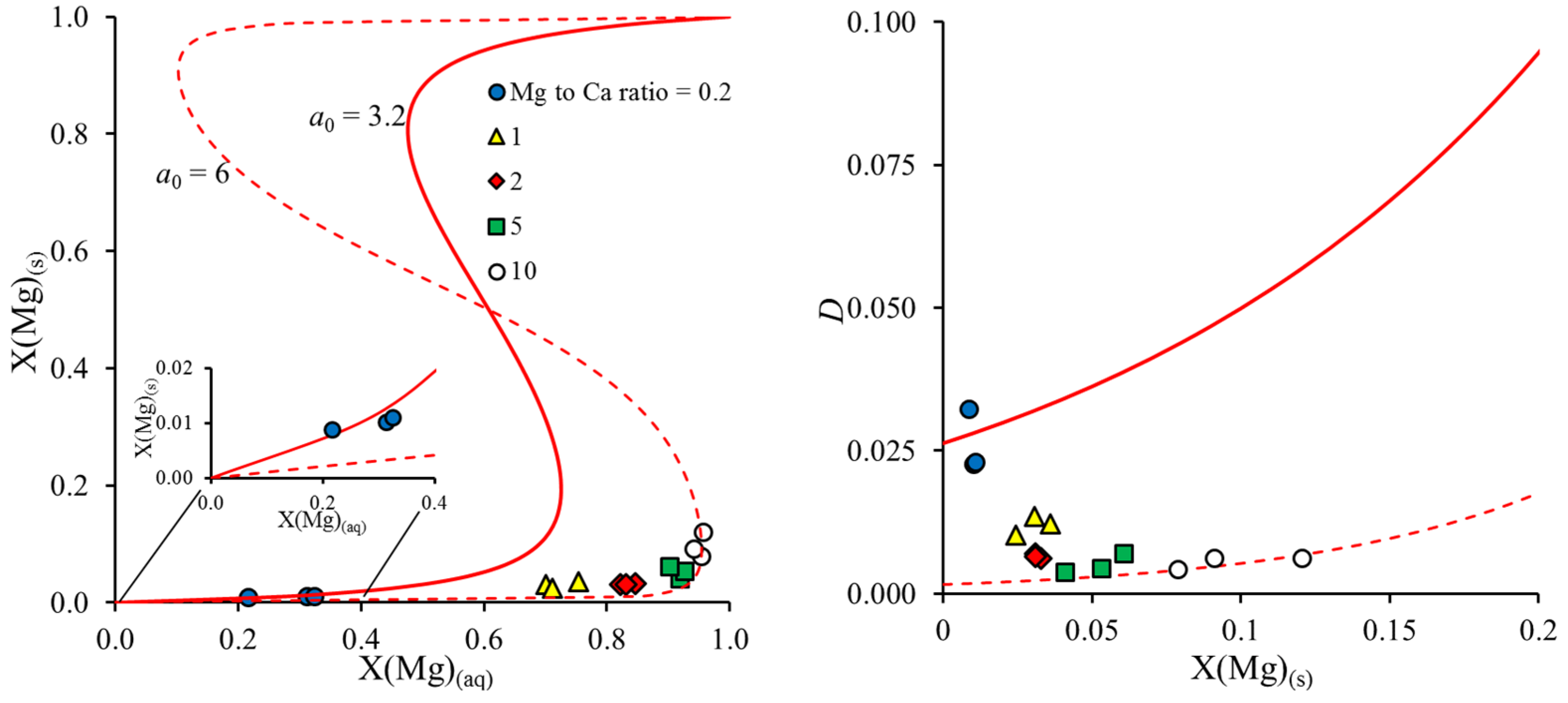
In general, a decrease in the partition coefficient with the increasing Mg content of the solid disagrees with the expected trend of thermodynamic solid solution models, as indicated in
Figure 5 (right graph, solid line). The data plotted in
Figure 4 indicate that partition coefficients at elevated Mg to Ca ratios showed a stronger influence on supersaturation, while, at the lowest Mg to Ca ratios, comparable values around
D = 0.03 were obtained in this study, by Mucci and Morse [
9], and by Busenberg and Plummer [
27] (i.e., for their Group I Mg-calcites in NaCl solution), independent of the degree of supersaturation. Thus, we suggest that the highest partition coefficients, obtained at the lowest Mg to Ca ratio, provide the best estimate for a thermodynamic value. Correspondingly, we used the value of
a0 = 3.3 ± 0.2 for further thermodynamic calculations. As pointed out previously [
27], this parameter is only applicable for dilute Mg-calcites (<ca. 1 mole % Mg). The corresponding thermodynamic model predicts a large miscibility gap from 5 to 95% MgCO
3 in calcite and a spinodal gap from 19 to 81% MgCO
3 in calcite. Experiments at higher initial Mg to Ca ratios in a solution mostly fall into the spinodal gap, as indicated in the Rozeboom diagram (
Figure 5). However, instead of the formation of a Mg-rich solid solution, aragonite formation is favored under these conditions, and Mg partitioning into Mg-calcite decreases. As this is opposed to the expected thermodynamic trend, it must be kinetically controlled, likely via processes at the Mg-calcite–water interface (as mentioned above) in competition with aragonite formation. The precipitation of the Mg-rich endmember from the solution is kinetically suppressed except for extreme salt brines, as has been pointed out before [
27].
Since it is not possible to capture the dependence of
D on the Mg to Ca ratio in a solution by a simple thermodynamic model, we derived the parameters for an empirical relation in the form of Equation (16) to describe the dependence of
D on the solution composition.
The optimized values for a, b, and c are presented in
Table 4. The corresponding model curves are included in
Figure 4.
In the presence of kaolinite, very similar results are obtained compared to the absence of kaolinite. Correspondingly, we conclude that the presence of kaolinite has only a moderate impact on the formation of Mg-calcite and other calcium carbonate polymorphs. The presence of bentonite reduces the reaction rate. So far, we can only speculate about the responsible processes. The effects could be related to the sorption capacity of bentonite for Ca
2+ and Mg
2+ ions, which may impact the aqueous ion concentrations. Moreover, dissolved silica in solution originating from the dissolution of amorphous silica (which may be a minor constituent of bentonite) could impact the crystal growth of calcium carbonate [
33].









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}