The ER Stress/UPR Axis in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis

 , , , ,
, , , ,  , ,
, ,  , , , and
, , , and 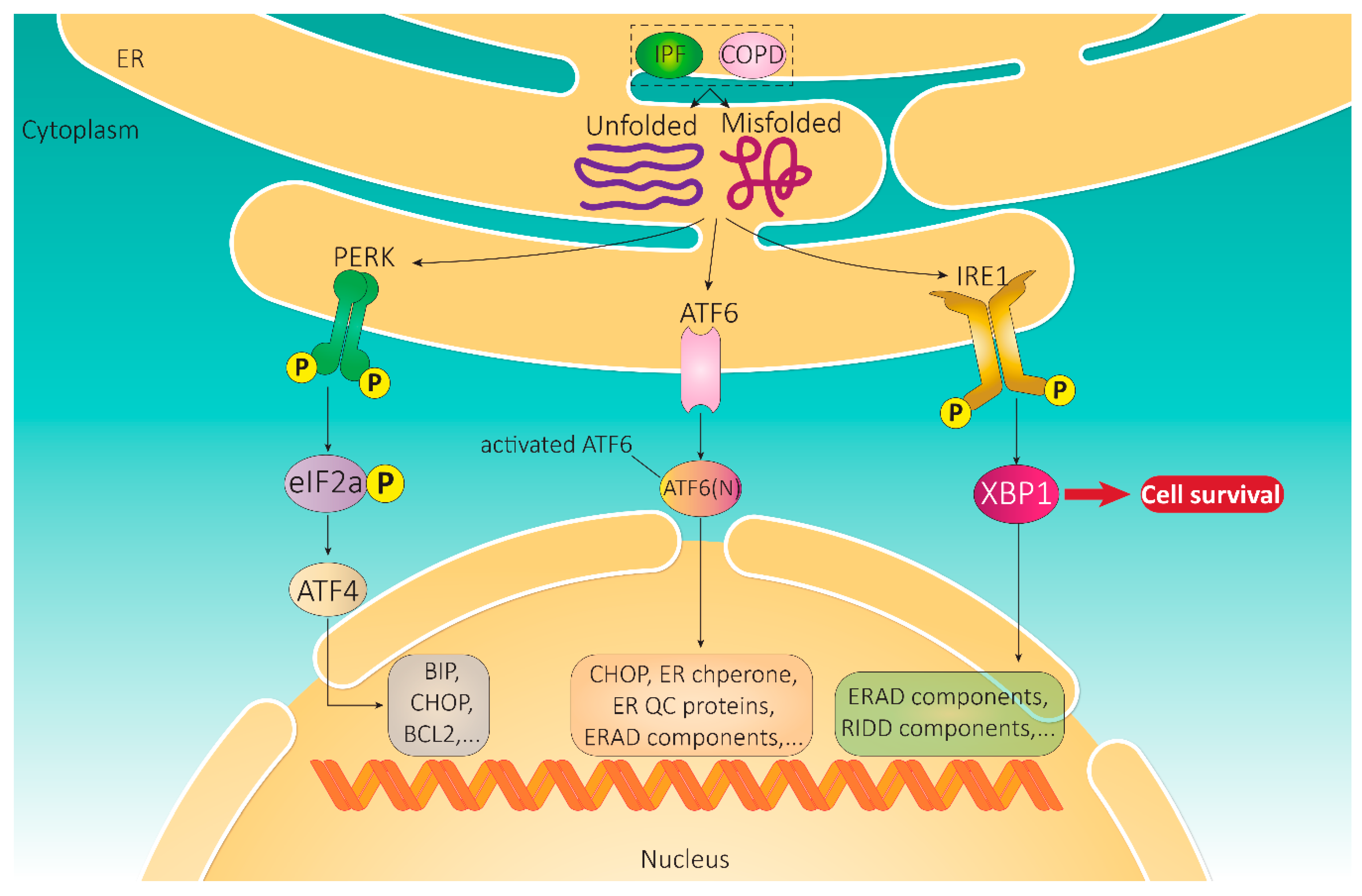{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Unfolded Protein Response
2.1. Integral UPR Pathway Proteins
2.1.1. IRE1
2.1.2. ATF6
2.1.3. PERK
2.2. Signaling Pathways Activated by UPR
3. Chronic Obstructive Pulmonary Disease
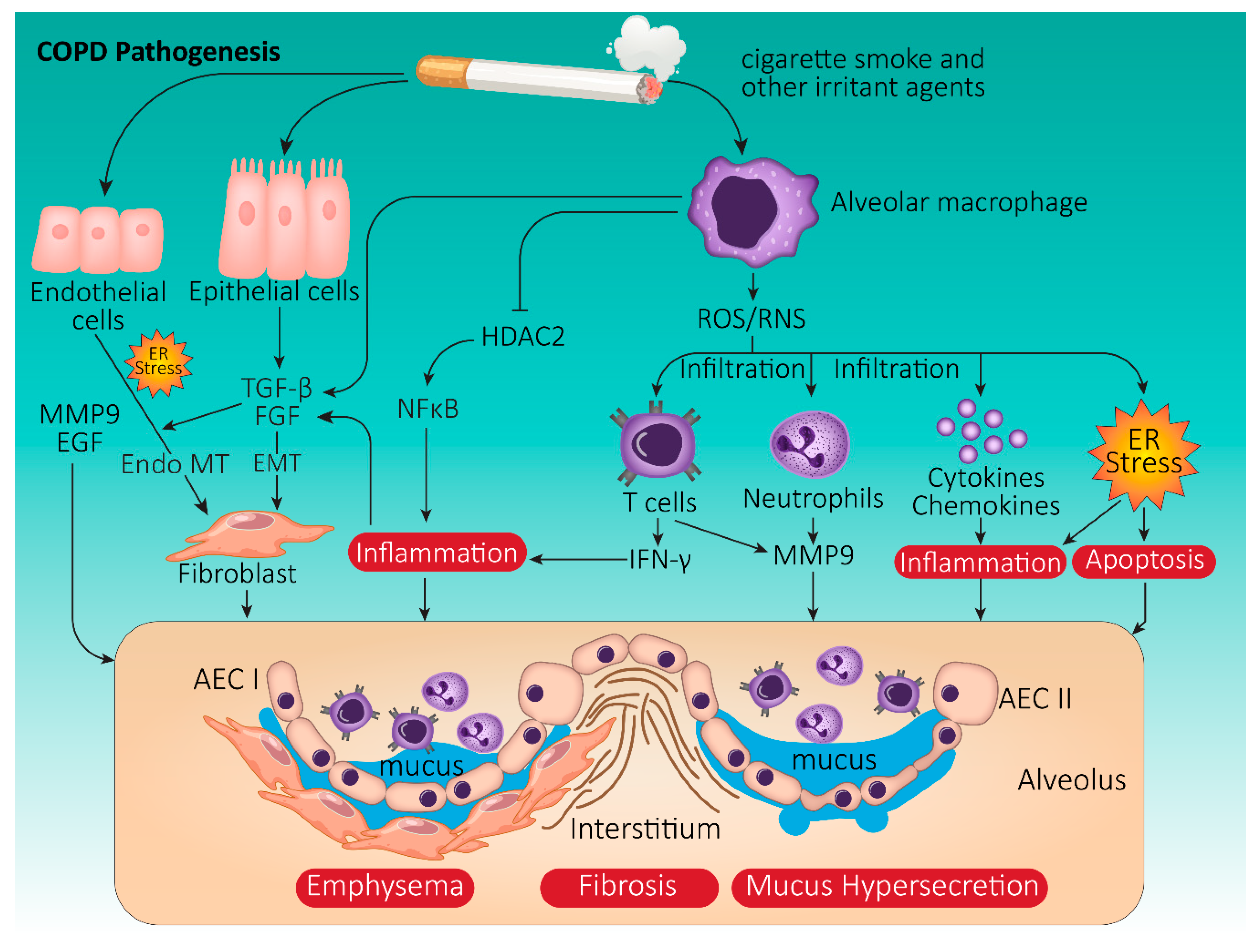
3.1. COPD Pathophysiology
3.2. Onset and Progression of COPD: The Role of Cigarette Smoke
3.3. COPD and UPR: Cellular Crosstalk Mechanisms
3.3.1. Oxidative Stress in COPD
3.3.2. CS-Induced UPR Signaling in COPD
3.3.3. UPR and Autophagy Signaling in COPD
3.3.4. UPR-Associated Diagnostic Markers in COPD
3.3.5. Non-Coding RNAs and UPR in COPD
4. Idiopathic Pulmonary Fibrosis (IPF)
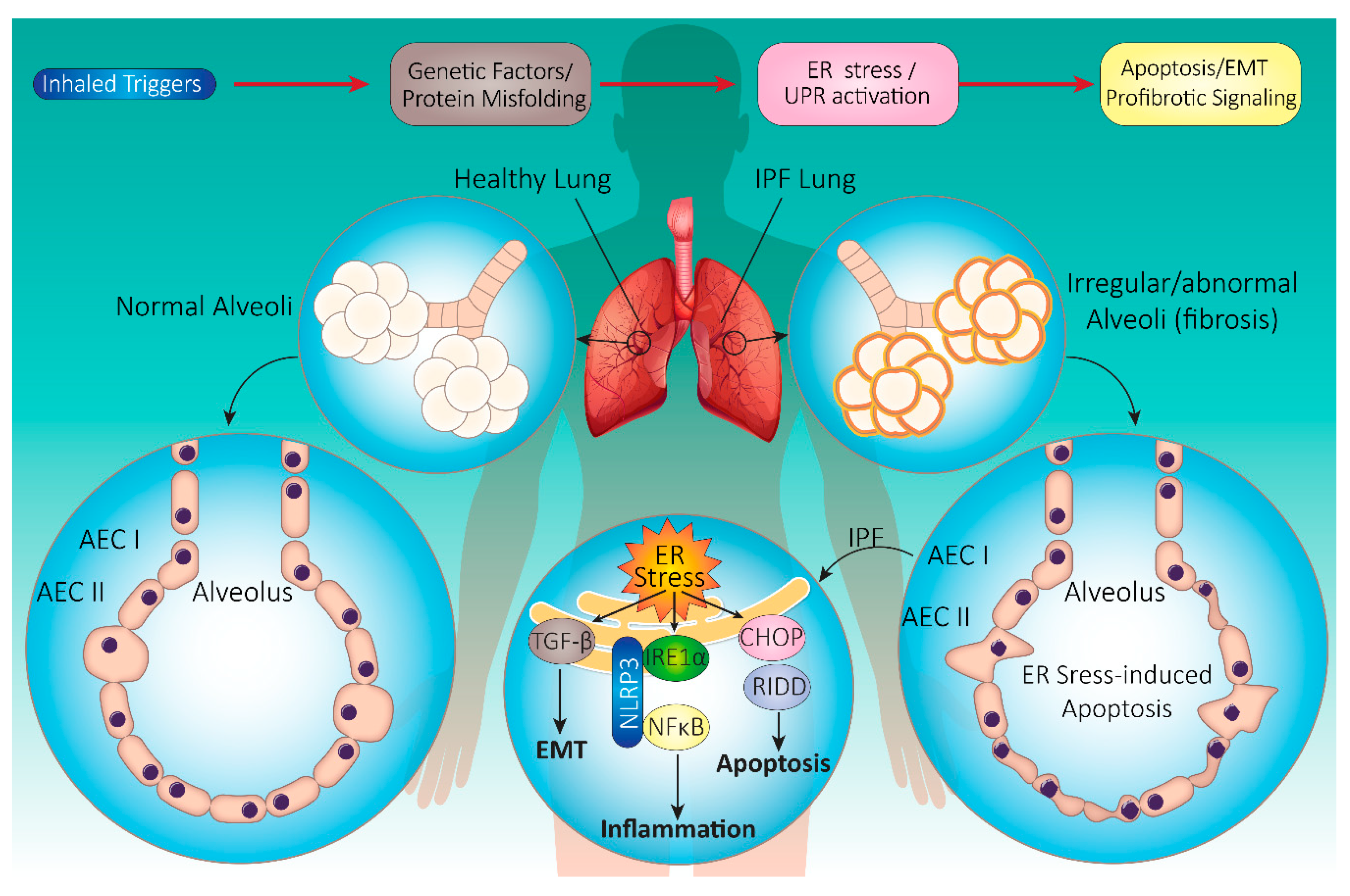
4.1. IPF Pathophysiology
4.2. Onset and Progression of IPF
4.3. IPF and UPR: Cellular Mechanisms
4.3.1. UPR Signaling in IPF
4.3.2. Exogenous Stimuli Contributing to ER Stress in IPF
Smoke
Viruses
UPR-Associated Diagnostic Markers in IPF
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Dastghaib, S.; Shojaei, S.; Mostafavi-Pour, Z.; Sharma, P.; Patterson, J.B.; Samali, A.; Mokarram, P.; Ghavami, S. Simvastatin Induces Unfolded Protein Response and Enhances Temozolomide-Induced Cell Death in Glioblastoma Cells. Cells 2020, 9, 2339. [Google Scholar] [CrossRef]
- Dastghaib, S.; Kumar, P.S.; Aftabi, S.; Damera, G.; Dalvand, A.; Sepanjnia, A.; Kiumarsi, M.; Aghanoori, M.R.; Sohal, S.S.; Ande, S.R.; et al. Mechanisms Targeting the Unfolded Protein Response in Asthma. Am. J. Respir. Cell Mol. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Madden, E.C.; Gorman, A.M.; Logue, S.E.; Samali, A. Tumour Cell Secretome in Chemoresistance and Tumour Recurrence. Trends Cancer 2020, 6, 489–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barua, D.; Abbasi, B.; Gupta, A.; Gupta, S. XBP1 increases transactivation of somatic mutants of ESR1 and loss of XBP1 reverses endocrine resistance conferred by gain-of-function Y537S ESR1 mutation. Heliyon 2020, 6, 05217. [Google Scholar] [CrossRef] [PubMed]
- Barua, D.; Gupta, A.; Gupta, S. Targeting the IRE1-XBP1 axis to overcome endocrine resistance in breast cancer: Opportunities and challenges. Cancer Lett. 2020, 486, 29–37. [Google Scholar] [CrossRef]
- Maitra, M.; Wang, Y.; Gerard, R.D.; Mendelson, C.R.; Garcia, C.K. Surfactant Protein A2 Mutations Associated with Pulmonary Fibrosis Lead to Protein Instability and Endoplasmic Reticulum Stress. J. Biol. Chem. 2010, 285, 22103–22113. [Google Scholar] [CrossRef] [Green Version]
- Naiel, S.; Tat, V.; Padwal, M.; Vierhout, M.; Mekhael, O.; Yousof, T.; Ayoub, A.; Abed, S.; Dvorkin-Gheva, A.; Ask, K. Protein Misfolding and Endoplasmic Reticulum Stress in Chronic Lung Disease: Will Cell-Specific Targeting Be the Key to the Cure? Chest 2020, 157, 1207–1220. [Google Scholar] [CrossRef]
- Wei, J.; Rahman, S.; Ayaub, E.A.; Dickhout, J.G.; Ask, K. Protein Misfolding and Endoplasmic Reticulum Stress in Chronic Lung Disease. Chest 2013, 143, 1098–1105. [Google Scholar] [CrossRef]
- Barreiro, E.; Salazar-Degracia, A.; Sancho-Muñoz, A.; Gea, J. Endoplasmic reticulum stress and unfolded protein response profile in quadriceps of sarcopenic patients with respiratory diseases. J. Cell. Physiol. 2019, 234, 11315–11329. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ribeiro, C.M.P.; Sun, L.; Okuda, K.; Kato, T.; Gilmore, R.C.; Martino, M.B.; Dang, H.; Abzhanova, A.; Lin, J.M.; et al. XBP1S Regulates MUC5B in a Promoter Variant-Dependent Pathway in Idiopathic Pulmonary Fibrosis Airway Epithelia. Am. J. Respir. Crit. Care Med. 2019, 200, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Arsene, F.; Tomoyasu, T.; Bukau, B. The heat shock response of Escherichia coli. Int. J. Food Microbiol. 2000, 55, 3–9. [Google Scholar] [CrossRef]
- Anckar, J.; Sistonen, L. Regulation of HSF1 function in the heat stress response: Implications in aging and disease. Annu. Rev. Biochem. 2011, 80, 1089–1115. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeganeh, B.; Rezaei Moghadam, A.; Tran, A.T.; Rahim, M.N.; Ande, S.R.; Hashemi, M.; Coombs, K.M.; Ghavami, S. Asthma and influenza virus infection: Focusing on cell death and stress pathways in influenza virus replication. Iran. J. Allergy Asthma Immunol. 2013, 12, 1–17. [Google Scholar] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Oikawa, D.; Kimata, Y.; Kohno, K.; Iwawaki, T. Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp. Cell Res. 2009, 315, 2496–2504. [Google Scholar] [CrossRef]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Kopp, M.C.; Ali, M.M. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.M.; Sher, A.A.; Coombs, K.M.; et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef] [Green Version]
- Marzban, H.; Del Bigio, M.R.; Alizadeh, J.; Ghavami, S.; Zachariah, R.M.; Rastegar, M. Cellular commitment in the developing cerebellum. Front. Cell Neurosci. 2014, 8, 450. [Google Scholar] [CrossRef] [Green Version]
- Veyron, S.; Peyroche, G.; Cherfils, J. FIC proteins: From bacteria to humans and back again. Pathog. Dis. 2018, 76, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preissler, S.; Rato, C.; Perera, L.A.; Saudek, V.; Ron, D. FICD acts bifunctionally to AMPylate and de-AMPylate the endoplasmic reticulum chaperone BiP. Nat. Struct. Mol. Biol. 2017, 24, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ham, H.; Woolery, A.R.; Tracy, C.; Stenesen, D.; Krämer, H.; Orth, K. Unfolded protein response-regulated Drosophila Fic (dFic) protein reversibly AMPylates BiP chaperone during endoplasmic reticulum homeostasis. J. Biol. Chem. 2014, 289, 36059–36069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moehlman, A.T.; Casey, A.K.; Servage, K.; Orth, K.; Krämer, H. Regulation of the unfolded protein response by BiP AMPylation protects photoreceptors from light-dependent degeneration. bioRxiv 2018, 348797. [Google Scholar] [CrossRef]
- Preissler, S.; Ron, D. Early Events in the Endoplasmic Reticulum Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 2019, 11, a033894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, A.K.; Moehlman, A.T.; Zhang, J.; Servage, K.A.; Krämer, H.; Orth, K. Fic-mediated deAMPylation is not dependent on homodimerization and rescues toxic AMPylation in flies. J. Biol. Chem. 2017, 292, 21193–21204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimata, Y.; Ishiwata-Kimata, Y.; Ito, T.; Hirata, A.; Suzuki, T.; Oikawa, D.; Takeuchi, M.; Kohno, K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J. Cell Biol. 2007, 179, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef] [Green Version]
- Yeganeh, B.; Rezaei Moghadam, A.; Alizadeh, J.; Wiechec, E.; Alavian, S.M.; Hashemi, M.; Geramizadeh, B.; Samali, A.; Bagheri Lankarani, K.; Post, M.; et al. Hepatitis B and C virus-induced hepatitis: Apoptosis, autophagy, and unfolded protein response. World J. Gastroenterol. 2015, 21, 13225–13239. [Google Scholar] [CrossRef]
- Fu, J.; Tao, T.; Li, Z.; Chen, Y.; Li, J.; Peng, L. The roles of ER stress in epilepsy: Molecular mechanisms and therapeutic implications. Biomed. Pharm. 2020, 131, 110658. [Google Scholar] [CrossRef]
- Sundaram, A.; Plumb, R.; Appathurai, S.; Mariappan, M. The Sec61 translocon limits IRE1alpha signaling during the unfolded protein response. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaram, A.; Appathurai, S.; Plumb, R.; Mariappan, M. Dynamic changes in complexes of IRE1alpha, PERK, and ATF6alpha during endoplasmic reticulum stress. Mol. Biol. Cell 2018, 29, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowski, D.T.; Arnold, S.M.; Miller, C.N.; Wu, J.; Li, J.; Gunnison, K.M.; Mori, K.; Sadighi Akha, A.A.; Raden, D.; Kaufman, R.J. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006, 4, e374. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [Green Version]
- Deegan, S.; Saveljeva, S.; Gorman, A.M.; Samali, A. Stress-induced self-cannibalism: On the regulation of autophagy by endoplasmic reticulum stress. Cell. Mol. Life Sci. 2013, 70, 2425–2441. [Google Scholar] [CrossRef]
- Iwawaki, T.; Akai, R.; Yamanaka, S.; Kohno, K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. USA 2009, 106, 16657–16662. [Google Scholar] [CrossRef] [Green Version]
- Tsuru, A.; Fujimoto, N.; Takahashi, S.; Saito, M.; Nakamura, D.; Iwano, M.; Iwawaki, T.; Kadokura, H.; Ron, D.; Kohno, K. Negative feedback by IRE1beta optimizes mucin production in goblet cells. Proc. Natl. Acad. Sci. USA 2013, 110, 2864–2869. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [Green Version]
- Yanagitani, K.; Imagawa, Y.; Iwawaki, T.; Hosoda, A.; Saito, M.; Kimata, Y.; Kohno, K. Cotranslational targeting of XBP1 protein to the membrane promotes cytoplasmic splicing of its own mRNA. Mol. Cell 2009, 34, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghavami, S.; Sharma, P.; Yeganeh, B.; Ojo, O.O.; Jha, A.; Mutawe, M.M.; Kashani, H.H.; Los, M.J.; Klonisch, T.; Unruh, H.; et al. Airway mesenchymal cell death by mevalonate cascade inhibition: Integration of autophagy, unfolded protein response and apoptosis focusing on Bcl2 family proteins. Biochim. Biophys. Acta 2014, 1843, 1259–1271. [Google Scholar] [CrossRef] [Green Version]
- Ghavami, S.; Yeganeh, B.; Stelmack, G.L.; Kashani, H.H.; Sharma, P.; Cunnington, R.; Rattan, S.; Bathe, K.; Klonisch, T.; Dixon, I.M.; et al. Apoptosis, autophagy and ER stress in mevalonate cascade inhibition-induced cell death of human atrial fibroblasts. Cell Death Dis. 2012, 3, e330. [Google Scholar] [CrossRef] [Green Version]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef]
- Bultynck, G.; Kiviluoto, S.; Henke, N.; Ivanova, H.; Schneider, L.; Rybalchenko, V.; Luyten, T.; Nuyts, K.; De Borggraeve, W.; Bezprozvanny, I.; et al. The C terminus of Bax inhibitor-1 forms a Ca2+-permeable channel pore. J. Biol. Chem. 2012, 287, 2544–2557. [Google Scholar] [CrossRef] [Green Version]
- Varadarajan, S.; Bampton, E.T.; Smalley, J.L.; Tanaka, K.; Caves, R.E.; Butterworth, M.; Wei, J.; Pellecchia, M.; Mitcheson, J.; Gant, T.W.; et al. A novel cellular stress response characterised by a rapid reorganisation of membranes of the endoplasmic reticulum. Cell Death. Differ. 2012, 19, 1896–1907. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Watanabe, N.; Nagano, M.; Kawai-Yamada, M.; Lam, E. Bax inhibitor-1: A highly conserved endoplasmic reticulum-resident cell death suppressor. Cell Death. Differ. 2011, 18, 1271–1278. [Google Scholar] [CrossRef] [Green Version]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Gaddam, D.; Stevens, N.; Hollien, J. Comparison of mRNA localization and regulation during endoplasmic reticulum stress in Drosophila cells. Mol. Biol. Cell 2013, 24, 14–20. [Google Scholar] [CrossRef] [PubMed]
- McGrath, E.P.; Logue, S.E.; Mnich, K.; Deegan, S.; Jager, R.; Gorman, A.M.; Samali, A. The Unfolded Protein Response in Breast Cancer. Cancers 2018, 10, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, V.; Korhonen, L.; Lindholm, D. The Unfolded Protein Response and Autophagy as Drug Targets in Neuropsychiatric Disorders. Front. Cell Neurosci. 2020, 14, 554548. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Toivonen, S.; Igoillo-Esteve, M.; Salpea, P. Endoplasmic reticulum stress and eIF2alpha phosphorylation: The Achilles heel of pancreatic beta cells. Mol. Metab. 2017, 6, 1024–1039. [Google Scholar] [CrossRef]
- Okamura, K.; Kimata, Y.; Higashio, H.; Tsuru, A.; Kohno, K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem. Biophys. Res. Commun. 2000, 279, 445–450. [Google Scholar] [CrossRef]
- Amin-Wetzel, N.; Neidhardt, L.; Yan, Y.; Mayer, M.P.; Ron, D. Unstructured regions in IRE1α specify BiP-mediated destabilisation of the luminal domain dimer and repression of the UPR. Elife 2019, 8, e50793. [Google Scholar] [CrossRef]
- Wang, X.Z.; Ron, D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science 1996, 272, 1347–1349. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.J.; Ryu, S.W.; Song, B.J. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J. Biol. Chem. 2006, 281, 21256–21265. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schinzel, A.; Glimcher, L.H.; et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 2006, 312, 572–576. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Roy, B.; Lee, A.S. The mammalian endoplasmic reticulum stress response element consists of an evolutionarily conserved tripartite structure and interacts with a novel stress-inducible complex. Nucleic. Acids Res. 1999, 27, 1437–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Hou, Y.C.; Hedvat, M.; Correa, R.G.; Shu, C.W.; Krajewska, M.; Diaz, P.W.; Tamble, C.M.; Quarato, G.; Gottlieb, R.A.; et al. Endoplasmic reticulum protein BI-1 regulates Ca(2)(+)-mediated bioenergetics to promote autophagy. Genes Dev. 2012, 26, 1041–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavener, D.R.; Gupta, S.; McGrath, B.C. PERK in beta cell biology and insulin biogenesis. Trends Endocrinol. Metab. 2010, 21, 714–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delepine, M.; Nicolino, M.; Barrett, T.; Golamaully, M.; Lathrop, G.M.; Julier, C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat. Genet. 2000, 25, 406–409. [Google Scholar] [CrossRef]
- Deng, X.; Xiao, L.; Lang, W.; Gao, F.; Ruvolo, P.; May, W.S., Jr. Novel role for JNK as a stress-activated Bcl2 kinase. J. Biol. Chem. 2001, 276, 23681–23688. [Google Scholar] [CrossRef] [Green Version]
- Wek, R.C.; Cavener, D.R. Translational control and the unfolded protein response. Antioxid. Redox Signal. 2007, 9, 2357–2371. [Google Scholar] [CrossRef]
- Mahameed, M.; Wilhelm, T.; Darawshi, O.; Obiedat, A.; Tommy, W.S.; Chintha, C.; Schubert, T.; Samali, A.; Chevet, E.; Eriksson, L.A.; et al. The unfolded protein response modulators GSK2606414 and KIRA6 are potent KIT inhibitors. Cell Death Dis. 2019, 10, 300. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Yan, X.; Wintergerst, K.A.; Cai, L.; Keller, B.B.; Tan, Y. Nrf2: Redox and Metabolic Regulator of Stem Cell State and Function. Trends Mol. Med. 2020, 26, 185–200. [Google Scholar] [CrossRef] [Green Version]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainbolt, T.K.; Saunders, J.M.; Wiseman, R.L. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab. 2014, 25, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Shutt, T.E.; McBride, H.M. Staying cool in difficult times: Mitochondrial dynamics, quality control and the stress response. Biochim. Biophys. Acta 2013, 1833, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wai, T.; Langer, T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK Arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell Rep. 2018, 22, 2827–2836. [Google Scholar] [CrossRef] [Green Version]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D.; et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojaei, S.; Suresh, M.; Klionsky, D.J.; Labouta, H.I.; Ghavami, S. Autophagy and SARS-CoV-2 infection: Apossible smart targeting of the autophagy pathway. Virulence 2020, 11, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Sureda, A.; Alizadeh, J.; Nabavi, S.F.; Berindan-Neagoe, I.; Cismaru, C.A.; Jeandet, P.; Los, M.J.; Clementi, E.; Nabavi, S.M.; Ghavami, S. Endoplasmic reticulum as a potential therapeutic target for covid-19 infection management? Eur. J. Pharmacol. 2020, 882, 173288. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [Green Version]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [Green Version]
- Castillo, K.; Rojas-Rivera, D.; Lisbona, F.; Caballero, B.; Nassif, M.; Court, F.A.; Schuck, S.; Ibar, C.; Walter, P.; Sierralta, J.; et al. BAX inhibitor-1 regulates autophagy by controlling the IRE1alpha branch of the unfolded protein response. EMBO J. 2011, 30, 4465–4478. [Google Scholar] [CrossRef] [Green Version]
- Eapen, M.S.; Kota, A.; Vindin, H.; McAlinden, K.D.; Xenaki, D.; Oliver, B.G.; Deshpande, D.A.; Sohal, S.S.; Sharma, P. Apoptosis signal-regulating kinase 1 (ASK1) inhibition attenuates human airway smooth muscle growth and migration in chronic obstructive pulmonary disease (COPD). Clin. Sci. 2018. [Google Scholar] [CrossRef] [Green Version]
- Urra, H.; Dufey, E.; Lisbona, F.; Rojas-Rivera, D.; Hetz, C. When ER stress reaches a dead end. Biochim. Biophys. Acta 2013, 1833, 3507–3517. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B. Cell biology: Autophagy and cancer. Nature 2007, 446, 745–747. [Google Scholar] [CrossRef]
- Ghavami, S.; Yeganeh, B.; Zeki, A.A.; Shojaei, S.; Kenyon, N.J.; Ott, S.; Samali, A.; Patterson, J.; Alizadeh, J.; Moghadam, A.R.; et al. Autophagy and the unfolded protein response promote profibrotic effects of TGF-beta1 in human lung fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L493–L504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.R.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef] [PubMed]
- Hazari, Y.; Bravo-San Pedro, J.M.; Hetz, C.; Galluzzi, L.; Kroemer, G. Autophagy in hepatic adaptation to stress. J. Hepatol. 2020, 72, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Emami, A.; Shojaei, S.; da Silva Rosa, S.C.; Aghaei, M.; Samiei, E.; Vosoughi, A.R.; Kalantari, F.; Kawalec, P.; Thliveris, J.; Sharma, P.; et al. Mechanisms of simvastatin myotoxicity: The role of autophagy flux inhibition. Eur. J. Pharmacol. 2019, 862, 172616. [Google Scholar] [CrossRef]
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 749–768. [Google Scholar] [CrossRef]
- Alizadeh, J.; Shojaei, S.; Sepanjnia, A.; Hashemi, M.; Eftekharpour, E.; Ghavami, S. Simultaneous detection of autophagy and epithelial to mesenchymal transition in the non-small cell lung cancer cells. Methods Mol. Biol. 2019, 1854, 87–103. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [Green Version]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haberzettl, P.; Hill, B.G. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biol. 2013, 1, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Bock, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandic, A.; Hansson, J.; Linder, S.; Shoshan, M.C. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J. Biol. Chem. 2003, 278, 9100–9106. [Google Scholar] [CrossRef] [Green Version]
- Van de Craen, M.; Vandenabeele, P.; Declercq, W.; Van den Brande, I.; Van Loo, G.; Molemans, F.; Schotte, P.; Van Criekinge, W.; Beyaert, R.; Fiers, W. Characterization of seven murine caspase family members. FEBS Lett. 1997, 403, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Khaoustov, V.I.; Chung, C.C.; Sohn, J.; Krishnan, B.; Lewis, D.E.; Yoffe, B. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology 2002, 36, 592–601. [Google Scholar] [CrossRef]
- Morishima, N.; Nakanishi, K.; Tsuchiya, K.; Shibata, T.; Seiwa, E. Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J. Biol. Chem. 2004, 279, 50375–50381. [Google Scholar] [CrossRef] [Green Version]
- Morishima, N.; Nakanishi, K.; Takenouchi, H.; Shibata, T.; Yasuhiko, Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 2002, 277, 34287–34294. [Google Scholar] [CrossRef] [Green Version]
- Hitomi, J.; Katayama, T.; Taniguchi, M.; Honda, A.; Imaizumi, K.; Tohyama, M. Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12. Neurosci. Lett. 2004, 357, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, M.; Takahashi, G.; Abe, K.; Horinouchi, T.; Itoyama, Y.; Tabayashi, K. Endoplasmic reticulum stress induced in motor neurons by transient spinal cord ischemia in rabbits. J. Thorac. Cardiovasc. Surg. 2005, 130, 640–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinhofer, I.; Anether, G.; Senfter, M.; Pfaller, K.; Bernhard, D.; Hara, M.; Greil, R. Stressful death of T-ALL tumor cells after treatment with the anti-tumor agent Tetrocarcin-A. FASEB J. 2002, 16, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.; O’Hare, P. Transmembrane bZIP transcription factors in ER stress signaling and the unfolded protein response. Antioxid. Redox Signal. 2007, 9, 2305–2321. [Google Scholar] [CrossRef] [PubMed]
- Vekich, J.A.; Belmont, P.J.; Thuerauf, D.J.; Glembotski, C.C. Protein disulfide isomerase-associated 6 is an ATF6-inducible ER stress response protein that protects cardiac myocytes from ischemia/reperfusion-mediated cell death. J. Mol. Cell. Cardiol. 2012, 53, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Belmont, P.J.; Chen, W.J.; Thuerauf, D.J.; Glembotski, C.C. Regulation of microRNA expression in the heart by the ATF6 branch of the ER stress response. J. Mol. Cell. Cardiol. 2012, 52, 1176–1182. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [Green Version]
- Vestbo, J.; Hurd, S.S.; Agusti, A.G.; Jones, P.W.; Vogelmeier, C.; Anzueto, A.; Barnes, P.J.; Fabbri, L.M.; Martinez, F.J.; Nishimura, M.; et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: Gold executive summary. Am. J. Respir. Crit. Care Med. 2013, 187, 347–365. [Google Scholar] [CrossRef]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Author, N. Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease. American Thoracic Society. Am. J. Respir. Crit. Care Med. 1995, 152, S77–S121. [Google Scholar]
- Maltais, F.; Decramer, M.; Casaburi, R.; Barreiro, E.; Burelle, Y.; Debigare, R.; Dekhuijzen, P.N.; Franssen, F.; Gayan-Ramirez, G.; Gea, J.; et al. An official American Thoracic Society/European Respiratory Society statement: Update on limb muscle dysfunction in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2014, 189, e15–e62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, A. Pathology and pathophysiology of chronic obstructive pulmonary disease. Intern. Med. 2002, 41, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sussan, T.E.; Biswal, S. Smoking and COPD and other respiratory diseases. In Cigarette Smoke Toxicity: Linking Individual Chemicals to Human Diseases; Wiley-VCH: Weinheim, Germany, 2011; pp. 167–190. [Google Scholar]
- Ito, K.; Ito, M.; Elliott, W.M.; Cosio, B.; Caramori, G.; Kon, O.M.; Barczyk, A.; Hayashi, S.; Adcock, I.M.; Hogg, J.C. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N. Engl. J. Med. 2005, 352, 1967–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Hanazawa, T.; Tomita, K.; Barnes, P.; Adcock, I.M. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: Role of tyrosine nitration. Biochem. Biophys. Res. Commun. 2004, 315, 240–245. [Google Scholar] [CrossRef]
- Barnes, P.J. Role of HDAC2 in the pathophysiology of COPD. Annu. Rev. Physiol. 2009, 71, 451–464. [Google Scholar] [CrossRef]
- Caramori, G.; Kirkham, P.; Barczyk, A.; Di Stefano, A.; Adcock, I. Molecular pathogenesis of cigarette smoking induced stable COPD. Ann. N. Y. Acad. Sci. 2015, 1340, 55–64. [Google Scholar] [CrossRef]
- Cohen, B.H.; Ball, W.C., Jr.; Brashears, S.; Diamond, E.L.; Kreiss, P.; Levy, D.A.; Menkes, H.A.; Permutt, S.; Tockman, M.S. Risk factors in chronic obstructive pulmonary disease (COPD). Am. J. Epidemiol. 1977, 105, 223–232. [Google Scholar] [CrossRef]
- Montserrat-Capdevila, J.; Seminario, M.A.; Godoy, P.; Marsal, J.R.; Ortega, M.; Pujol, J.; Castan, M.T.; Alseda, M.; Betriu, A.; Lecube, A.; et al. Prevalence of chronic obstructive pulmonary disease (COPD) not diagnosed in a population with cardiovascular risk factors. Med. Clin. 2018. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The role of the Reactive Oxygen Species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxidative Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Harding, H.P.; Zhang, Y.; Jolicoeur, E.M.; Kuroda, M.; Ron, D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998, 17, 5708–5717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.; Yao, J.; Zeng, W.; Mizuno, Y.; Kamm, K.E.; Stull, J.T.; Harding, H.P.; Ron, D.; Muallem, S. ER stress disrupts Ca2+-signaling complexes and Ca2+ regulation in secretory and muscle cells from PERK-knockout mice. J. Cell Sci. 2006, 119, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.R.; Lefcoe, N.M.; Hamilton, J.T. Studies of the action of nicotine in guinea-pig tracheal smooth muscle: Interaction with beta-adrenoceptor antagonists. Eur. J. Pharmacol. 1980, 67, 53–64. [Google Scholar] [CrossRef]
- Pryor, W.A.; Stone, K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann. N. Y. Acad. Sci. 1993, 686, 12–27, discussion 27-18. [Google Scholar] [CrossRef]
- Eiserich, J.P.; van der Vliet, A.; Handelman, G.J.; Halliwell, B.; Cross, C.E. Dietary antioxidants and cigarette smoke-induced biomolecular damage: A complex interaction. Am. J. Clin. Nutr. 1995, 62, 1490S–1500S. [Google Scholar] [CrossRef]
- Frei, B.; Forte, T.M.; Ames, B.N.; Cross, C.E. Gas phase oxidants of cigarette smoke induce lipid peroxidation and changes in lipoprotein properties in human blood plasma. Protective effects of ascorbic acid. Biochem. J. 1991, 277 Pt 1, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Eiserich, J.P.; Vossen, V.; O’Neill, C.A.; Halliwell, B.; Cross, C.E.; van der Vliet, A. Molecular mechanisms of damage by excess nitrogen oxides: Nitration of tyrosine by gas-phase cigarette smoke. FEBS Lett. 1994, 353, 53–56. [Google Scholar] [CrossRef] [Green Version]
- Min, T.; Bodas, M.; Mazur, S.; Vij, N. Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J. Mol. Med. 2011, 89, 577–593. [Google Scholar] [CrossRef] [Green Version]
- Tran, I.; Ji, C.; Ni, I.; Min, T.; Tang, D.; Vij, N. Role of cigarette smoke-induced aggresome formation in chronic obstructive pulmonary disease-emphysema pathogenesis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Kenche, H.; Baty, C.J.; Vedagiri, K.; Shapiro, S.D.; Blumental-Perry, A. Cigarette smoking affects oxidative protein folding in endoplasmic reticulum by modifying protein disulfide isomerase. FASEB J. 2013, 27, 965–977. [Google Scholar] [CrossRef] [PubMed]
- van Rijt, S.H.; Keller, I.E.; John, G.; Kohse, K.; Yildirim, A.O.; Eickelberg, O.; Meiners, S. Acute cigarette smoke exposure impairs proteasome function in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L814–L823. [Google Scholar] [CrossRef] [PubMed]
- Somborac-Bacura, A.; van der Toorn, M.; Franciosi, L.; Slebos, D.J.; Zanic-Grubisic, T.; Bischoff, R.; van Oosterhout, A.J. Cigarette smoke induces endoplasmic reticulum stress response and proteasomal dysfunction in human alveolar epithelial cells. Exp. Physiol. 2013, 98, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Groskreutz, D.J.; Babor, E.C.; Monick, M.M.; Varga, S.M.; Hunninghake, G.W. Respiratory syncytial virus limits alpha subunit of eukaryotic translation initiation factor 2 (eIF2alpha) phosphorylation to maintain translation and viral replication. J. Biol. Chem. 2010, 285, 24023–24031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monick, M.M.; Powers, L.S.; Walters, K.; Lovan, N.; Zhang, M.; Gerke, A.; Hansdottir, S.; Hunninghake, G.W. Identification of an autophagy defect in smokers’ alveolar macrophages. J. Immunol. 2010, 185, 5425–5435. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Chen, X.; Lee, A.H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef]
- Woo, C.W.; Kutzler, L.; Kimball, S.R.; Tabas, I. Toll-like receptor activation suppresses ER stress factor CHOP and translation inhibition through activation of eIF2B. Nat. Cell Biol. 2012, 14, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Kelsen, S.G.; Duan, X.; Ji, R.; Perez, O.; Liu, C.; Merali, S. Cigarette smoke induces an unfolded protein response in the human lung: A proteomic approach. Am. J. Respir. Cell Mol. Biol. 2008, 38, 541–550. [Google Scholar] [CrossRef]
- Aksoy, M.O.; Kim, V.; Cornwell, W.D.; Rogers, T.J.; Kosmider, B.; Bahmed, K.; Barrero, C.; Merali, S.; Shetty, N.; Kelsen, S.G. Secretion of the endoplasmic reticulum stress protein, GRP78, into the BALF is increased in cigarette smokers. Respir. Res. 2017, 18, 78. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, D.; Thimmulappa, R.; Vij, N.; Navas-Acien, A.; Sussan, T.; Merali, S.; Zhang, L.; Kelsen, S.G.; Myers, A.; Wise, R.; et al. Heightened endoplasmic reticulum stress in the lungs of patients with chronic obstructive pulmonary disease: The role of Nrf2-regulated proteasomal activity. Am. J. Respir. Crit. Care Med. 2009, 180, 1196–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, T.; Carroll, T.P.; Buckley, P.G.; Cummins, R.; O’Neill, S.J.; McElvaney, N.G.; Greene, C.M. miR-199a-5p silencing regulates the unfolded protein response in chronic obstructive pulmonary disease and alpha1-antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 2014, 189, 263–273. [Google Scholar] [CrossRef]
- Merali, S.; Barrero, C.A.; Bowler, R.P.; Chen, D.E.; Criner, G.; Braverman, A.; Litwin, S.; Yeung, A.; Kelsen, S.G. Analysis of the plasma proteome in COPD: Novel low abundance proteins reflect the severity of lung remodeling. COPD 2014, 11, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.M.; Seeger, W.; et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappelletti, C.; Galbardi, B.; Kapetis, D.; Vattemi, G.; Guglielmi, V.; Tonin, P.; Salerno, F.; Morandi, L.; Tomelleri, G.; Mantegazza, R.; et al. Autophagy, inflammation and innate immunity in inflammatory myopathies. PLoS ONE 2014, 9, e111490. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Zeki, A.A.; Yeganeh, B.; Kenyon, N.J.; Post, M.; Ghavami, S. Autophagy in airway diseases: A new frontier in human asthma? Allergy 2016, 71, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Brun, P.; Tarricone, E.; Di Stefano, A.; Mattiuzzo, E.; Mehrbod, P.; Ghavami, S.; Leonardi, A. The regulatory activity of autophagy in conjunctival fibroblasts and its possible role in vernal keratoconjunctivitis. J. Allergy Clin. Immunol. 2020, 146, 1210–1213. [Google Scholar] [CrossRef]
- Chen, Z.H.; Kim, H.P.; Sciurba, F.C.; Lee, S.J.; Feghali-Bostwick, C.; Stolz, D.B.; Dhir, R.; Landreneau, R.J.; Schuchert, M.J.; Yousem, S.A.; et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS ONE 2008, 3, e3316. [Google Scholar] [CrossRef] [Green Version]
- Bodas, M.; Patel, N.; Silverberg, D.; Walworth, K.; Vij, N. Master autophagy regulator transcription factor EB regulates cigarette smoke-induced autophagy impairment and chronic obstructive pulmonary disease-emphysema pathogenesis. Antioxid. Redox Signal. 2017, 27, 150–167. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Lee, S.J.; Choi, A.M. Autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. Expert Rev. Respir. Med. 2010, 4, 573–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.; Wang, Z.; Tao, L.; Wang, Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010, 6, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appenzeller-Herzog, C.; Hall, M.N. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012, 22, 274–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombroski, B.A.; Nayak, R.R.; Ewens, K.G.; Ankener, W.; Cheung, V.G.; Spielman, R.S. Gene expression and genetic variation in response to endoplasmic reticulum stress in human cells. Am. J. Hum. Genet. 2010, 86, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slack, F.J.; Chinnaiyan, A.M. The role of non-coding RNAs in oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Xuan, W.; Wang, H.; Sun, F.; Zhang, C.; Gong, Q.; Ge, S. miR-200b regulates cellular senescence and inflammatory responses by targeting ZEB2 in pulmonary emphysema. Artif. Cells Nanomed. Biotechnol. 2020, 48, 656–663. [Google Scholar] [CrossRef] [Green Version]
- Mei, J.; Zhang, Y.; Lu, S.; Wang, J. Long non-coding RNA NNT-AS1 regulates proliferation, apoptosis, inflammation and airway remodeling of chronic obstructive pulmonary disease via targeting miR-582-5p/FBXO11 axis. Biomed. Pharm. 2020, 129, 110326. [Google Scholar] [CrossRef]
- Liu, K.; Gao, L.; Ma, X.; Huang, J.-J.; Chen, J.; Zeng, L.; Ashby, C.R.; Zou, C.; Chen, Z.-S. Long non-coding RNAs regulate drug resistance in cancer. Mol. Cancer 2020, 19, 54. [Google Scholar] [CrossRef]
- Beermann, J.; Piccoli, M.T.; Viereck, J.; Thum, T. Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef] [Green Version]
- Yaman Agaoglu, F.; Kovancilar, M.; Dizdar, Y.; Darendeliler, E.; Holdenrieder, S.; Dalay, N.; Gezer, U. Investigation of miR-21, miR-141, and miR-221 in blood circulation of patients with prostate cancer. Tumor Biol. 2011, 32, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Toiyama, Y.; Takahashi, M.; Hur, K.; Nagasaka, T.; Tanaka, K.; Inoue, Y.; Kusunoki, M.; Boland, C.R.; Goel, A. Serum miR-21 as a diagnostic and prognostic biomarker in colorectal cancer. J. Natl. Cancer Inst. 2013, 105, 849–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, A.A. Treating disease at the RNA level with oligonucleotides. N. Engl. J. Med. 2019, 380, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Wen, J.; Ren, Z.; Zhang, G. Non-coding RNAs: New biomarkers and therapeutic targets for esophageal cancer. Oncotarget 2017, 8, 43571–43578. [Google Scholar] [CrossRef] [Green Version]
- De Smet, E.G.; Van Eeckhoutte, H.P.; Avila Cobos, F.; Blomme, E.; Verhamme, F.M.; Provoost, S.; Verleden, S.E.; Venken, K.; Maes, T.; Joos, G.F.; et al. The role of miR-155 in cigarette smoke-induced pulmonary inflammation and COPD. Mucosal. Immunol. 2020, 13, 423–436. [Google Scholar] [CrossRef] [Green Version]
- Chi, Y.; Di, Q.; Han, G.; Li, M.; Sun, B. Mir-29b mediates the regulation of Nrf2 on airway epithelial remodeling and Th1/Th2 differentiation in COPD rats. Saudi J. Biol. Sci. 2019, 26, 1915–1921. [Google Scholar] [CrossRef]
- Velasco-Torres, Y.; Ruiz, V.; Montaño, M.; Pérez-Padilla, R.; Falfán-Valencia, R.; Pérez-Ramos, J.; Pérez-Bautista, O.; Ramos, C. Participation of the miR-22-HDAC4-DLCO Axis in Patients with COPD by Tobacco and Biomass. Biomolecules 2019, 9, 837. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lyu, X.; Wu, X.; Yu, L.; Hu, K. Long non-coding RNA PVT1, a novel biomarker for chronic obstructive pulmonary disease progression surveillance and acute exacerbation prediction potentially through interaction with microRNA-146a. J. Clin. Lab. Anal. 2020, 34, e23346. [Google Scholar] [CrossRef]
- Zhou, A.Y.; Zhao, Y.Y.; Zhou, Z.J.; Duan, J.X.; Zhu, Y.Z.; Cai, S.; Chen, P. Microarray Analysis of Long Non-Coding RNAs in Lung Tissues of Patients with COPD and HOXA-AS2 Promotes HPMECs Proliferation via Notch1. Int. J. Chron. Obs. Pulmon. Dis. 2020, 15, 2449–2460. [Google Scholar] [CrossRef]
- Zhou, S.; Jiang, H.; Li, M.; Wu, P.; Sun, L.; Liu, Y.; Zhu, K.; Zhang, B.; Sun, G.; Cao, C.; et al. Circular RNA hsa_circ_0016070 Is Associated with Pulmonary Arterial Hypertension by Promoting PASMC Proliferation. Mol. Ther. Nucleic Acids 2019, 18, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Chen, J.; Jing, G.; Grayson, T.B.; Shalev, A. miR-204 Targets PERK and Regulates UPR Signaling and β-Cell Apoptosis. Mol. Endocrinol. 2016, 30, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Yang, Z.; Yang, X.; Li, T.; Liu, M.; Tang, H. miR-346 functions as a pro-survival factor under ER stress by activating mitophagy. Cancer Lett. 2018, 413, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.o.; Flaherty, K.R.; Lasky, J.A. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Suda, T. Idiopathic pulmonary fibrosis: Diagnosis and clinical manifestations. Clin. Med. Insights Circ. Respir. Pulm. Med. 2016, 9, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Caminati, A.; Lonati, C.; Cassandro, R.; Elia, D.; Pelosi, G.; Torre, O.; Zompatori, M.; Uslenghi, E.; Harari, S. Comorbidities in idiopathic pulmonary fibrosis: An underestimated issue. Eur. Respir. Rev. 2019, 28. [Google Scholar] [CrossRef]
- Garcia-Sancho, C.; Buendia-Roldan, I.; Fernandez-Plata, M.R.; Navarro, C.; Perez-Padilla, R.; Vargas, M.H.; Loyd, J.E.; Selman, M. Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respir. Med. 2011, 105, 1902–1907. [Google Scholar] [CrossRef] [Green Version]
- Marshall, R.P.; Puddicombe, A.; Cookson, W.O.C.; Laurent, G.J. Adult familial cryptogenic fibrosing alveolitis in the United Kingdom. Thorax 2000, 55, 143–146. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, U.; Laitinen, T.; Tukiainen, P. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: Evidence of founder effect among multiplex families in Finland. Thorax 2002, 57, 338–342. [Google Scholar] [CrossRef] [Green Version]
- Bitterman, P.B.; Rennard, S.I.; Keogh, B.A.; Wewers, M.D.; Adelberg, S.; Crystal, R.G. Familial idiopathic pulmonary fibrosis. N. Engl. J. Med. 1986, 314, 1343–1347. [Google Scholar] [CrossRef]
- Borie, R.; Kannengiesser, C.; Nathan, N.; Tabèze, L.; Pradère, P.; Crestani, B. Familial pulmonary fibrosis. Rev. Mal. Respir. 2015, 32, 413–434. [Google Scholar] [CrossRef]
- Barros, A.; Oldham, J.; Noth, I. Genetics of Idiopathic Pulmonary Fibrosis. Am. J. Med. Sci. 2019, 357, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Mathai, S.K.; Schwartz, D.A. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front. Med. (Lausanne) 2017, 4, 154. [Google Scholar] [CrossRef] [PubMed]
- Douglas, W.W.; Ryu, J.H.; Schroeder, D.R. Idiopathic pulmonary fibrosis: Impact of oxygen and colchicine, prednisone, or no therapy on survival. Am. J. Respir. Crit. Care Med. 2000, 161, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Bjoraker, J.A.; Ryu, J.H.; Edwin, M.K.; Myers, J.L.; Tazelaar, H.D.; Schroeder, D.R.; Offord, K.P. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1998, 157, 199–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cokkinides, V.; Albano, J.; Samuels, A.; Ward, M.E.; Thum, J.M. American cancer society: Cancer facts and figures. Atlanta Am. Cancer Soc. 2005. [Google Scholar]
- Spagnolo, P.; Sverzellati, N.; Rossi, G.; Cavazza, A.; Tzouvelekis, A.; Crestani, B.; Vancheri, C. Idiopathic pulmonary fibrosis: An update. Ann. Med. 2015, 47, 15–27. [Google Scholar] [CrossRef]
- Dempsey, O.J.; Kerr, K.M.; Gomersall, L.; Remmen, H.; Currie, G.P. Idiopathic pulmonary fibrosis: An update. J. Assoc. Physicians 2006, 99, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef]
- Sauleda, J.; Nunez, B.; Sala, E.; Soriano, J.B. Idiopathic Pulmonary Fibrosis: Epidemiology, Natural History, Phenotypes. Med. Sci. (Basel) 2018, 6, 110. [Google Scholar] [CrossRef] [Green Version]
- King Jr, T.E.; Tooze, J.A.; Schwarz, M.I.; Brown, K.R.; Cherniack, R.M. Predicting survival in idiopathic pulmonary fibrosis: Scoring system and survival model. Am. J. Respir. Crit. Care Med. 2001, 164, 1171–1181. [Google Scholar] [CrossRef]
- Gribbin, J.; Hubbard, R.B.; Le Jeune, I.; Smith, C.J.P.; West, J.; Tata, L.J. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax 2006, 61, 980–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swigris, J.J.; Kuschner, W.G.; Jacobs, S.S.; Wilson, S.R.; Gould, M.K. Health-related quality of life in patients with idiopathic pulmonary fibrosis: A systematic review. Thorax 2005, 60, 588–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottin, V.; Hirani, N.A.; Hotchkin, D.L.; Nambiar, A.M.; Ogura, T.; Otaola, M.; Skowasch, D.; Park, J.S.; Poonyagariyagorn, H.K.; Wuyts, W.; et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Luckhardt, T.; Hecker, L.; Zhou, Y.; Liu, G.; Antony, V.B.; de Andrade, J.; Thannickal, V.J. New insights into the pathogenesis and treatment of idiopathic pulmonary fibrosis. Drugs 2011, 71, 981–1001. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.L.; Swigris, J.J. Idiopathic pulmonary fibrosis: Diagnosis and epidemiology. Clin. Chest Med. 2012, 33, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, T.; Usuki, J.; Azuma, A.; Nakagawa, T.; Kudoh, S. Diabetes Mellitus May Increase Risk for Idiopathic Pulmonary Fibrosisa. Chest 2003, 123, 2007–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gribbin, J.; Hubbard, R.; Smith, C. Role of diabetes mellitus and gastro-oesophageal reflux in the aetiology of idiopathic pulmonary fibrosis. Respir. Med. 2009, 103, 927–931. [Google Scholar] [CrossRef] [Green Version]
- Matsuse, T.; Ohga, E.; Teramoto, S.; Fukayama, M.; Nagai, R.; Horiuchi, S.; Ouchi, Y. Immunohistochemical localisation of advanced glycation end products in pulmonary fibrosis. J. Clin. Pathol 1998, 51, 515–519. [Google Scholar] [CrossRef] [Green Version]
- Myers, J.L. Histopathology of IPF and related disorders. In Idiopathic Pulmonary Fibrosis; Springer: Berlin/Heidelberg, Germany, 2014; pp. 35–54. [Google Scholar]
- Gross, T.J.; Hunninghake, G.W. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2001, 345, 517–525. [Google Scholar] [CrossRef]
- American Thoracic, S. Idiopathic pulmonary fibrosis: Diagnosis and treatment: International consensus statement. Am. J. Respir. Crit. Care Med. 2000, 161, 646–664. [Google Scholar]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King Jr, T.E.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.s.; Wells, A.U. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Churg, A.M.; Myers, J.L.; Tazelaar, H.D. Thurlbeck’s Pathology of the Lung; Thieme: New York, NY, USA, 2005; p. 1157. [Google Scholar]
- Gotoh, T.; Terada, K.; Oyadomari, S.; Mori, M. hsp70-DnaJ chaperone pair prevents nitric oxide-and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004, 11, 390. [Google Scholar] [CrossRef] [PubMed]
- Gauldie, J.; Kolb, M.; Sime, P.J. A new direction in the pathogenesis of idiopathic pulmonary fibrosis? Respir. Res. 2001, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Selman, M.s. Idiopathic pulmonary fibrosis: New insights in its pathogenesis. Int. J. Biochem. Cell Biol. 2002, 34, 1534–1538. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Thwaite, E.L.; Kazerooni, E.A.; Gross, B.H.; Toews, G.B.; Colby, T.V.; Travis, W.D.; Mumford, J.A.; Murray, S.; Flint, A. Radiological versus histological diagnosis in UIP and NSIP: Survival implications. Thorax 2003, 58, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumikawa, H.; Johkoh, T.; Colby, T.V.; Ichikado, K.; Suga, M.; Taniguchi, H.; Kondoh, Y.; Ogura, T.; Arakawa, H.; Fujimoto, K. Computed tomography findings in pathological usual interstitial pneumonia: Relationship to survival. Am. J. Respir. Crit. Care Med. 2008, 177, 433–439. [Google Scholar] [CrossRef]
- Nishimura, K.; Kitaichi, M.; Izumi, T.; Nagai, S.; Kanaoka, M.; Itoh, H. Usual interstitial pneumonia: Histologic correlation with high-resolution CT. Radiology 1992, 182, 337–342. [Google Scholar] [CrossRef]
- Johkoh, T.; Müller, N.L.; Cartier, Y.; Kavanagh, P.V.; Hartman, T.E.; Akira, M.; Ichikado, K.; Ando, M.; Nakamura, H. Idiopathic interstitial pneumonias: Diagnostic accuracy of thin-section CT in 129 patients. Radiology 1999, 211, 555–560. [Google Scholar] [CrossRef]
- Hansell, D.M.; Bankier, A.A.; MacMahon, H.; McLoud, T.C.; Muller, N.L.; Remy, J. Fleischner Society: Glossary of terms for thoracic imaging. Radiology 2008, 246, 697–722. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.-H.; Misumi, S.; Sahin, H.; Brown, K.K.; Newell, J.D.; Lynch, D.A. Computed tomographic features of idiopathic fibrosing interstitial pneumonia: Comparison with pulmonary fibrosis related to collagen vascular disease. J. Comput. Assist. Tomogr. 2009, 33, 410–415. [Google Scholar] [CrossRef]
- Souza, C.A.; Müller, N.L.; Lee, K.S.; Johkoh, T.; Mitsuhiro, H.; Chong, S. Idiopathic interstitial pneumonias: Prevalence of mediastinal lymph node enlargement in 206 patients. Am. J. Roentgenol. 2006, 186, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Kekevian, A.; Gershwin, M.E.; Chang, C. Diagnosis and classification of idiopathic pulmonary fibrosis. Autoimmun. Rev. 2014, 13, 508–512. [Google Scholar] [CrossRef]
- Watanabe, K.; Ishii, H.; Kiyomi, F.; Terasaki, Y.; Hebisawa, A.; Kawabata, Y.; Johkoh, T.; Sakai, F.; Kondoh, Y.; Inoue, Y.; et al. Criteria for the diagnosis of idiopathic pleuroparenchymal fibroelastosis: A proposal. Respir. Investig. 2019, 57, 312–320. [Google Scholar] [CrossRef]
- Noble, P.W.; Homer, R.J. Back to the future: Historical perspective on the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2005, 33, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.; O’Connor, R. Idiopathic pulmonary fibrosis: Current understanding of the pathogenesis and the status of treatment. Can. Med. Assoc. J. 2004, 171, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.D.; Hua, J.; Mui, A.; O’Connor, R.; Grotendorst, G.; Khalil, N. Release of biologically active TGF-beta1 by alveolar epithelial cells results in pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L527–L539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adkins, J.M.; Collard, H.R. Idiopathic pulmonary fibrosis. Semin. Respir. Crit. Care Med. 2012, 33, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef] [Green Version]
- du Bois, R.M.; Nathan, S.D.; Richeldi, L.; Schwarz, M.I.; Noble, P.W. Idiopathic pulmonary fibrosis: Lung function is a clinically meaningful endpoint for phase III trials. Am. J. Respir. Crit. Care Med. 2012, 186, 712–715. [Google Scholar] [CrossRef]
- Velnar, T.; Bailey, T.; Smrkolj, V. The wound healing process: An overview of the cellular and molecular mechanisms. J. Int. Med. Res. 2009, 37, 1528–1542. [Google Scholar] [CrossRef]
- Imokawa, S.; Sato, A.; Hayakawa, H.; Kotani, M.; Urano, T.; Takada, A. Tissue factor expression and fibrin deposition in the lungs of patients with idiopathic pulmonary fibrosis and systemic sclerosis. Am. J. Respir. Crit. Care Med. 1997, 156, 631–636. [Google Scholar] [CrossRef] [Green Version]
- Andrew, A.S.; Klei, L.R.; Barchowsky, A. AP-1-dependent induction of plasminogen activator inhibitor-1 by nickel does not require reactive oxygen. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L616–L623. [Google Scholar] [CrossRef]
- Wuyts, W.A.; Wijsenbeek, M.; Bondue, B.; Bouros, D.; Bresser, P.; Robalo Cordeiro, C.; Hilberg, O.; Magnusson, J.; Manali, E.D.; Morais, A.; et al. Idiopathic Pulmonary Fibrosis: Best Practice in Monitoring and Managing a Relentless Fibrotic Disease. Respiration 2019, 99, 1–10. [Google Scholar] [CrossRef]
- Corrin, B.; Dewar, A. Pathogenesis of idiopathic interstitial pulmonary fibrosis. Ultrastruct. Pathol. 1996, 20, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Thannickal, V.J.; Pardo, A.; Zisman, D.A.; Martinez, F.J.; Lynch Iii, J.P. Idiopathic pulmonary fibrosis: Pathogenesis and therapeutic approaches. Drugs 2004, 64, 405–431. [Google Scholar] [CrossRef] [PubMed]
- Hoo, Z.H.; Whyte, M.K.B. Idiopathic pulmonary fibrosis. Thorax 2012, 67, 742–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, F.; Lunardi, F.; Giacometti, C.; Marulli, G.; Gnoato, M.; Pontisso, P.; Saetta, M.; Valente, M.; Rea, F.; Perissinotto, E.; et al. Overexpression of squamous cell carcinoma antigen in idiopathic pulmonary fibrosis: Clinicopathological correlations. Thorax 2008, 63, 795–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Bian, W.; Gu, X.H.; Shen, C. Differing Expression of Cytokines and Tumor Markers in Combined Pulmonary Fibrosis and Emphysema Compared to Emphysema and Pulmonary Fibrosis. COPD 2017, 14, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Battinger, E.P. TGF-a and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.; Jones, M.G.; Davies, D.E.; Wang, Y. Epithelial-mesenchymal transition contributes to pulmonary fibrosis via aberrant epithelial/fibroblastic cross-talk. J. Lung Health Dis. 2019, 3, 31–35. [Google Scholar] [CrossRef] [Green Version]
- Bridges, R.S.; Kass, D.; Loh, K.; Glackin, C.; Borczuk, A.C.; Greenberg, S. Gene expression profiling of pulmonary fibrosis identifies Twist1 as an antiapoptotic molecular “rectifier” of growth factor signaling. Am. J. Pathol. 2009, 175, 2351–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayachandran, A.; Königshoff, M.; Yu, H.; Rupniewska, E.; Hecker, M.; Klepetko, W.; Seeger, W.; Eickelberg, O. SNAI transcription factors mediate epithelial-mesenchymal transition in lung fibrosis. Thorax 2009, 64, 1053–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandbo, N. Mechanisms of fibrosis in IPF. In Idiopathic Pulmonary Fibrosis; Meyer, K., Steven, D.N., Eds.; Humana Press: Totowa, NJ, USA, 2019; pp. 133–182. [Google Scholar]
- Coward, W.R.; Saini, G.; Jenkins, G. The pathogenesis of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2010, 4, 367–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1119–L1126. [Google Scholar] [CrossRef] [Green Version]
- Nogee, L.M.; Dunbar, A.E.; Wert, S.E.; Askin, F.; Hamvas, A.; Whitsett, J.A. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N. Engl. J. Med. 2001, 344, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Degryse, A.L.; Li, B.; Han, W.; Sherrill, T.P.; Plieth, D.; Neilson, E.G.; Blackwell, T.S. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2009, 180, 657–665. [Google Scholar] [CrossRef] [Green Version]
- Lawson, W.E.; Cheng, D.-S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; Lane, K.B. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kropski, J.A.; Blackwell, T.S. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J. Clin. Investig. 2018, 128, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridges, J.P.; Wert, S.E.; Nogee, L.M.; Weaver, T.E. Expression of a human surfactant protein C mutation associated with interstitial lung disease disrupts lung development in transgenic mice. J. Biol. Chem. 2003, 278, 52739–52746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conkright, J.J.; Na, C.-L.; Weaver, T.E. Overexpression of surfactant protein-C mature peptide causes neonatal lethality in transgenic mice. Am. J. Respir. Cell Mol. Biol. 2002, 26, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Zhou, B.; Ann, D.K.; Minoo, P.; Liu, Y.; Banfalvi, A.; Krishnaveni, M.S.; Dubourd, M.; Demaio, L.; Willis, B.C. Role of endoplasmic reticulum stress in epithelial and mesenchymal transition of alveolar epithelial cells: Effects of misfolded surfactant protein. Am. J. Respir. Cell. Mol. Biol. 2011, 45, 498–509. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, Y.; Zhang, Z.; He, L.; Zhu, J.; Zhang, M.; He, X.; Cheng, Z.; Ao, Q.; Cao, Y. Chop deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production. Mol. Ther. 2016, 24, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Ishitsuka, Y.; Hayasaka, M.; Yamada, Y.; Miyata, K.; Endo, M.; Kondo, Y.; Moriuchi, H.; Irikura, M.; Tanaka, K.-i. The exacerbating roles of CCAAT/enhancer-binding protein homologous protein (CHOP) in the development of bleomycin-induced pulmonary fibrosis and the preventive effects of tauroursodeoxycholic acid (TUDCA) against pulmonary fibrosis in mice. Pharmacol. Res. 2015, 99, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Ayaub, E.A.; Kolb, P.S.; Mohammed-Ali, Z.; Tat, V.; Murphy, J.; Bellaye, P.S.; Shimbori, C.; Boivin, F.J.; Lai, R.; Lynn, E.G. GRP78 and CHOP modulate macrophage apoptosis and the development of bleomycin-induced pulmonary fibrosis. J. Pathol. 2016, 239, 411–425. [Google Scholar] [CrossRef]
- Baek, H.A.; Kim, D.S.; Park, H.S.; Jang, K.Y.; Kang, M.J.; Lee, D.G.; Moon, W.S.; Chae, H.J.; Chung, M.J. Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2012, 46, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Fassio, A.; Sitia, R. Formation, isomerisation and reduction of disulphide bonds during protein quality control in the endoplasmic reticulum. Histochem. Cell Biol. 2002, 117, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, P.L.; Maher, T.M. The role of infection in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2013, 22, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergnon, J.M.; Weynants, P.; Vincent, M.; Mornex, J.F.; Brune, J. Cryptogenic fibrosing alveolitis and Epstein-Barr virus: An association? Lancet 1984, 324, 768–771. [Google Scholar] [CrossRef]
- Isler, J.A.; Skalet, A.H.; Alwine, J.C. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 2005, 79, 6890–6899. [Google Scholar] [CrossRef] [Green Version]
- Doran, P.; Egan, J.J. Herpesviruses: A cofactor in the pathogenesis of idiopathic pulmonary fibrosis? Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L709–L710. [Google Scholar] [CrossRef] [Green Version]
- Dickens, J.A.; Malzer, E.; Chambers, J.E.; Marciniak, S.J. Pulmonary endoplasmic reticulum stress: Scars, smoke and suffocation. FEBS J. 2018, 286, 322–341. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 2020, 8, 807–815. [Google Scholar] [CrossRef]
- Taz, T.A.; Ahmed, K.; Paul, B.K.; Kawsar, M.; Aktar, N.; Mahmud, S.M.H.; Moni, M.A. Network-based identification genetic effect of SARS-CoV-2 infections to Idiopathic pulmonary fibrosis (IPF) patients. Brief. Bioinform. 2020. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.; Lambrecht, B.; Janssens, S. The UPR and lung disease. In Proceedings of the Semin Immunopathol; Springer: Berlin/Heidelberg, Germany, 2013; pp. 293–306. [Google Scholar]
- Marcinak, S.J.; Ron, D. The unfolded protein response in lung disease. Proc. Am. Thorac. Soc. 2010, 7, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoszewski, R.; Rab, A.; Fu, L.; Bartoszewska, S.; Collawn, J.; Bebok, Z. CFTR expression regulation by the unfolded protein response. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2011; Volume 491, pp. 3–24. [Google Scholar]
- Kelsen, S.G. The unfolded protein response in chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 2016, 13, S138–S145. [Google Scholar]
- Parfrey, H.; Moseley, E.; Beardsley, B.; Knight, J.; Marciniak, S.; Rassl, D. S74 Endoplasmic stress is associated with fibrosis in interstitial lung disease. Thorax 2017, 72, A47. [Google Scholar]
- Burman, A.; Tanjore, H.; Blackwell, T.S. Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. 2018, 68, 355–365. [Google Scholar] [CrossRef]
- Mahmood, S.H. Role of the IRE/XBP-1 Pathway in Cigarette Smoke Affected Macrophage Polarization In Vitro; McMaster University: Hamilton, ON, Canada, 2017. [Google Scholar]
- Lenna, S.; Trojanowska, M. The role of endoplasmic reticulum stress and the unfolded protein response in fibrosis. Curr. Opin. Rheumatol. 2012, 24. [Google Scholar] [CrossRef]
- Bucala, R.; Spiegel, L.A.; Chesney, J.; Hogan, M.; Cerami, A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1994, 1, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Gundersen, S.G.; Haagensen, I.; Jonassen, T.O.; Figenschau, K.J.; de Jonge, N.; Deelder, A.M. Magnetic bead antigen capture enzyme-linked immunoassay in microtitre trays for rapid detection of schistosomal circulating anodic antigen. J. Immunol. Methods 1992, 148, 1–8. [Google Scholar] [CrossRef]
- Moeller, A.; Gilpin, S.E.; Ask, K.; Cox, G.; Cook, D.; Gauldie, J.; Margetts, P.J.; Farkas, L.; Dobranowski, J.; Boylan, C.; et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Dupin, I.; Contin-Bordes, C.; Berger, P. Fibrocytes in Asthma and Chronic Obstructive Pulmonary Disease: Variations on the Same Theme. Am. J. Respir. Cell Mol. Biol. 2018, 58, 288–298. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Klicka, K.; Paskal, W.; Dudkiewicz, J.; Wejman, J.; Pyzlak, M.; Włodarski, P.K. miR-410-3p is induced by vemurafenib via ER stress and contributes to resistance to BRAF inhibitor in melanoma. PLoS ONE 2020, 15, e0234707. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Xu, T.; Chen, Y.; Qu, W.; Sun, D.; Liu, X.; Sun, L. MiR-185-5p ameliorates endoplasmic reticulum stress and renal fibrosis by downregulation of ATF6. Lab. Investig. 2020, 100, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.; Liu, R.; Wang, L.; Tang, M.; Li, S.R.; Hu, X. LncRNA RPPH1 attenuates Aβ(25-35)-induced endoplasmic reticulum stress and apoptosis in SH-SY5Y cells via miR-326/PKM2. Int. J. Neurosci. 2020. [Google Scholar] [CrossRef]
- Deng, W.; Fu, J.; Wang, T.; Chen, J.X.; Fu, L.B.; Peng, W. Hsa_circRNA_101036 acts as tumor-suppressor in oral squamous cell carcinoma cells via inducing endoplasmic reticulum stress. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6111–6121. [Google Scholar] [CrossRef]
- Tang, G.H.; Chen, X.; Ding, J.C.; Du, J.; Lin, X.T.; Xia, L.; Lian, J.B.; Ye, F.; He, X.S.; Liu, W. LncRNA LUCRC Regulates Colorectal Cancer Cell Growth and Tumorigenesis by Targeting Endoplasmic Reticulum Stress Response. Front. Genet. 2019, 10, 1409. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, A.; Khansarinejad, B.; Hosseinkhani, S.; Ghanei, M.; Mowla, S.J. miR-199a-5p and miR-495 target GRP78 within UPR pathway of lung cancer. Gene 2017, 620, 15–22. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aghaei, M.; Dastghaib, S.; Aftabi, S.; Aghanoori, M.-R.; Alizadeh, J.; Mokarram, P.; Mehrbod, P.; Ashrafizadeh, M.; Zarrabi, A.; McAlinden, K.D.; et al. The ER Stress/UPR Axis in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Life 2021, 11, 1. https://0-doi-org.brum.beds.ac.uk/10.3390/life11010001
Aghaei M, Dastghaib S, Aftabi S, Aghanoori M-R, Alizadeh J, Mokarram P, Mehrbod P, Ashrafizadeh M, Zarrabi A, McAlinden KD, et al. The ER Stress/UPR Axis in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Life. 2021; 11(1):1. https://0-doi-org.brum.beds.ac.uk/10.3390/life11010001
Chicago/Turabian StyleAghaei, Mahmoud, Sanaz Dastghaib, Sajjad Aftabi, Mohamad-Reza Aghanoori, Javad Alizadeh, Pooneh Mokarram, Parvaneh Mehrbod, Milad Ashrafizadeh, Ali Zarrabi, Kielan Darcy McAlinden, and et al. 2021. "The ER Stress/UPR Axis in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis" Life 11, no. 1: 1. https://0-doi-org.brum.beds.ac.uk/10.3390/life11010001