Vascular Wall Reactions to Coronary Stents—Clinical Implications for Stent Failure

Department of Cardiology, University Medical Center Mainz and DZHK Standort Rhein-Main, 55131 Mainz, Germany
Life 2021, 11(1), 63; https://0-doi-org.brum.beds.ac.uk/10.3390/life11010063
Submission received: 20 December 2020
/
Revised: 14 January 2021
/
Accepted: 15 January 2021
/
Published: 17 January 2021
(This article belongs to the Special Issue 2020: A 10 Years Journey—Advances in Life Sciences)
Abstract
:Coronary stents belong to the most commonly implanted devices worldwide. A number of different types of stent exist, with very different mechanical and biochemical characteristics that influence their interactions with vascular tissues. Inappropriate inflammatory reactions are the major cause of the two major complications that follow implantation of stents in a percentage as high as 5–20%. It is therefore important to understand these reactions and how different they are among different generations of stents.
1. Introduction
With several million implantations per year, coronary artery stents are among the most commonly used devices for the treatment of a disease that still represents the major cause of mortality and morbidity worldwide [1]. As compared to bare metal stents (BMS), drug eluting stents (DES) have reduced rates of restenosis and target lesion revascularization, profoundly modifying the interventional treatment of coronary artery disease [2]. However, DES failure remains a problem that affects, depending on a number of factors, up to 20% of the devices implanted [1]. In particular, stent thrombosis is a complication associated with a very variable, but not negligible, mortality (from 3.8% in the DESERT (International Drug-Eluting Stent Event Registry of Thrombosis) registry to 45% in the registry by Iakovou et al.) [3,4,5,6,7,8]. While being less malign, stent restenosis is also associated with an acute presentation (unstable angina or myocardial infarction) in about two-thirds of the cases, without difference across stent generations. Our understanding of the pathophysiology of stent failure remains incomplete and these phenomena are surely multifactorial. Patient, procedural and plaque factors obviously play an important role in stent failure [9]; however, it is also clear from observations in both BMS and early-generation DES that the biological reactions induced by the implantation trauma and by the exposure to the stent components are crucial. Far from being only passive supports for the vascular architecture, coronary stents interact with vascular biology and stimulate a number of cellular reactions. Ideally, healing processes should cover the struts with a thin layer of functioning neo-endothelium and minimal or no extracellular matrix that lead to the restitution of vascular function re-establishing laminar flow. Following coronary angioplasty, however, a number of unwanted mechanisms are also activated, as witnessed by systemic increases in c-reactive protein, inflammatory cytokines, and blood cell parameters, all of which are predictors of stent failure during follow-up [10,11,12].
Thus, while the multifactorial nature of stent failure is acknowledged [13], the present review will focus on the biological responses that follow stent implantation, and how these may lead to stent failure across the different generations of stents.
2. Stent Complications and Stent Pathology among Different Device Generations
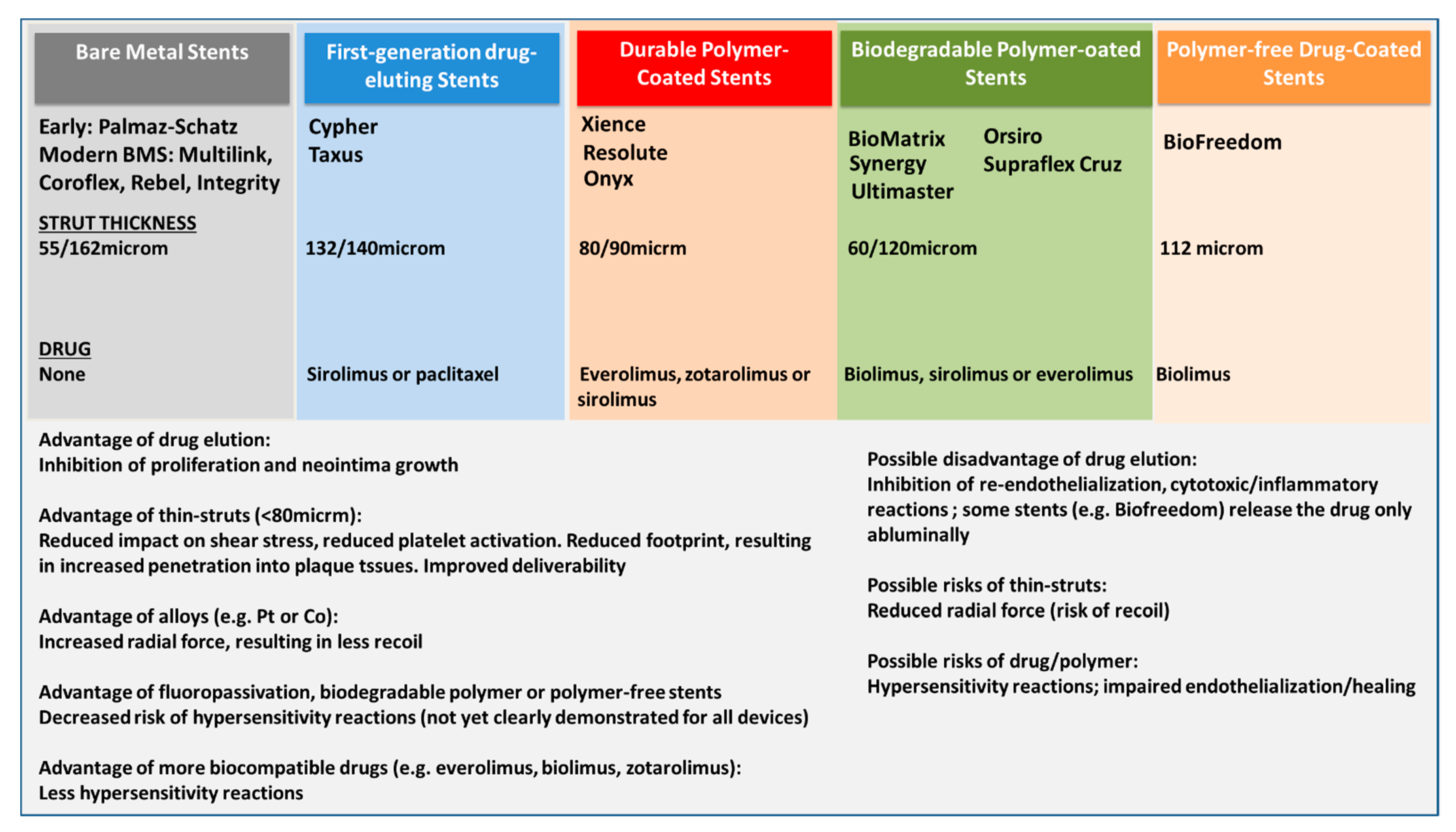
With the constant development of newer device types, the incidence and pattern of their short- and long-term complications have also changed (Figure 1). As compared to balloon-only angioplasty, BMS solved the issue of acute recoil. However, the application of these devices in complex lesions (such as small vessels, bifurcation lesions, diabetic patients, etc.) resulted in a risk of restenosis. Bare metal stent restenosis was characterized by progressive, slow growth of a fibrotic extracellular matrix typically occurring within the first 6–9 months after implantation [2]. While first-generation DES such as Cypher sirolimus-eluting stents (SES, Cordis Corp., Miami Lakes, FL, USA) and Taxus paclitaxel-eluting stents (PES, Boston Scientific, Natick, MA, USA) reduced the incidence of in-stent restenosis, an increase in the incidence of late in-stent thrombosis due to delayed arterial healing and adverse hypersensitivity reactions compromised the safety of these devices [14,15]. Although in-stent thrombosis was not a new phenomenon, its characteristics in patients who had received implantation of BMS where completely different. In BMS, stent thrombosis had typically an earlier onset, and it was almost invariably caused by the hemodynamic changes caused by the growth of neointima [9]. In contrast, in first-generation DES, stent thrombosis occurred later (one or more years after implantation) and it was characterized by evidence of delayed arterial healing, lack of endothelialization, persistent fibrin deposition as well as hypersensitivity reactions with eosinophilic/heterophilic infiltration of the arterial wall as pathognomonic findings [16,17,18,19]. Further, another new clinical entity, in-stent neoatherosclerosis, was described as a long-term consequence of bare metal and particularly first-generation DES: neoatherogenesis represents today one of the most common causes of late and very late stent failure [20]. Its incidence appears to be twice as high in DES-treated segments lesions than in lesions treated with BMS (31% vs. 16%), and it appears to occur earlier after DES than BMS (420 days (361–683 days) versus 2160 (1800–2880) days). As discussed below, late (1 month to 12 months after implantation) and very late (beyond 1 year) [21] stent thrombosis as well as neoatherogenesis appear to be caused (also) by pathological responses to one of the component of the DES.
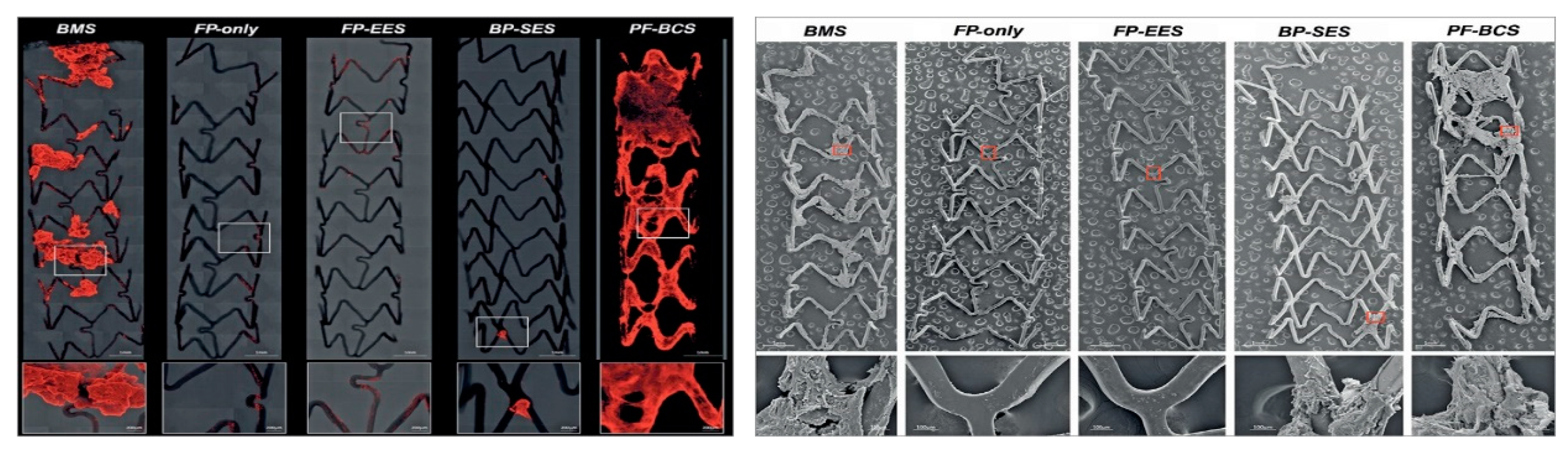
Second-generation DES were developed explicitly to address these issues of biocompatibility. They feature a thinner strut platform with streamlined strut geometries to decrease prothrombotic fibrin deposition and a thromboresistant polymer as well as different (possibly less toxic) drugs such as everolimus, biolimus, or zotarolimus, which do not inhibit endothelial cell adhesion as compared to tacrolimus [22]. Of importance, while first-generation DES were designed to delay cell proliferation including re-endothelization processes to reduce the risk of restenosis, newer-generation DES were developed to promote a faster re-endothelization through their enhanced biocompatibility. In particular, the introduction of fluoropolymer coatings appears to confer (in a dose-dependent manner) thromboresistance by reducing platelet activation [23,24], an effect that is believed to result from adhesion and retention of albumin on the polymer surface. This albumin-rich protein layer may in turn inhibit the adhesion of prothrombotic proteins such as fibrinogen. A high hydrophobicity, high bond strength, low polarizability and a high degree of fluorination appear to be responsible for these advantages. As well, fluoropolymer-coated everolimus-eluting stents were associated with significantly lower inflammatory cell adhesion as compared to BMS with the same stent platform [25] (Figure 2). Additional features of some modern stents include polymers allowing slow release of the drug (to avoid toxic reactions), drug coating limited to the abluminal side (to avoid impairment of endothelialization), antibodies for the adhesion of endothelial progenitor cells, elution of antioxidant drugs.
Meta-analysis studies confirmed a lower target-lesion revascularization and incidence of stent thrombosis with newer-generation devices as compared to 1st generation DES and BMS [26,27]. The safety profile of second generation DES was also confirmed by intravascular imaging [28], angioscopy and pathology studies confirming improved stent coverage by neoendothelium associated with less aggregates of red blood cells, less peri-strut fibrin deposition, and a lower inflammation score [28,29]. Further evolutions to improve the biocompatibility of implants included the development of antithrombotic polymers (so-called fluoropassivation [25]), bioresorbable polymers (which however do not seem to improve significantly patients outcomes [30]), and completely bioresorbable scaffolds (whose mechanical characteristics, particularly strut thickness however represented a limitation [13,31,32]).
3. The Mechanisms of Restenosis
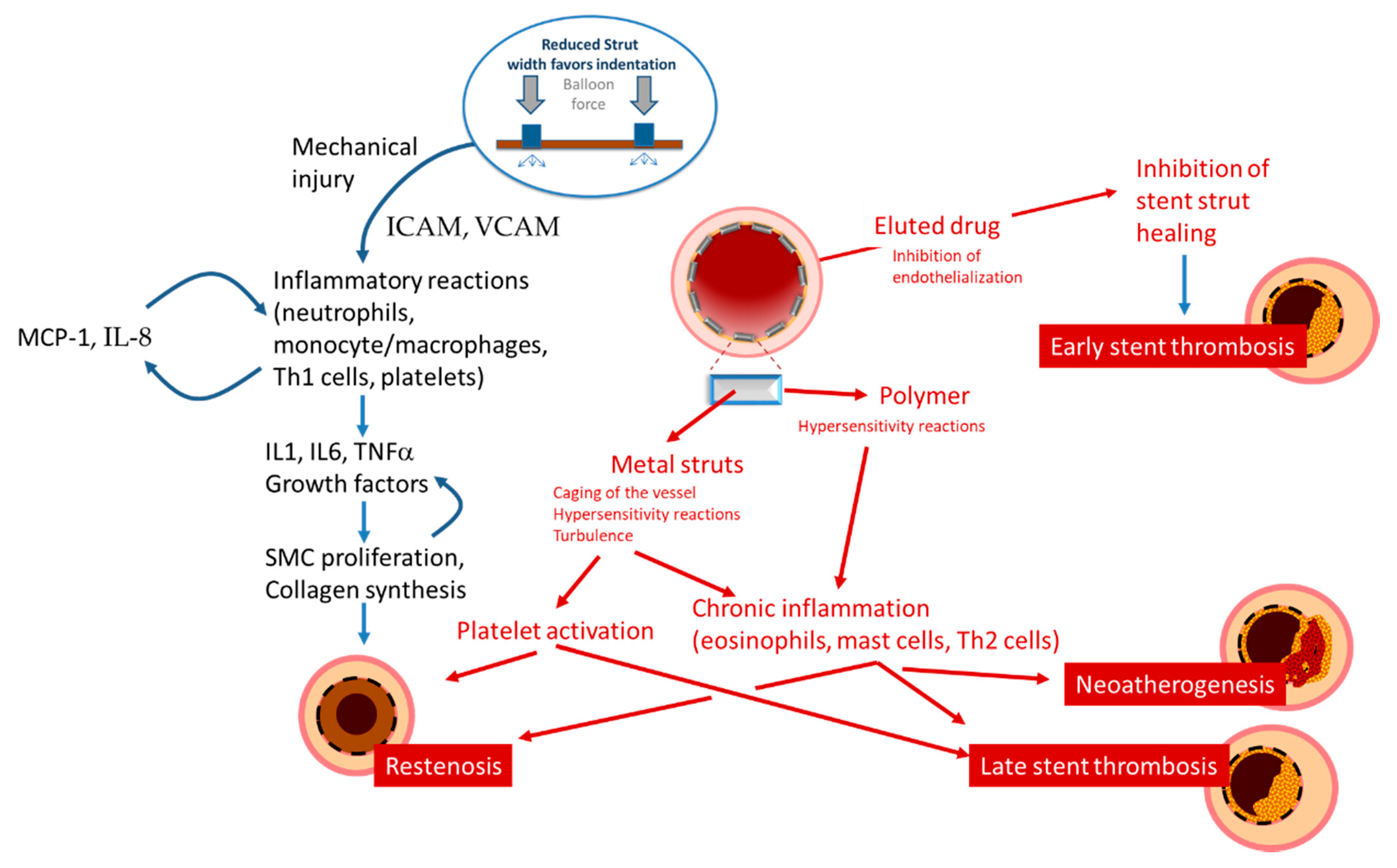
Restenosis is defined as the reduction in lumen size of an artery after intra-arterial intervention. While restenosis prior to the advent of coronary stents was mostly caused by recoil, in-stent restenosis is a response-to-injury process caused by formation of a tissue called neointima. For BMS, the theory postulated that this neointima is mostly composed of fibrotic tissue with a gradual and progressive onset over months, which led to the concept that in-stent restenosis is a benign phenomenon. In contrast, reports on the presentation of restenosis have consistently shown—without differences between BMS and DES—that more than 50% of the patients with in-stent restenosis present with unstable angina and myocardial infarction [33]. The initiating stimulus for restenosis is endothelial denudation and/or mechanical injury to the vessel wall, triggering an inflammatory response which can also be measured in terms of increase in circulatory markers such as c-reactive protein, amyloid A and fibrinogen [34,35]. Upon implantation, endothelial denudation and the exposure of components of the vascular extracellular matrix like collagen causes activation and adhesion of platelets. As well, the stent struts cause, proportionally to their thickness and dependent on the design of the stent, a disturbance in the flow that triggers the formation of large fibrin clots, further causing flow disturbances close to the struts and activating homeostatic reactions characterized by phagocyte invasion. In this context, the immunosuppressive action of the drug eluted might have paradoxically negative effects, inhibiting tissue repair reaction and fibrin removal. These processes might explain the acute presentation of restenosis, as the activation of the thrombotic cascade ensuing from incomplete thrombin removal, although not causing occlusive thrombus, activates the recruitment of inflammatory cells such as monocytes, T cells, neutrophils via expression of adhesion molecules (ICAM, VCAM, intercellular, and vascular adhesion molecules), production of chemoattractant molecules (MCP-1 or monocyte chemoattractant protein-1, Interleukin (IL)-8) by endothelial, and smooth muscle cells and the production of growth factor such as PDGF (platelet-derived growth factor), bFGF (fibroblast growth factor), TGF (transforming growth factor)-beta, IGF (insulin-like growth factor), VEGF (vascular endothelial growth factor, and thrombin (Table 1, Figure 3).
The cytokines involved in these processes include IL-1, IL-6, and TNF-alpha [36]. This cascade is believed to activate a self-sustaining positive feedback loop of autocrine/paracrine messages that leads to stimulation of vascular smooth muscle cells growth and the production of extracellular matrix components. Matrix metalloproteinases activated by plasmin and their tissue inhibitors are also important mediators of this remodeling process. The formation of an endothelium is a critical step in these processes: an efficient endothelium may inhibit smooth muscle cell proliferation and neointimal growth; impaired neoendothelialization is associated with increased neointima proliferation [37]. However, studies have shown that stenting with BMS, and even more with of first generation DES, inhibits endothelialization [38,39]. Several lines of evidence also suggest that the above signaling cascades following coronary damage and stent implantation may also mobilize bone marrow-derived CD34-postive stem cells, which may then differentiate along endothelial or smooth muscle cell lines, in a balance between endothelial-like stem cell responses that favor reendothelialization and smooth muscle-like stem cell responses that promote restenosis [40].
A number of potential therapies have attempted to address or reduce the incidence of restenosis. Based on the concept that oxidative stress may play an important regulatory role in inflammatory states (also supported by the fact that elevated systemic level of oxidative stress markers predict restenosis [41]), a number of trials have tested the effects of antioxidants on the incidence of restenosis. Probucol, a lipid-lowering agent with strong antioxidant properties, has shown to decrease the risk of restenosis and major adverse events by 41% and 31% in a meta-analysis of 15 studies and 859 patients [42]. Probucol-eluting stents have also been tested, but they did not show superiority as compared to newer-generation DES in a large randomized trial [43]. The use of other drugs, such as sirolimus, colchicine, prednisolone and other reviewed in [44] has shown variable results and will need to be further investigated.
4. The Mechanisms of Stent Thrombosis
The pathophysiology of in-stent thrombosis is complex and multifactorial, and the vascular responses to stent implantation are only one of the players [9]. An incomplete healing and an impaired endothelization [5] resulting from the toxic effect of stent drug and/or polymer are however important risk factor.
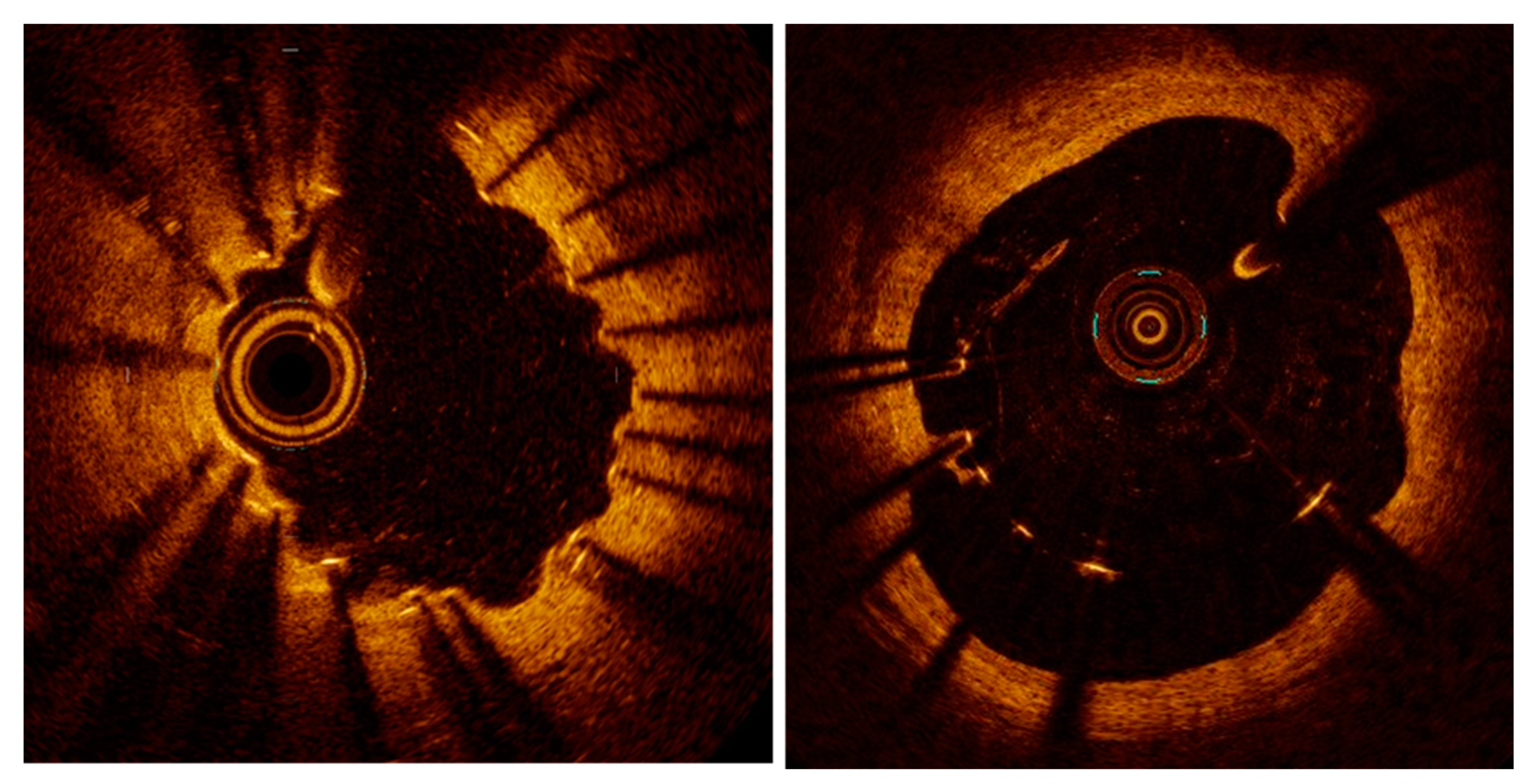
As described above, local and systemic inflammation promote neointimal proliferation, resulting in the pathogenesis of in-stent restenosis. More importantly, chronic low-grade inflammation is also felt to play a primary role in the pathogenesis of stent thrombosis [45,46,47,48]. In particular, as originally observed by the group of Virmani, type IV hypersensitivity, or foreign body-induced activation may be recruited, resulting in a delayed or inefficient stent reendothelialization (Figure 3). These inflammatory processes cause vascular positive remodeling (vessel wall enlargement) and subsequent stent malapposition [47,49], which, in turn, cause flow turbulence and platelet activation [9] (Figure 4).
The histology underlying these phenomena has been investigated in several studies [49]. Eosinophil infiltrates surrounding stent struts may result from a reaction against the metal of the stent, usually 316L stainless steel in the case of BMS and first-generation DES [50]. To this regard, modern stents based on alloys such as platinum chromium or cobalt chromium have a lower nickel content, and other materials, such as zinc-based scaffolds, are being tested with promising results [51]. Further, triggers for hypersensitivity reactions may be the antirestenotic drug and/or the polymer carrying the drug. Polymers in particular have been shown to produce hypersensitivity reactions in humans and porcine models and several case series demonstrating inflammation around a DES have been published [52,53]. These eosinophilic infiltrates have been proposed to result in a specific form arteritis with tissue necrosis and erosion around the stent struts, causing the typical appearance denominated “peri-strut low intensity areas”, positive vessel remodeling with aneurysma and evaginations (Figure 5).
The above processes may also lead to a direct activation of the coagulation cascade and platelet activation. An analysis of thrombus composition in the PRESTIGE cooperation (Prevention of Late Stent Thrombosis by an Interdisciplinary Global European Effort) provides important insights into the pathological processes leading to thrombus formation. In this study, a total of 253 thrombus specimens were analyzed, the majority (68.8%) from cases of late stent thrombosis. Histology showed leukocyte infiltration, neutrophil extracellular traps and eosinophils, with a higher incidence in late stent thromboses of DES [54]. In the RADAR (Research on Adverse Drug Events and Reports) database, DES-induced hypersensitivity reactions were also associated with systemic clinical manifestations suggestive of allergy, including nonurticarial rash, hives, dyspnea, myalgia/arthralgia, itching, and blisters. In a recent imaging study, putative causes of stent thrombosis that could be caused by an inappropriate remodeling process triggered by inflammation were found in 98% of patients with late stent thrombosis. The most frequent findings included strut malapposition (34.5%), neoatherosclerosis (27.6%), uncovered struts (12.1%), and stent under expansion (6.9%) [55].
While the subsequent development of bioresorbable polymers does not seem to have been associated with additional advantages as compared to the antithrombotic fluoropolymer [56,57,58], the progressive reduction of stent strut thickness has led to improved healing and reduced incidence of stent thrombosis [27,59,60,61]. Given the high mortality associated with stent thrombosis, further progress in understanding its mechanisms and its prevention are of primary importance. Medical therapy has a very important impact on the processes described above. Antiplatelet agents are clearly the mainstay in the prevention of thrombosis in the arterial tree. Complementing their effect on platelet aggregation, their anti-inflammatory and endothelium-protective “pleiotropic” effects, as recently demonstrated in the EST (Endothelium, Stent, and Thrombosis) trial and reviewed in [34,45], may have an important additional role. A similar effect of statins has been hypothesized based on observations from the BASKET (Basel Stent Kosten Effektivität Trial) and the Korean Acute Myocardial Infarction Registry [62,63], but remains to be tested in specific trials.
5. Neoatherogenesis: A Long-Term Complication
The chronic inflammation and endothelial dysfunction discussed above as causes of restenosis may also induce late de novo neoatherosclerosis. In a recent imaging study, lipid neointima was shown in 40% of the cases of in-stent restenosis, calcific lesions were reported in a similar incidence, and thin-cap fibroatheromas was observed in up to 15%. The frequency of these forms of neoatherosclerosis was significantly greater with DES than BMS (48.4% vs. 23.5%, p = 0.018), even though lesions in BMS had a thinner cap [64] (Figure 6).
Backscatter intravascular ultrasound evaluation confirmed that the proportion of restenotic lesions containing lipid pools is larger in DES as compared to BMS, suggesting that the formation of a neoplaque (neoatherosclerosis) may in part be proportionally more important in these devices [65]. Although not specific for DES versus BMS, neoatherosclerosis is therefore proportionally a larger issue for modern stents, which appear to be less at risk for fibrotic restenosis and stent thrombosis. Data from two large registry studies examining the characteristics and possible mechanisms of very late stent failure and comparing early- with new-generation DES confirm that the three most common causes of very late DES failure include strut malapposition (34.5%), neoatherosclerosis (27.6%), and uncovered struts (12.1%) [55]. Importantly, the time pattern of the mechanisms of restenosis appears to be influenced by its pathophysiology: while early (<1 year) in-stent restenosis in second generation DES appears to be (similar to that of BMS) mainly caused by intimal hyperplasia, late (>1 year) restenosis is most frequently caused by neotherosclerosis [66]. Specifically, this latter type of lesions was characterized by higher prevalence of lipid-laden neointima, thick- or thin-cap fibroatheroma, neovessels, microcalcifications, and macrophage pools. Of importance, the mechanisms that lead to the mid-term inhibition of tissue proliferation might actually contribute to the pathophysiology of long-term neoatherosclerosis. An impaired endothelialization leads to decreased nitric oxide production, loose intercellular junctions (with subsequent loss of barrier function) and decreased exposure of antithrombotic barriers, all of which promote infiltration, retention, and increased expression of foamy macrophages within the neointima [67]. As for native atherosclerosis, macrophage cells group to form a fibroatheromatous plaque (“intimal xanthoma”), which can then evolve into a rupture-prone thin-cap fibrous atheroma [68]. Apoptosis of macrophages may also results in the formation of a necrotic core and calcifications may also be present [69]. An impaired endothelialization (and thick stent struts) also cause blood flow turbulence, modifying wall shear stress and local hemodynamic forces and leading to a functionally impaired endothelium [70]. An ineffective endothelial barrier and the expression of adhesion molecules (failing the inhibition of the nitric oxide pathway) results the penetration of inflammatory cells lipoprotein and proteoglycans into the subendothelial space. Most of the drugs released by current DES devices inhibit the mammalian target of rapamycin (mTOR), a phosphatidylinositol 3-kinase-related kinase (PIKK) belonging to the family of serine/threonine protein kinases [71]. mTOR is involved in regulating a number of fundamental cell processes from protein synthesis to autophagy, and the mTORC1 (mTOR complex 1, consisting of mTOR and RAPTOR, regulatory associated protein of mTOR), mammalian LST8/G-protein β-subunit like protein and lethal with sec thirteen 8) regulates the activity of p70, which is vital for cellular proliferation and migration of both endothelial and smooth muscle. By inhibiting p70, mTOR inhibition leads to the prevention of restenosis after injury [72]. However, mTORC1 also promotes cell growth by suppressing protein catabolism, most notably autophagy. As well, mTOR also modulates inflammatory reactions as it blocks T-cell activation, and is also involved in adipogenesis and lipogenesis. Exposure to mTOR inhibitors increases vascular permeability, which might promote neoatherosclerosis [73]. Further, mTORC2 positively regulates Akt activity, whose function includes endothelial survival as well as transcriptional control of VE-cadherin [72]. Translocation of VE cadherin from the membrane to the intracellular space contributes to impaired endothelial barrier function (mechanisms described in [72]). Although these mechanisms still remain incompletely understood, it is however clear that they may depend on ancillary, off-target effects of the antiproliferative drug eluted by the stent. Evidence to this regard is summarized in recent reviews, which also discuss the chances offered by alternative, more specific, drugs [72].
6. Conclusions
Percutaneous coronary interventions cause mechanical injury and vascular inflammation which, along with the presence of a foreign body and the biochemical processes triggered by the polymer and the drug eluted by the stent, cause complex interactions between endothelial cells, smooth muscle cells, platelets, and inflammatory cells including neutrophils, monocytes, and lymphocytes. The activation of signaling cascades leads to repair and vascular healing but also adverse arterial remodeling, neointimal proliferation, and restenosis. Although effectively reduced after the introduction of DES, restenosis, and thrombosis remain important issues with important clinical implications. While the current generations of DES appear to have reached an innovation plateau that allows safe treatment of complex anatomies, future generations of devices, with a better mechanical profile and improved biocompatibility are still awaited.
Funding
T.G. is DZHK professor. There was no support for this specific work.
Conflicts of Interest
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Timmis, A.; Townsend, N.; Gale, C.; Grobbee, R.; Maniadakis, N.; Flather, M.; Wilkins, E.; Wright, L.; Vos, R.; Bax, J.; et al. European Society of Cardiology: Cardiovascular Disease Statistics 2017. Eur. Heart J. 2018, 39, 508–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moses, J.W.; Leon, M.B.; Popma, J.J.; Fitzgerald, P.J.; Holmes, D.R.; O‘Shaughnessy, C.; Caputo, R.P.; Kereiakes, D.J.; Williams, D.O.; Teirstein, P.S.; et al. Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery. N. Engl. J. Med. 2003, 349, 1315–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waksman, R.; Kirtane, A.J.; Torguson, R.; Cohen, D.J.; Ryan, T.; Raber, L.; Applegate, R.; Waxman, S.; Gordon, P.; Kaneshige, K.; et al. Correlates and outcomes of late and very late drug-eluting stent thrombosis: Results from DESERT (International Drug-Eluting Stent Event Registry of Thrombosis). JACC Cardiovasc. Interv. 2014, 7, 1093–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claessen, B.E.; Henriques, J.P.; Jaffer, F.A.; Mehran, R.; Piek, J.J.; Dangas, G.D. Stent thrombosis: A clinical perspective. JACC Cardiovasc. Interv. 2014, 7, 1081–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrado, J.; Buckley, L.; Duran, A.; Trujillo, P.; Toldo, S.; Valle Raleigh, J.; Abbate, A.; Biondi-Zoccai, G.; Guzman, L.A. Restenosis, Stent Thrombosis, and Bleeding Complications: Navigating Between Scylla and Charybdis. J. Am. Coll. Cardiol. 2018, 71, 1676–1695. [Google Scholar] [CrossRef] [PubMed]
- Iakovou, I.; Schmidt, T.; Bonizzoni, E.; Ge, L.; Sangiorgi, G.M.; Stankovic, G.; Airoldi, F.; Chieffo, A.; Montorfano, M.; Carlino, M.; et al. Incidence, predictors, and outcome of thrombosis after successful implantation of drug-eluting stents. JAMA 2005, 293, 2126–2130. [Google Scholar] [CrossRef] [PubMed]
- Claessen, B.E.; Dangas, G.D. The quest for the optimal treatment for in-stent restenosis. Catheter. Cardiovasc. Interv. 2018, 92, 300–301. [Google Scholar] [CrossRef]
- Magalhaes, M.A.; Minha, S.; Chen, F.; Torguson, R.; Omar, A.F.; Loh, J.P.; Escarcega, R.O.; Lipinski, M.J.; Baker, N.C.; Kitabata, H.; et al. Clinical presentation and outcomes of coronary in-stent restenosis across 3-stent generations. Circ. Cardiovasc. Interv. 2014, 7, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Gori, T.; Polimeni, A.; Indolfi, C.; Raber, L.; Adriaenssens, T.; Munzel, T. Predictors of stent thrombosis and their implications for clinical practice. Nat. Rev. Cardiol. 2019, 16, 243–256. [Google Scholar] [CrossRef]
- Kawamoto, R.; Hatakeyama, K.; Imamura, T.; Ishikawa, T.; Date, H.; Shibata, Y.; Takenaga, M.; Asada, Y.; Eto, T. Relation of C-reactive protein to restenosis after coronary stent implantation and to restenosis after coronary atherectomy. Am. J. Cardiol. 2004, 94, 104–107. [Google Scholar] [CrossRef]
- Yilmaz, S.; Sen, F.; Unal, S.; Yayla, C.; Ozeke, O.; Aras, D.; Topaloglu, S.; Aydogdu, S. Usefulness of the platelet-to-lymphocyte ratio in predicting bare-metal stent restenosis. Scand. Cardiovasc. J. 2015, 49, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, Y.; Jiang, H.; Ding, X.; Zhu, R.; Li, B.; Zhao, Y. Plasma levels of interleukin 18, interleukin 10, and matrix metalloproteinase-9 and -137G/C polymorphism of interleukin 18 are associated with incidence of in-stent restenosis after percutaneous coronary intervention. Inflammation 2013, 36, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, H.; Munzel, T.; Gori, T. Coronary Stent Thrombosis- Predictors and Prevention. Dtsch. Arztebl. Int. 2020, 117, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Tada, T.; Byrne, R.A.; Simunovic, I.; King, L.A.; Cassese, S.; Joner, M.; Fusaro, M.; Schneider, S.; Schulz, S.; Ibrahim, T.; et al. Risk of stent thrombosis among bare-metal stents, first-generation drug-eluting stents, and second-generation drug-eluting stents: Results from a registry of 18,334 patients. JACC Cardiovasc. Interv. 2013, 6, 1267–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmerini, T.; Biondi-Zoccai, G.; Della Riva, D.; Mariani, A.; Genereux, P.; Branzi, A.; Stone, G.W. Stent thrombosis with drug-eluting stents: Is the paradigm shifting? J. Am. Coll. Cardiol. 2013, 62, 1915–1921. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, A.; Jinnouchi, H.; Torii, S.; Virmani, R.; Finn, A.V. Understanding the Impact of Stent and Scaffold Material and Strut Design on Coronary Artery Thrombosis from the Basic and Clinical Points of View. Bioengineering 2018, 5, 71. [Google Scholar] [CrossRef] [Green Version]
- Vorpahl, M.; Yazdani, S.K.; Nakano, M.; Ladich, E.; Kolodgie, F.D.; Finn, A.V.; Virmani, R. Pathobiology of stent thrombosis after drug-eluting stent implantation. Curr. Pharm. Des. 2010, 16, 4064–4071. [Google Scholar] [CrossRef]
- Finn, A.V.; Nakazawa, G.; Joner, M.; Kolodgie, F.D.; Mont, E.K.; Gold, H.K.; Virmani, R. Vascular responses to drug eluting stents: Importance of delayed healing. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1500–1510. [Google Scholar] [CrossRef]
- Nakazawa, G.; Finn, A.V.; Vorpahl, M.; Ladich, E.R.; Kolodgie, F.D.; Virmani, R. Coronary responses and differential mechanisms of late stent thrombosis attributed to first-generation sirolimus- and paclitaxel-eluting stents. J. Am. Coll. Cardiol. 2011, 57, 390–398. [Google Scholar] [CrossRef] [Green Version]
- Borovac, J.A.; D‘Amario, D.; Vergallo, R.; Porto, I.; Bisignani, A.; Galli, M.; Annibali, G.; Montone, R.A.; Leone, A.M.; Niccoli, G.; et al. Neoatherosclerosis after drug-eluting stent implantation: A novel clinical and therapeutic challenge. Eur. Heart J. Cardiovasc. Pharmacother. 2019, 5, 105–116. [Google Scholar] [CrossRef]
- Garcia-Garcia, H.M.; McFadden, E.P.; Farb, A.; Mehran, R.; Stone, G.W.; Spertus, J.; Onuma, Y.; Morel, M.A.; van Es, G.A.; Zuckerman, B.; et al. Standardized End Point Definitions for Coronary Intervention Trials: The Academic Research Consortium-2 Consensus Document. Circulation 2018, 137, 2635–2650. [Google Scholar] [CrossRef] [PubMed]
- Kruger-Genge, A.; Hiebl, B.; Franke, R.P.; Lendlein, A.; Jung, F. Effects of Tacrolimus or Sirolimus on the adhesion of vascular wall cells: Controlled in-vitro comparison study. Clin. Hemorheol. Microcirc. 2017, 67, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Otsuka, F.; Gupta, A.; Jinnouchi, H.; Torii, S.; Harari, E.; Virmani, R.; Finn, A.V. Revisiting the role of durable polymers in cardiovascular devices. Expert Rev. Cardiovasc. Ther. 2017, 15, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Szott, L.M.; Irvin, C.A.; Trollsas, M.; Hossainy, S.; Ratner, B.D. Blood compatibility assessment of polymers used in drug eluting stent coatings. Biointerphases 2016, 11, 029806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torii, S.; Cheng, Q.; Mori, H.; Lipinski, M.J.; Acampado, E.; Perkins, L.E.L.; Hossainy, S.F.; Pacetti, S.D.; Kolodgie, F.D.; Virmani, R.; et al. Acute thrombogenicity of fluoropolymer-coated versus biodegradable and polymer-free stents. EuroIntervention 2019, 14, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Bangalore, S.; Toklu, B.; Patel, N.; Feit, F.; Stone, G.W. Newer-Generation Ultrathin Strut Drug-Eluting Stents Versus Older Second-Generation Thicker Strut Drug-Eluting Stents for Coronary Artery Disease. Circulation 2018, 138, 2216–2226. [Google Scholar] [CrossRef]
- Iantorno, M.; Lipinski, M.J.; Garcia-Garcia, H.M.; Forrestal, B.J.; Rogers, T.; Gajanana, D.; Buchanan, K.D.; Torguson, R.; Weintraub, W.S.; Waksman, R. Meta-Analysis of the Impact of Strut Thickness on Outcomes in Patients With Drug-Eluting Stents in a Coronary Artery. Am. J. Cardiol. 2018, 122, 1652–1660. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Otake, H.; Shinke, T.; Nakagawa, M.; Hariki, H.; Inoue, T.; Osue, T.; Hiranuma, N.; Nishio, R.; Kinutani, H.; et al. Two-year vessel healing after everolimus-eluting stent implantation: Serial assessment by optical coherence tomography. J. Cardiol. 2015, 65, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, R.; Hao, H.; Imanaka, T.; Shibuya, M.; Ueda, Y.; Tsujimoto, M.; Ishibashi-Ueda, H.; Hirota, S. Initial pathological responses of second-generation everolimus-eluting stents implantation in Japanese coronary arteries: Comparison with first-generation sirolimus-eluting stents. J. Cardiol. 2018, 71, 452–457. [Google Scholar] [CrossRef] [Green Version]
- Picard, F.; Pighi, M.; de Hemptinne, Q.; Airaksinen, J.; Vinco, G.; de Pommereau, A.; Biancari, F.; Varenne, O. Comparison of the biodegradable polymer everolimus-eluting stent with contemporary drug-eluting stents: A systematic review and meta-analysis. Int. J. Cardiol. 2019, 278, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Anadol, R.; Gori, T. The mechanisms of late scaffold thrombosis. Clin. Hemorheol. Microcirc. 2017, 67, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Kereiakes, D.J.; Onuma, Y.; Serruys, P.W.; Stone, G.W. Bioresorbable Vascular Scaffolds for Coronary Revascularization. Circulation 2016, 134, 168–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.S.; John, J.M.; Chew, D.P.; Lee, D.S.; Ellis, S.G.; Bhatt, D.L. Bare metal stent restenosis is not a benign clinical entity. Am. Heart J. 2006, 151, 1260–1264. [Google Scholar] [CrossRef] [PubMed]
- Schnorbus, B.; Daiber, A.; Jurk, K.; Warnke, S.; Koenig, J.; Lackner, K.J.; Munzel, T.; Gori, T. Effects of clopidogrel vs. prasugrel vs. ticagrelor on endothelial function, inflammatory parameters, and platelet function in patients with acute coronary syndrome undergoing coronary artery stenting: A randomized, blinded, parallel study. Eur. Heart J. 2020, 41, 3144–3152. [Google Scholar] [CrossRef]
- Gori, T. Endothelial Function: A Short Guide for the Interventional Cardiologist. Int. J. Mol. Sci. 2018, 19, 3838. [Google Scholar] [CrossRef] [Green Version]
- Donners, M.M.; Daemen, M.J.; Cleutjens, K.B.; Heeneman, S. Inflammation and restenosis: Implications for therapy. Ann. Med. 2003, 35, 523–531. [Google Scholar] [CrossRef]
- Christen, T.; Verin, V.; Bochaton-Piallat, M.; Popowski, Y.; Ramaekers, F.; Debruyne, P.; Camenzind, E.; van Eys, G.; Gabbiani, G. Mechanisms of neointima formation and remodeling in the porcine coronary artery. Circulation 2001, 103, 882–888. [Google Scholar] [CrossRef] [Green Version]
- Puricel, S.; Kallinikou, Z.; Espinola, J.; Arroyo, D.; Goy, J.J.; Stauffer, J.C.; Baeriswyl, G.; Smits, P.C.; Cook, S.; Togni, M. Comparison of endothelium-dependent and -independent vasomotor response after abluminal biodegradable polymer biolimus-eluting stent and persistent polymer everolimus-eluting stent implantation (COMPARE-IT). Int. J. Cardiol. 2016, 202, 525–531. [Google Scholar] [CrossRef]
- Togni, M.; Raber, L.; Cocchia, R.; Wenaweser, P.; Cook, S.; Windecker, S.; Meier, B.; Hess, O.M. Local vascular dysfunction after coronary paclitaxel-eluting stent implantation. Int. J. Cardiol. 2007, 120, 212–220. [Google Scholar] [CrossRef]
- Inoue, T.; Croce, K.; Morooka, T.; Sakuma, M.; Node, K.; Simon, D.I. Vascular inflammation and repair: Implications for re-endothelialization, restenosis, and stent thrombosis. JACC Cardiovasc. Interv. 2011, 4, 1057–1066. [Google Scholar] [CrossRef] [Green Version]
- Moldoveanu, E.; Mut-Vitcu, B.; Tanaseanu, G.R.; Marta, D.S.; Manea, G.; Kosaka, T.; Vidulescu, C.; Tanaseanu, C. Low basal levels of circulating adiponectin in patients undergoing coronary stenting predict in-stent restenosis, independently of basal levels of inflammatory markers: Lipoprotein associated phospholipase A2, and myeloperoxidase. Clin. Biochem. 2008, 41, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, M.; Lu, H.; Qiao, W.; Xi, D.; Luo, T.; Xiong, H.; Guo, Z. Effects of probucol on restenosis after percutaneous coronary intervention: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0124021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kufner, S.; Sorges, J.; Mehilli, J.; Cassese, S.; Repp, J.; Wiebe, J.; Lohaus, R.; Lahmann, A.; Rheude, T.; Ibrahim, T.; et al. Randomized Trial of Polymer-Free Sirolimus- and Probucol-Eluting Stents Versus Durable Polymer Zotarolimus-Eluting Stents: 5-Year Results of the ISAR-TEST-5 Trial. JACC Cardiovasc. Interv. 2016, 9, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Guerra, E.; Byrne, R.A.; Kastrati, A. Pharmacological inhibition of coronary restenosis: Systemic and local approaches. Expert Opin. Pharmacother. 2014, 15, 2155–2171. [Google Scholar] [CrossRef]
- Ullrich, H.; Gori, T. The pleiotropic effects of antiplatelet therapies. Clin. Hemorheol. Microcirc. 2019, 73, 29–34. [Google Scholar] [CrossRef]
- Cook, S.; Ladich, E.; Nakazawa, G.; Eshtehardi, P.; Neidhart, M.; Vogel, R.; Togni, M.; Wenaweser, P.; Billinger, M.; Seiler, C.; et al. Correlation of intravascular ultrasound findings with histopathological analysis of thrombus aspirates in patients with very late drug-eluting stent thrombosis. Circulation 2009, 120, 391–399. [Google Scholar] [CrossRef]
- Nebeker, J.R.; Virmani, R.; Bennett, C.L.; Hoffman, J.M.; Samore, M.H.; Alvarez, J.; Davidson, C.J.; McKoy, J.M.; Raisch, D.W.; Whisenant, B.K.; et al. Hypersensitivity cases associated with drug-eluting coronary stents: A review of available cases from the Research on Adverse Drug Events and Reports (RADAR) project. J. Am. Coll. Cardiol. 2006, 47, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Virmani, R.; Guagliumi, G.; Farb, A.; Musumeci, G.; Grieco, N.; Motta, T.; Mihalcsik, L.; Tespili, M.; Valsecchi, O.; Kolodgie, F.D. Localized hypersensitivity and late coronary thrombosis secondary to a sirolimus-eluting stent: Should we be cautious? Circulation 2004, 109, 701–705. [Google Scholar] [CrossRef] [Green Version]
- Chaabane, C.; Otsuka, F.; Virmani, R.; Bochaton-Piallat, M.L. Biological responses in stented arteries. Cardiovasc. Res. 2013, 99, 353–363. [Google Scholar] [CrossRef]
- Koster, R.; Vieluf, D.; Kiehn, M.; Sommerauer, M.; Kahler, J.; Baldus, S.; Meinertz, T.; Hamm, C.W. Nickel and molybdenum contact allergies in patients with coronary in-stent restenosis. Lancet 2000, 356, 1895–1897. [Google Scholar] [CrossRef]
- Hiebl, B.; Nennig, E.; Schiestel, S.; Kovacs, A.; Jung, F.; Fischer, H. Biocompatibility of a novel zinc stent with a closed-cell-design. Clin. Hemorheol. Microcirc. 2015, 61, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Jinnouchi, H.; Kutyna, M.; Torii, S.; Cheng, Q.; Sakamoto, A.; Guo, L.; Cornelissen, A.; Perkins, L.E.L.; Hossainy, S.F.; Pacetti, S.D.; et al. Comparison of Acute Thrombogenicity and Albumin Adsorption in Three Different Durable Polymer Coronary Drug-Eluting Stents. EuroIntervention 2020. [Google Scholar] [CrossRef]
- Mori, M.; Sakata, K.; Yokawa, J.; Nakanishi, C.; Murai, K.; Okada, H.; Shimojima, M.; Yoshida, S.; Yoshioka, K.; Takuwa, Y.; et al. Everolimus-Eluting Biodegradable Abluminal Coating Stent versus Durable Conformal Coating Stent: Termination of the Inflammatory Response Associated with Neointimal Healing in a Porcine Coronary Model. J. Interv. Cardiol. 2020, 2020, 1956015. [Google Scholar] [CrossRef] [PubMed]
- Riegger, J.; Byrne, R.A.; Joner, M.; Chandraratne, S.; Gershlick, A.H.; Ten Berg, J.M.; Adriaenssens, T.; Guagliumi, G.; Godschalk, T.C.; Neumann, F.J.; et al. Histopathological evaluation of thrombus in patients presenting with stent thrombosis. A multicenter European study: A report of the prevention of late stent thrombosis by an interdisciplinary global European effort consortium. Eur. Heart J. 2016, 37, 1538–1549. [Google Scholar] [CrossRef] [Green Version]
- Taniwaki, M.; Radu, M.D.; Zaugg, S.; Amabile, N.; Garcia-Garcia, H.M.; Yamaji, K.; Jorgensen, E.; Kelbaek, H.; Pilgrim, T.; Caussin, C.; et al. Mechanisms of Very Late Drug-Eluting Stent Thrombosis Assessed by Optical Coherence Tomography. Circulation 2016, 133, 650–660. [Google Scholar] [CrossRef] [Green Version]
- Polimeni, A.; Sorrentino, S.; Spaccarotella, C.; Mongiardo, A.; Sabatino, J.; De Rosa, S.; Gori, T.; Indolfi, C. Stent Thrombosis After Percutaneous Coronary Intervention: From Bare-Metal to the Last Generation of Drug-Eluting Stents. Cardiol. Clin. 2020, 38, 639–647. [Google Scholar] [CrossRef]
- de Winter, R.J.; Katagiri, Y.; Asano, T.; Milewski, K.P.; Lurz, P.; Buszman, P.; Jessurun, G.A.J.; Koch, K.T.; Troquay, R.P.T.; Hamer, B.J.B.; et al. A sirolimus-eluting bioabsorbable polymer-coated stent (MiStent) versus an everolimus-eluting durable polymer stent (Xience) after percutaneous coronary intervention (DESSOLVE III): A randomised, single-blind, multicentre, non-inferiority, phase 3 trial. Lancet 2018, 391, 431–440. [Google Scholar] [CrossRef]
- Cassese, S.; Ndrepepa, G.; Byrne, R.A.; Kufner, S.; Lahmann, A.L.; Mankerious, N.; Xhepa, E.; Laugwitz, K.L.; Schunkert, H.; Fusaro, M.; et al. Outcomes of patients treated with ultrathin-strut biodegradable polymer sirolimus-eluting stents versus fluoropolymer-based everolimus-eluting stents: A meta-analysis of randomised trials. EuroIntervention 2018, 14, 224–231. [Google Scholar] [CrossRef]
- El-Hayek, G.; Bangalore, S.; Casso Dominguez, A.; Devireddy, C.; Jaber, W.; Kumar, G.; Mavromatis, K.; Tamis-Holland, J.; Samady, H. Meta-Analysis of Randomized Clinical Trials Comparing Biodegradable Polymer Drug-Eluting Stent to Second-Generation Durable Polymer Drug-Eluting Stents. JACC Cardiovasc. Interv. 2017, 10, 462–473. [Google Scholar] [CrossRef]
- Yamaji, K.; Zanchin, T.; Zanchin, C.; Stortecky, S.; Koskinas, K.C.; Hunziker, L.; Praz, F.; Blochlinger, S.; Moro, C.; Moschovitis, A.; et al. Unselected Use of Ultrathin Strut Biodegradable Polymer Sirolimus-Eluting Stent Versus Durable Polymer Everolimus-Eluting Stent for Coronary Revascularization. Circ. Cardiovasc. Interv. 2018, 11, e006741. [Google Scholar] [CrossRef]
- Zhu, P.; Zhou, X.; Zhang, C.; Li, H.; Zhang, Z.; Song, Z. Safety and efficacy of ultrathin strut biodegradable polymer sirolimus-eluting stent versus durable polymer drug-eluting stents: A meta-analysis of randomized trials. BMC Cardiovasc. Disord. 2018, 18, 170. [Google Scholar] [CrossRef] [PubMed]
- Jeger, R.V.; Brunner-La Rocca, H.P.; Bertel, O.; Kiowski, W.; Pfisterer, M.E.; Kaiser, C.A.; Investigators, B. Stent thrombosis after coronary stent implantation: A protective effect of high-dose statin therapy? Cardiology 2013, 126, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.C.; Ahn, Y.; Hong, Y.J.; Kim, J.H.; Jeong, M.H.; Kim, Y.J.; Chae, S.C.; Cho, M.C.; Other Korea Acute Myocardial Infarction Registry, I. Statin therapy to reduce stent thrombosis in acute myocardial infarction patients with elevated high-sensitivity C-reactive protein. Int. J. Cardiol. 2013, 167, 1848–1853. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, D.; Yasumura, K.; Nakamura, H.; Matsuhiro, Y.; Yasumoto, K.; Tanaka, A.; Matsunaga-Lee, Y.; Yano, M.; Yamato, M.; Egami, Y.; et al. Different Neoatherosclerosis Patterns in Drug-Eluting-and Bare-Metal Stent Restenosis—Optical Coherence Tomography Study. Circ. J. 2019, 83, 313–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, H.; Amano, T.; Takashima, H.; Harada, K.; Kitagawa, K.; Suzuki, A.; Kunimura, A.; Shimbo, Y.; Harada, K.; Yoshida, T.; et al. Differences in tissue characterization of restenotic neointima between sirolimus-eluting stent and bare-metal stent: Integrated backscatter intravascular ultrasound analysis for in-stent restenosis. Eur. Heart J. Cardiovasc. Imaging 2013, 14, 996–1001. [Google Scholar] [CrossRef] [Green Version]
- Jinnouchi, H.; Kuramitsu, S.; Shinozaki, T.; Tomoi, Y.; Hiromasa, T.; Kobayashi, Y.; Domei, T.; Soga, Y.; Hyodo, M.; Shirai, S.; et al. Difference of Tissue Characteristics Between Early and Late Restenosis After Second-Generation Drug-Eluting Stents Implantation- An Optical Coherence Tomography Study. Circ. J. 2017, 81, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Joner, M.; Nakazawa, G.; Finn, A.V.; Quee, S.C.; Coleman, L.; Acampado, E.; Wilson, P.S.; Skorija, K.; Cheng, Q.; Xu, X.; et al. Endothelial cell recovery between comparator polymer-based drug-eluting stents. J. Am. Coll. Cardiol. 2008, 52, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Yahagi, K.; Kolodgie, F.D.; Otsuka, F.; Finn, A.V.; Davis, H.R.; Joner, M.; Virmani, R. Pathophysiology of native coronary, vein graft, and in-stent atherosclerosis. Nat. Rev. Cardiol. 2016, 13, 79–98. [Google Scholar] [CrossRef]
- Otsuka, F.; Byrne, R.A.; Yahagi, K.; Mori, H.; Ladich, E.; Fowler, D.R.; Kutys, R.; Xhepa, E.; Kastrati, A.; Virmani, R.; et al. Neoatherosclerosis: Overview of histopathologic findings and implications for intravascular imaging assessment. Eur. Heart J. 2015, 36, 2147–2159. [Google Scholar] [CrossRef] [Green Version]
- Parry, T.J.; Brosius, R.; Thyagarajan, R.; Carter, D.; Argentieri, D.; Falotico, R.; Siekierka, J. Drug-eluting stents: Sirolimus and paclitaxel differentially affect cultured cells and injured arteries. Eur. J. Pharmacol. 2005, 524, 19–29. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Jinnouchi, H.; Guo, L.; Sakamoto, A.; Sato, Y.; Cornelissen, A.; Kawakami, R.; Mori, M.; Torii, S.; Kuntz, S.; Harari, E.; et al. Advances in mammalian target of rapamycin kinase inhibitors: Application to devices used in the treatment of coronary artery disease. Future Med. Chem. 2020, 12, 1181–1195. [Google Scholar] [CrossRef] [PubMed]
- Harari, E.; Guo, L.; Smith, S.L.; Paek, K.H.; Fernandez, R.; Sakamoto, A.; Mori, H.; Kutyna, M.D.; Habib, A.; Torii, S.; et al. Direct Targeting of the mTOR (Mammalian Target of Rapamycin) Kinase Improves Endothelial Permeability in Drug-Eluting Stents-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2217–2224. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.Y.; Kim, J.S.; Shin, D.H.; Kim, B.K.; Ko, Y.G.; Choi, D.; Jang, Y.; Hong, M.K. Favorable effect of optimal lipid-lowering therapy on neointimal tissue characteristics after drug-eluting stent implantation: Qualitative optical coherence tomographic analysis. Atherosclerosis 2015, 242, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Liu, Y.; Zhao, F.; Shi, D.; Chen, K. Neoatherosclerosis after Drug-Eluting Stent Implantation: Roles and Mechanisms. Oxid. Med. Cell. Longev. 2016, 2016, 5924234. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Schematic presentation of the different stent generations, from bare metal stents (BMS) to newer generation drug eluting stents (DES). Only some of the several hundred trademark brands are listed, each device type has individual characteristics, the reader is directed to the manufacturer website or specific publications.
Figure 1.
Schematic presentation of the different stent generations, from bare metal stents (BMS) to newer generation drug eluting stents (DES). Only some of the several hundred trademark brands are listed, each device type has individual characteristics, the reader is directed to the manufacturer website or specific publications.

Figure 2.
Representative confocal microscopic (left panels) and scanning electron microscope images (right panels) describing the thrombogenicity of different stent types images of (BMS = bare metal stents; FP: fluoropolymer; FP-EES: fluoro-polymer and everolimus-eluting; BP-SES: bioresorbable polymer with sirolimus; PF-BCS: polymer-free Biolimus A9-coated stents). Left panel: images are obtained using immunofluorescent staining against dual platelet markers (CD61/CD42b) in a swine shunt model. Stents with fluoropolymer (FP-only and FP-EES) were associated with less platelet adhesion. Right panels show evidence of platelet aggregation clot on BMS, BP-SES, and PF-BCS. Image reproduced from [25], with permission.
Figure 2.
Representative confocal microscopic (left panels) and scanning electron microscope images (right panels) describing the thrombogenicity of different stent types images of (BMS = bare metal stents; FP: fluoropolymer; FP-EES: fluoro-polymer and everolimus-eluting; BP-SES: bioresorbable polymer with sirolimus; PF-BCS: polymer-free Biolimus A9-coated stents). Left panel: images are obtained using immunofluorescent staining against dual platelet markers (CD61/CD42b) in a swine shunt model. Stents with fluoropolymer (FP-only and FP-EES) were associated with less platelet adhesion. Right panels show evidence of platelet aggregation clot on BMS, BP-SES, and PF-BCS. Image reproduced from [25], with permission.

Figure 3.
Schematic presentation of the mechanisms leading to restenosis, thrombosis, and neoatherosclerosis.
Figure 3.
Schematic presentation of the mechanisms leading to restenosis, thrombosis, and neoatherosclerosis.

Figure 4.
Examples of peri-strut evaginations (left) and malapposition (right) resulting from positive remodeling (lumen expansion) after stent implantation.
Figure 4.
Examples of peri-strut evaginations (left) and malapposition (right) resulting from positive remodeling (lumen expansion) after stent implantation.

Figure 5.
An optical coherence tomography image showing one-year result after implantation of a bioresorbable scaffold. Malapposed struts (3 o’clock), evaginations (8 to 10 o’clock) and peri-strut low-intensity areas (stars) are evident.
Figure 5.
An optical coherence tomography image showing one-year result after implantation of a bioresorbable scaffold. Malapposed struts (3 o’clock), evaginations (8 to 10 o’clock) and peri-strut low-intensity areas (stars) are evident.

Figure 6.
Optical coherence tomography images on neoatherogenesis. (left): low-backscattering areas (~) with high attenuation suggestive of atheroma within the stent borders (stent struts are marked with *). (right): a calcific plaque (+).
Figure 6.
Optical coherence tomography images on neoatherogenesis. (left): low-backscattering areas (~) with high attenuation suggestive of atheroma within the stent borders (stent struts are marked with *). (right): a calcific plaque (+).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Mechanisms and mediators involved in restenosis.
| Mechanism(s) Involved | Mediators and Signaling | |
|---|---|---|
| Response to Damage | ||
| Proliferation | Migration of smooth muscle cells, production of membrane metalloproteinases | Cytokines (IL-1, IL-6, TNF-alpha) Growth factors (PDGF, IGF, FGF, VEGF) |
| Remodeling | Remodeling of the neointima, deposition of extracellular matrix, neoatherogenesis | Macrophages/Foam cells Cytokines (IFNgamma) Growth factors (PDGF, TGFbeta, IGF, VEGF) |
| Thrombus Formation | Adhesion and activation of platelets Recruitment and diapedesis of Inflammatory cells | Expression of vWF and TF Turbolent flow Nitric oxide, Thrombin Adhesion molecules Chemotactic factors (IL-8, MCP-1) Cytokines (IL-1, IL-6, TNF-alpha) Growth factors (PDGF, Thrombin) |
TNF: tumor necrosis factor; vWF: von Willebrand factor; IFN: interferon; FGF: fibroblast growth factor; all other abbreviations as in text.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gori, T. Vascular Wall Reactions to Coronary Stents—Clinical Implications for Stent Failure. Life 2021, 11, 63. https://0-doi-org.brum.beds.ac.uk/10.3390/life11010063
AMA Style
Gori T. Vascular Wall Reactions to Coronary Stents—Clinical Implications for Stent Failure. Life. 2021; 11(1):63. https://0-doi-org.brum.beds.ac.uk/10.3390/life11010063
Chicago/Turabian StyleGori, Tommaso. 2021. "Vascular Wall Reactions to Coronary Stents—Clinical Implications for Stent Failure" Life 11, no. 1: 63. https://0-doi-org.brum.beds.ac.uk/10.3390/life11010063
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.