
The Eastern Nebraska Salt Marsh Microbiome Is Well Adapted to an Alkaline and Extreme Saline Environment


College of Science and Technology, Bellevue University, Bellevue, NE 68005, USA
*
Author to whom correspondence should be addressed.
Life 2021, 11(5), 446; https://0-doi-org.brum.beds.ac.uk/10.3390/life11050446
Submission received: 25 March 2021
/
Revised: 28 April 2021
/
Accepted: 12 May 2021
/
Published: 15 May 2021
(This article belongs to the Section Microbiology)
Abstract
:The Eastern Nebraska Salt Marshes contain a unique, alkaline, and saline wetland area that is a remnant of prehistoric oceans that once covered this area. The microbial composition of these salt marshes, identified by metagenomic sequencing, appears to be different from well-studied coastal salt marshes as it contains bacterial genera that have only been found in cold-adapted, alkaline, saline environments. For example, Rubribacterium was only isolated before from an Eastern Siberian soda lake, but appears to be one of the most abundant bacteria present at the time of sampling of the Eastern Nebraska Salt Marshes. Further enrichment, followed by genome sequencing and metagenomic binning, revealed the presence of several halophilic, alkalophilic bacteria that play important roles in sulfur and carbon cycling, as well as in nitrogen fixation within this ecosystem. Photosynthetic sulfur bacteria, belonging to Prosthecochloris and Marichromatium, and chemotrophic sulfur bacteria of the genera Sulfurimonas, Arcobacter, and Thiomicrospira produce valuable oxidized sulfur compounds for algal and plant growth, while alkaliphilic, sulfur-reducing bacteria belonging to Sulfurospirillum help balance the sulfur cycle. This metagenome-based study provides a baseline to understand the complex, but balanced, syntrophic microbial interactions that occur in this unique inland salt marsh environment.
1. Introduction
Over 100 million years ago, Nebraska was covered by a vast sea. Now, all that is left of that are widespread salt marshes, which makes this one of the few places in the U.S. where the naturally occurring groundwater is saline [1]. The alkaline and saline marshes in Nebraska are part of a rare wetland type that occurs in the western Sandhills, the North Platte Valley, and the valley of Salt Creek and Little Salt Creek [2]. In these areas, the salinity is so high that it creates a unique natural community in the middle of the tallgrass prairie that is selective to the growth of only salt-adapted species. For example, the Salt Creek tiger beetle cannot be found anywhere else on Earth [3,4], and Saltwort (Salicornia rubra) is a state-listed endangered plant species that, in Nebraska, is only found in these saline wetlands [5,6].
However, urban expansion and consequential changes to the hydrological systems have endangered the continuing existence of these unique salt marshes. Only about 4000 acres remain scattered throughout the region of the estimated 20,000 acres that once existed [1].
In 2003, the Nature Conservancy helped form the Saline Wetlands Conservation Partnership as a way to work collaboratively among organizations concerned with preserving the last remnants of Nebraska’s saline wetlands. One of the ongoing scientific efforts is to understand exactly how these wetlands became salty, besides studying the larger ecological impact of these unique habitats. In particular, the smaller salt marsh area of the Salt Creek valley, near Lincoln, NE, is the most endangered by growing urban development. Most of the nearby surrounding area of that lower Salt Creek valley area has been used for the development of roads, industrial and residential properties, or agricultural expansion (Figure 1A). The Marsh Wren saline wetland restoration project was started to conserve and restore approximately 150 acres containing saline wetlands and other habitats of the lower Salt Creek valley and was completed in 2017 [7]. A combination of traditional restoration methods and physical manipulation of hydrology through pumping of saline groundwater to the wetland surface was used.
The bacterial composition of coastal salt marshes has been well studied on a global scale [8,9,10]. On the contrary, these Nebraska salt marshes are thousands of miles away from any coast and have not been part of a larger saline body of water for apparently millions of years. Very little is known about the microbial composition of these salt marshes, how this compares to other coastal or marine environments, or how the bacterial species and their metabolism potentially impact the rest of the local ecosystem. An initial study of the microbial composition of these systems could also be used as a baseline and future indicator of the success of conservation and restoration efforts in these areas. We therefore set out to study the microbial composition of these land-locked saltmarshes that are a relic of ancient oceans that once covered the middle of North America.
2. Materials and Methods
2.1. Environmental Sampling
We collected samples from three areas near the crossing of Small Salt Creek and Salt Creek near Lincoln, NE, in early October 2020 (Lat. 40°52′54.42″ N; Lon. 96°39′35.82″ W) (Figure 1A). Two samples were taken from a larger pond area (Figure 1B: SM2B and SM2C) approximately 6 m apart, while two others were taken from nearby puddles with a visibly different ecosystem: SM2A showed high levels of insect activity and plant decay, while SM2E had visible indications of white sulfur deposits (Figure 1). All sampling sites had a salinity of >38 PPT (measured with a handheld salinity hydrometer (Fluvial Sea)), nitrate levels of ~5 mg/L (measured with the Nitrate Freshwater and Saltwater Test Kit from API), but differed in pH values: SM2B/C pH 9–9.5; SM2E pH 8.0, and SM2A pH 6.3. Samples were collected in sterile collection tubes and immediately transferred to the lab where they were stored at 4 °C the same day. The next day, 8 mL of each sample were centrifuged for 15 min at 16,000× g to form a biomass pellet.
2.2. Nucleic Acid Extraction and 16S rRNA Amplicon Sequencing
Total DNA was extracted using the PureLink Microbiome DNA Purification Kit (Invitrogen). Utilizing Qubit and NanoDrop, we determined the quality and quantity of DNA, showing an absorbance ratio of 260/280 between 1.83 (SM2E) and 2.00 (SM2A). A 16S rRNA amplicon sequencing library was prepared for each sample, following the 16S Metagenomic Sequencing Library Preparation protocol (Illumina, San Diego, CA, USA). Amplicon primers targeting the V3 and V4 region [11], including the Illumina adapter overhang sequences, are described in the Illumina library prep protocol and were synthesized by Sigma Aldrich. The samples were sequenced using a 1.8 pM library with an Illumina MiniSeq. Paired-end (2 × 150 bp) sequencing generated 1,191,674 reads (SM2A), 1,298,748 reads (SM2B), 1,292,958 reads (SM2C), and 1,141,458 reads (SM2E).
2.3. Sequence Read Analysis
The primer sequences were removed, and reads with low quality scores (average score < 20) were filtered out using the FASTQ Toolkit within BaseSpace (Illumina, Version 2.2.0). The 16S Metagenomics app within BaseSpace (Version 1.0.1) was used to perform a taxonomic classification, using an Illumina-curated taxonomic database RefSeq RDP 16S v3 [12] and the RDP naive Bayes taxonomic classification algorithm with an accuracy of >98.2% at the species level [13]. Default parameters were used for all software unless otherwise noted.
2.4. Enrichment Cultivation Strategy
We used RCVB media, which contains DL-malic acid as the organic carbon source, for the enrichment of photosynthetic bacteria in the SM2B salt marsh sample [14]. The NaCl concentration in the media was increased to 35 g/L to reflect the salinity of the sampling sites. After autoclaving, the RCVB media was supplemented with niacin (1 mg/L), thiamine (1 mg/L), vitamin B12 (10 mg/L), and sodium sulfide (0.03% Na2S.9H20), and the pH was adjusted to pH 9.0. Five milliliters of the original sample were used to inoculate 45 mL of media in a 50 mL falcon tube and sealed off tightly. The tubes were placed at room temperature in front of a window for several weeks. This sample was labeled SM_Orange-Green, and two milliliters were used for genomic DNA extraction.
2.5. Whole-Genome Sequencing
A 2 mL sample of the enrichment cultures (SME_Orange-Green) was precipitated by centrifugation (10 min at 5000× g). DNA was isolated from these pellets using the GeneJet DNA purification kit (Thermo Scientific). In the case of the pink SM2E fraction that grew in the original isolation tube (SME_Pink), cells were scraped off from the tube wall using a sterile loop and resuspended in 180 µL of the GeneJet lysis buffer. Purified DNA samples were analyzed for purity and concentration using a Nanodrop and Qubit, showing an absorbance ratio of 260/280 of 1.85 (SME_Orange-Green) and 1.77 (SME_Pink).
The DNA libraries were prepared with the Nextera DNA Flex Library Prep Kit (Illumina). All genomes were sequenced using 500 µL of a 1.8 pM library, with an Illumina MiniSeq instrument, using paired-end sequencing (2 × 150 bp). Quality control of the reads was performed using FASTQC within BaseSpace (Illumina, Version 1.0.0), using a k-mer size of 5 and contamination filtering.
2.6. 16S rRNA Amplification
Degenerate primers were used for the 16S rRNA PCR reaction, described by Weisberg et al. [15], for the initial identification of the pink-enriched SM2E fraction (SME_Pink). The FD1 forward and the RP1 reverse primers were used. Amplified fragments were gel purified using the PureLink Quick Gel Extraction & PCR purification kit (Invitrogen) and sequenced by Sanger sequencing using the respective FD1 and RP1 sequencing primers (at the Iowa State University DNA core facility). The degenerate primers were designed to amplify most bacterial 16S rRNA, and given the known impurities in the SM2E sample, this could result in the amplification of 16S rRNA from different species; however, both of the sequencing reactions gave the same single result. Forward and reverse sequences were assembled (using a CLUSTALW alignment of the sequenced fragments) into one 16S rRNA sequence of 718 bp.
2.7. Metagenomic Binning Analysis
The sequencing reads of SME_Orange-Green and SME_Pink were used to perform a metagenomic binning using the Metagenomic Binning service within PATRIC [16]. Paired-end reads were used as the input, and default parameters were used. Sets of contig bins were constructed, with hits against contigs that have less than fourfold coverage or are less than 400 bp in length being removed. The contig pool was split into bins using reference genomes. Quality control of each bin was performed using checkM [17]. Each bin was automatically annotated using RAST within PATRIC [16], and consistency checks of the annotation were performed, producing a coarse score (percentage of roles that are correctly present or absent) and a fine score (percentage of roles that are correctly absent or present in the correct number). Identified genomes were ranked based on their coarse score, fine score, and completeness.
2.8. Whole-Genome Comparison
Average percentage nucleotide identity (ANIb) between the whole genomes was calculated using JSpecies [18]. A whole-genome-based phylogenetic tree was generated using the CodonTree method within PATRIC [16], which used PGFams as homology groups. For Prosthecochloris, 648 PGFams were found among these selected genomes using the CodonTree analysis, while 698 were found in the case of Marichromatium. The aligned proteins and coding DNA from single-copy genes were used for RAxML analysis [19,20]. iTOL was used for tree visualization [21].
3. Results and Discussion
3.1. Metagenomic Analysis: Taxonomic Overview
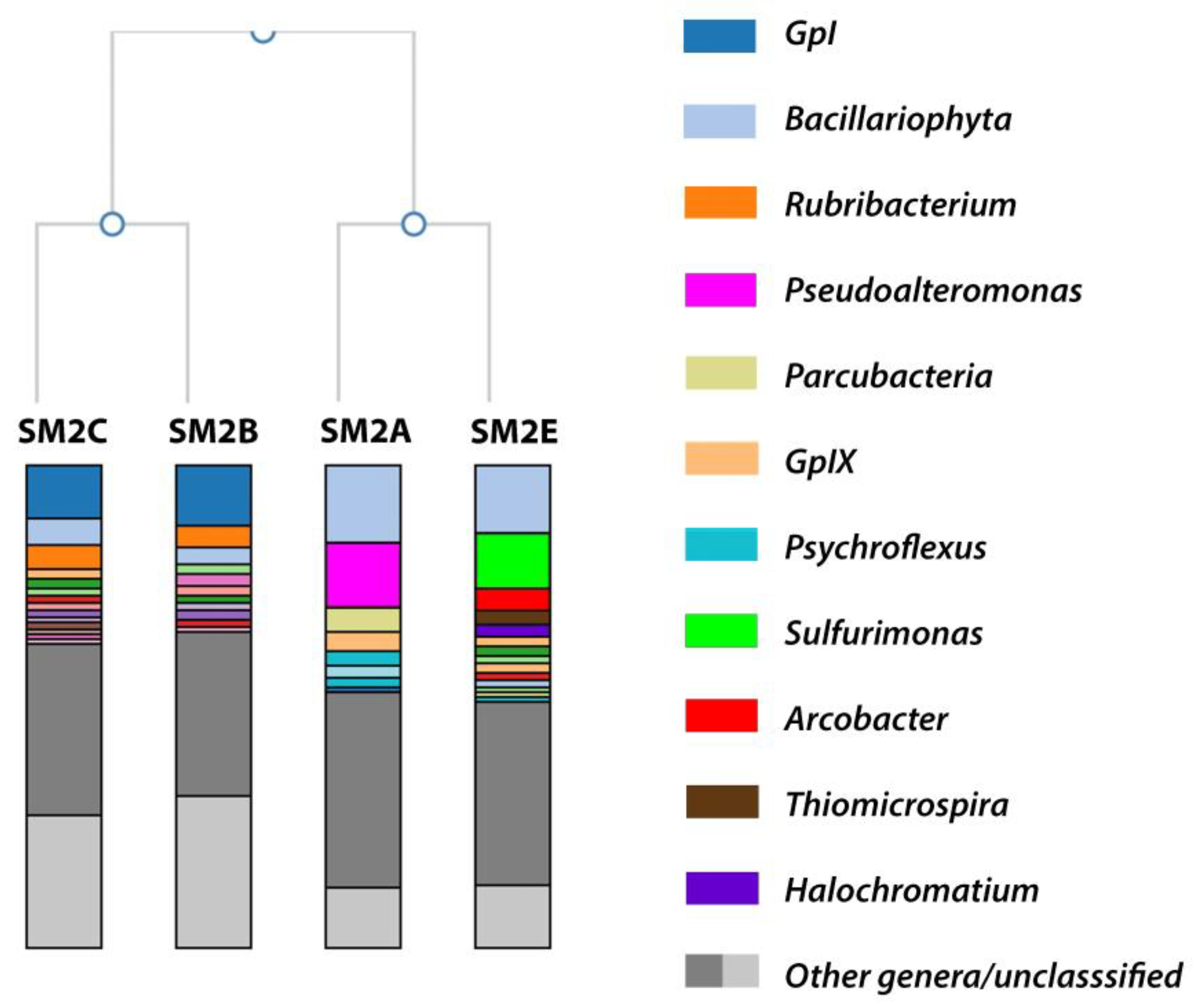
According to a Shannon species diversity analysis (run within the 16S Metagenomics analysis) [22,23], each of the samples contained the following total potential species hits and Shannon diversity index (SDi): SM2A 2495 species (SDi 2.92); SM2B 1470 species (SDi 2.39); SM2C 1843 species (SDi 2.73), and SM3E 2646 (SDi 3.20). An aggregate comparison and hierarchal clustering of the taxonomic classification results of all four samples was performed within the 16S Metagenomic analysis (Figure 2). Samples SM2A and SM2E had more than 87% of the reads identified at the genus level, while sample SM2B and SM2C had ~70% of the reads identified at the genus level.
Table 1 provides an overview of the relative abundance (as a percentage of the total sequencing reads for each sample) of the most abundant genera from Figure 2. It was clear that a large portion of the species identified in all of the samples belonged to diatomic algae (Bacillariophyta, identified based on their chloroplast 16S rRNA) or cyanobacteria (GpI and GpIX), which were likely responsible for the observed green color of the sampling sites. However, the samples from the different locations showed substantial differences in the rest of the microbiome composition. At the phylum level, the majority of bacteria in each sample belonged to the Proteobacteria: SM2A 41.3%; SM2B 26.2%; SM2C 28.2%; and SM2E 51.3% of total reads. However, the phylum in SM2A was mainly composed of Gammaproteobacteria (28.7% of total reads) and Alphaproteobacteria (6.4%), while SM2B and SM2C had more Alphaproteobacteria (20.7 and 19.7%) and less Gammaproteobacteria (4.1 and 5.6%). SM2E on the other hand mainly contained Epsilonproteobacteria (23.0%) and Gammaproteobacteria (18.0%).
3.1.1. SM2B/C Analysis
The reads identified from the SM2B and SM2C samples were very similar and contained abundant representation of the bacterial genus Rubribacterium (~5% of total reads), the cyanobacterial genus GpI (13% of total reads), and the algal genus Bacillariophyta (5% of total reads) (Table 1). Cyanobacteria and algae are commonly found in marine or coastal salt marshes, so it was not surprising that these were well represented here. They were likely responsible for the green coloration of the sampling sites. Bacillariophyta are diatoms commonly found in oceans, waterways, and soils around the world [24,25]. Together with cyanobacteria, these species enrich the bacterial ecosystem with organic matter and nutrients through oxygenic photosynthesis [26,27]. Rubribacterium is an alkaliphilic purple nonsulfur bacterium, belonging to the family of Rhodobacteraceae. The single species of this genus has only been isolated from an Eastern Siberian soda lake [28]. Purple nonsulfur bacteria have been found in weakly and moderately mineralized soda lakes [29,30,31]; however, only three alkaliphilic species are known. Two belong to the genus Rhodobaca [32,33], and one is Rubribacterium polymorphum [28]. It was intriguing that this rare Rubribacterium had the highest bacterial representation in the Nebraska Salt Marsh sample. Even though the two locations where Rubribacterium was found are several thousands of miles apart, there are certain similarities between the isolation sites. The Eastern Siberia soda lake had a pH of 9.5, identical to the pH of the SM2B/C samples. Both locations had a high mineralization of the water (22 g/L of the Eastern Siberia lake vs. 38 g/L for the Nebraska Salt Marsh samples), and in both locations, the water had high amounts of green algae and cyanobacteria ([28] and this study). Rubribacterium grows well in the dark on organic substrates with a pH optimum of 8.5–9.5 and an optimal NaCl concentration of 10 g/L, although it has been shown to grow up to 40 g/L [28].
3.1.2. SM2A Analysis
Both SM2A and SM2E also contained a substantial representation of Bacillariophyta (~17% of total reads) and the cyanobacterial genus GpIX (4 and 2% of total reads, respectively) (Table 1). However, the bacterial representation was substantially different. SM2A mainly contained Pseudoalteromonas (14%), Parcubacteria (7.3%), and Psychroflexus (6.3%) and a minor fraction of Salinivibrio (2%) (Table 1 and Figure 2).
Pseudoalteromonas contains marine species that are found to be associated with higher plant and insect organisms, and several species prevent the fouling of aquatic environments with their bacteriolytic and algacidal activity [34]. This has been found to promote the survival of other marine organisms. Parcubacteria have been identified as symbiotic bacteria from a range of anoxic environments [35,36]. Members of this genus have limited mechanisms for energy and nutrient conservation (for example, lacking genes for the TCA cycle and electron transport), making them dependent on symbiosis for survival. They have been implicated in environmental hydrogen and sulfur cycling in anoxic environments [37]. The observation of both Pseudoalteromonas and Parcubacteria coincides well with the observed presence of higher insect activity and decaying plants in the SM2A sampling location (Figure 1B). Psychroflexus is a psychrophilic genus belonging to the Flavobacteriaceae and has been isolated from Antarctic sea ice [38], hypersaline lakes and sea basins [39,40,41], and the microbial consortium on the surface of Austrian cheese [42]. Similarly, Salinivibrio consists of halophilic bacteria commonly found in hypersaline aquatic habitats and salted foods [43,44]. Finding these species is consistent with the cold, saline environment of the Nebraska Salt Marshes in the fall.
3.1.3. SM2E Analysis
SM2E on the other hand contained a substantial bacterial representation of Sulfurimonas (16.3% of total reads) and smaller fractions of Arcobacter (5%), Thiomicrospira (3.5%), and Halochromatium (2%) (Table 1 and Figure 2). Sulfurimonas species have been identified in distinct environments such as hydrothermal deep-sea vents, marine sediment, and coastal estuaries [45,46,47]. Sulfurimonas are sulfur-oxidizing bacteria, and it has been shown recently that Sulfurimonas species play an important role in coastal mangrove environments for the oxidation of the toxic sulfide produced by sulfur-reducing bacteria, like Sulfurospirillum [46,47].
Arcobacter are Campylobacter-like organisms that have been found in both animal and environmental sources, but compared to Campylobacter, they can grow at lower temperatures and are aerotolerant. Arcobacter has been isolated from a wider range of environments. One halophilic species, A. halophilus, has been collected from a hypersaline lagoon in Hawaii [48]. A. nitrofigilis is a nitrogen-fixing bacterium isolated from the roots of the salt marsh plant Spartina alterniflora [49,50]. Based on a provisional identification, it appears that the dominant plant species in the NE Salt Marsh sampling area is the invasive Phragmites sp. Further studies will be necessary to determine whether or not this Arcobacter species is also important for nitrogen fixation of Phragmites sp., similar to what was found for the salt marsh Spartina alterniflora [49,50]. Arcobacter sulfidicus is an obligate microaerophile that oxidizes sulfides and is a producer of filamentous sulfur [51]. Large populations of this bacterium produce mats of this solid, white sulfur filament [52]. This is consistent with the observed white sulfur deposits at the SM2E sampling site (Figure 1B).
Thiomicrospira is another colorless sulfur bacterium that derives energy from the oxidation of reduced sulfur compounds. The genus Thiomicrospira contains sixteen species [53]. Many of these, for example Tms. aerophila, Tms. microaerophila, Tms. cyclica, and Tms. sibirica, have been isolated from alkaline environments: Mono Lake (U.S.), Soap Lake (U.S.), and a soda lake in northern Russia [54,55,56]. The fact that these are obligatory alkalophilic and obligately chemolithoautotrophic sulfur-oxidizing bacteria is consistent with the alkaline conditions of the NE Salt Marsh area.
Halochromatium bacteria occur in hypersaline habitats. They belong to the Chromatiaceae, which is the main family of the purple sulfur bacteria [57,58]. Purple sulfur bacteria use reduced sulfur (e.g., sulfide or thiosulfate) as an electron donor in their photosynthetic pathways, thereby often producing granules of elemental sulfur [57,58]. After about two weeks of storing the SM2E sample at 4 °C, a significant pink growth occurred on the wall of the original sampling tube. A sample was taken from the pink substrate for genomic DNA extraction and used for 16S rRNA PCR and genome sequencing. The result showed that this was a species of Halochromatium, most closely related to Halochromatium roseum JA134, based on the 16S rRNA comparison (NCBI BLAST identity 99%; 710/718 bp). The whole genome was only partially completed and (as expected) showed significant contamination (>1000 contigs, estimated 40% contamination), but metagenomic binning further confirmed that this species belonged to the Chromatiaceae.
The heavy presence of sulfur-oxidizing and filamentous sulfur-producing species in the SM2E sample was consistent with the visible white sulfur precipitates in that sample area. There appeared to be a low abundance of sulfur-reducing species, for example Sulfurospirillum was found in 0.38% of total reads, while only two genera of Desulfobacteraceaea (Desulfonema and Desulfotignum) were found at 0.65 and 1.0%, respectively. This low abundance and limited plant growth in that area suggested that the homeostasis in the SM2E sampling area was possibly disrupted and an excess of oxidized sulfur components was being produced. The SM2E area appears to have been formed after the main salt marsh pond retracted, possibly due to seasonal fluctuations.
3.2. Genomic Analysis of Enrichment Cultures
Since the 16S rRNA amplicon metagenomic data showed that many of the bacterial representations were found to be from less characterized or rare photosynthetic bacteria, we decided to attempt an enrichment in order to obtain whole-genome or partial-genome data that could be used for a better identification and taxonomic classification of these species.
SM2B was used to inoculate an alkaline (pH 9.0), saline, sulfide-containing medium for photosynthetic sulfur bacteria cultivation. After about two weeks of anaerobic incubation with a night-dark light cycle, the culture showed a faint orange-brown color, which turned dark green in the following two weeks. A two milliliter sample of this (SM_Orange-Green) was used for genomic DNA extraction and genome sequencing. Since we did not expect this to be a single pure culture, we used the sequencing reads to perform a metagenomic binning within PATRIC [16]. We obtained two valuable bins: one genome related to Prosthecochloris sp. (97.9% coarse consistency; 96.4% fine consistency), the other to Marichromatium sp. (96.4% coarse consistency; 92.3% fine consistency), both with 100% completeness. These strains were designated as Prosthecochloris sp. SM2 and Marichromatium sp. SM2, for “Salt Marsh Location 2”. We also obtained three bins containing partial, incomplete genomes, all three identified as Sulfurospirillum species. Although Prosthecochloris and Marichromatium were not found in high abundance in the 16S rRNA amplicon analysis (see above), both species were found to be present in the SM2B sample. Prosthecochloris was found be present in 1.7% of the total reads in sample SM2B, while Marichromatium was found to be at 0.8%. However, 16S rRNA amplicon analysis limits resolution at the species and sometimes at the genus level since it only uses a fragment of the 16S rRNA gene for identification. The actual representation of these species might be higher since we found the family of Chlorobiaceae in 2.5% and the Chromatiaceae in 1.6% of the total reads. The anaerobic, high sulfide, and halophilic conditions of the enrichment cultivation were certainly favorable for these photosynthetic species.
3.2.1. Prosthecochloris sp. SM2
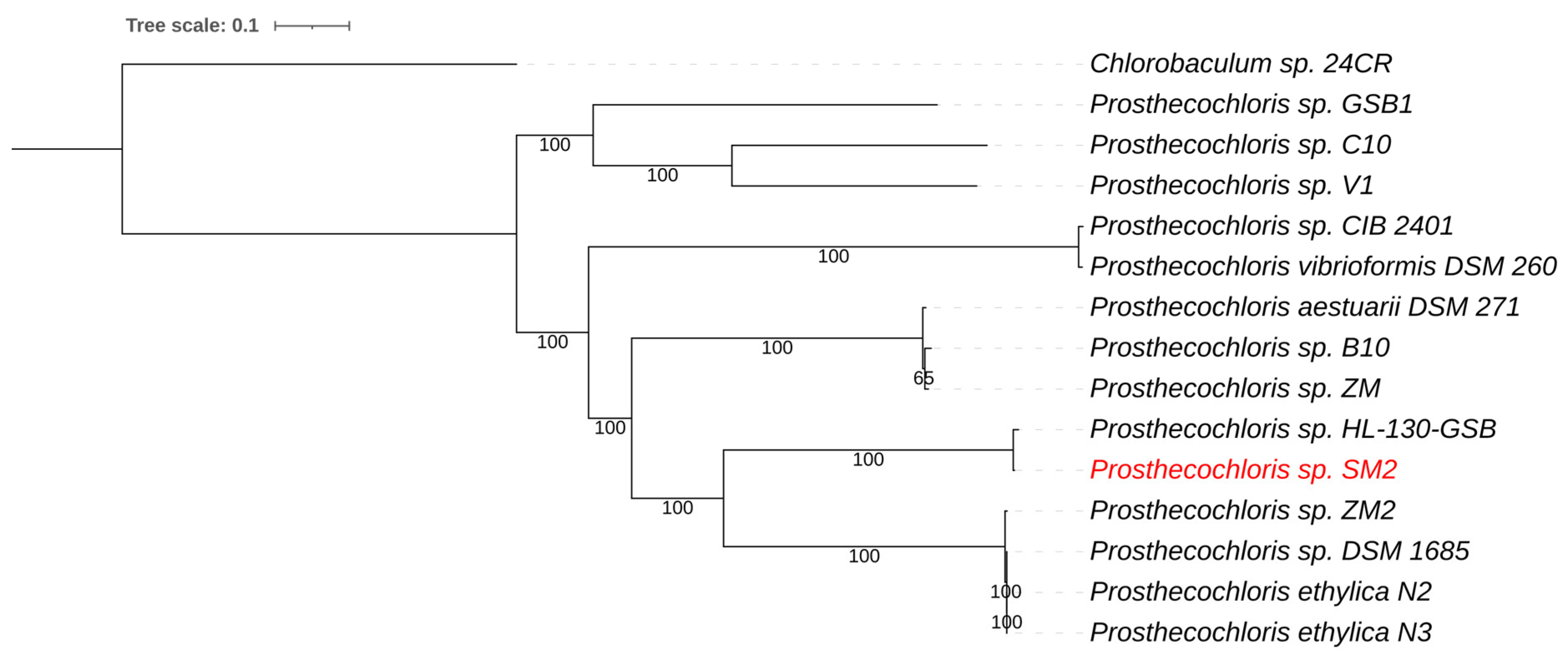
Prosthecochloris are green sulfur bacteria (Chlorobiaceae) that are anoxygenic phototrophic bacteria. The sulfur metabolism of green sulfur bacteria involves the oxidation of sulfide and the deposition of elemental sulfur globules outside the cells [59]. The Prosthecochloris genome was assembled into 112 contigs with a total genome size of 2.43 Mb and a G+C content of 51.8%. Average nucleotide identity (ANIb) comparison showed that the genome sequence of the NE Salt Marsh Prosthecochloris was most similar to strain HL130-GSB, with an average nucleotide identity (ANI) of 98.4%. This ANI value was above 95%, which is the arbitrary cutoff value for species differentiation [18], indicating that these strains likely belonged to the same species. A whole genome-based phylogenetic tree for Prosthecochloris placed the green component as the closest relative to strain HL-130-GSB (Figure 3). This was consistent with the ANI comparisons mentioned above. Prosthecochloris HL-130-GSB was isolated from a cyanobacterial mat obtained from Hot Lake, a saline, high-sulfate, meromictic lake in WA, USA [60].
Not surprisingly, the Prosthecochloris sp. SM2 genome contains homologues of the genes for sulfur oxidation, SoxB and SoxYZ, the latter of which is genetically clustered with a flavocytochrome c:sulfide dehydrogenase encoding gene. This indicates that Prosthecochloris sp. SM2 is indeed capable of sulfur oxidation. We recently showed that several Prosthecochloris strains that form syntrophic interactions with sulfur-reducing bacteria contain very large agglutination proteins [61]. These agglutination proteins function as excreted adhesion proteins that are important for the formation of syntrophic interactions and larger bacterial consortia [61,62]. The formation of these interactions aids in the mutual exchange of metabolites. At this point, it is not known if this new strain also forms syntrophic complexes; however, we were able to identify the same large agglutination proteins: a structural toxin protein RtxA homologue and a large outer membrane adhesion protein (“tandem-95 repeat” protein), followed by a smaller gene encoding an agglutination protein (TolC family type I secretion outer membrane protein), an ABC transmembrane transporter (type I secretion system ATPase), and a HlyD homologous protein (type I secretion membrane fusion protein). This indicates that this new Prosthecochloris sp. SM2 is also capable of producing the adhesion complex that has been found to be important for the formation of close interspecies interactions.
3.2.2. Marichromatium sp. SM2
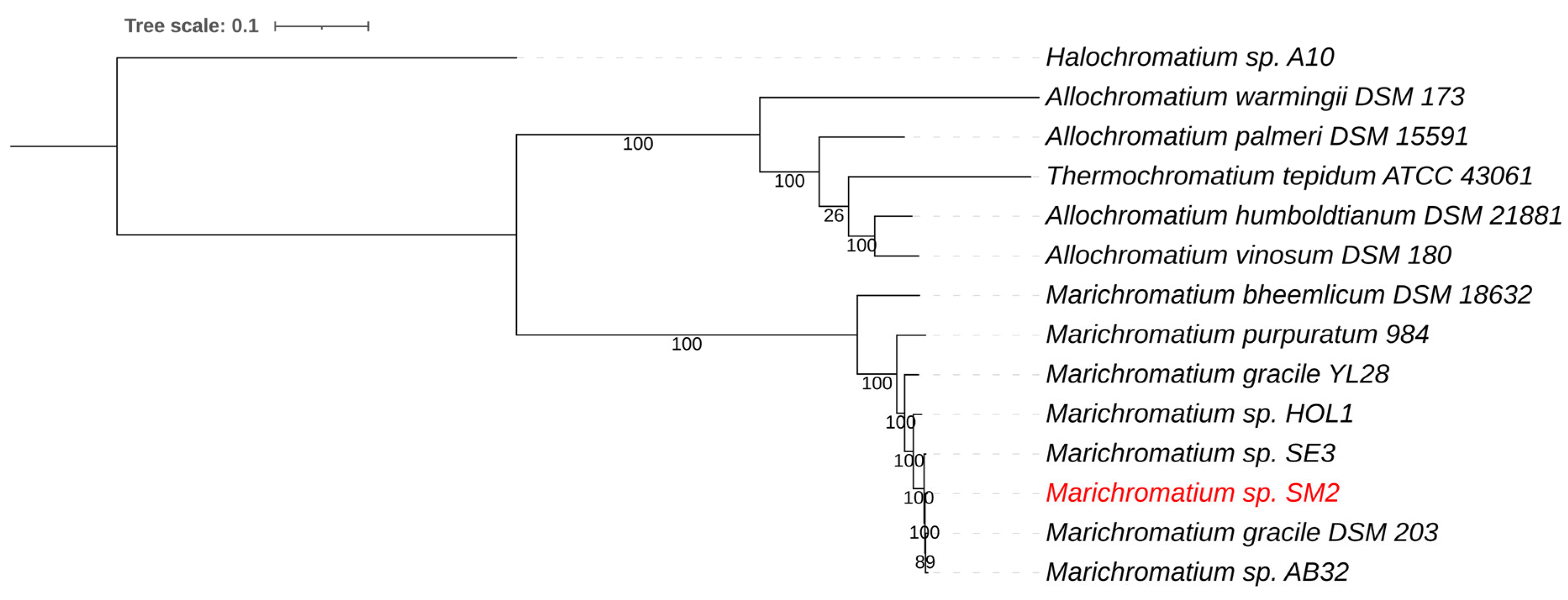
Marichromatium is a small genus belonging to the purple sulfur bacteria (Chromatiaceae) [57]. It was found that the rRNAs of Chromatiaceae form two major lines of descent, one composed primarily of halophilic and the other of mainly fresh-water origin. Marichromatium is placed within the halophilic branch of the Chromatiaceae [57]. Several strains of Marichromatium are known, and the genus appears to be commonly found throughout the world; however, there does not seem to be much variation in the group as a whole. The Marichromatium genome from the enriched NE Salt Marsh sample was assembled into 293 contigs with a total genome size of 3.94 Mb and a G+C content of 67.6%. Average nucleotide identity (ANIb) comparison showed that the genome sequence was most similar to Marichromatium gracile DSM203T, with an average nucleotide identity (ANI) of 98.4%. A whole genome-based phylogenetic tree for Marichromatium, including Halochromatium, Allochromatium, and Thermochromatium genomes, placed the green component as the closest relative to Marichromatium gracile (Figure 4). Marichromatium gracile DSM203T was isolated form marine waters at Headley Harbour, Massachusetts, USA [64].
As expected, the Marichromatium sp. SM2 genome also contains the SoxB and SoxYZ genes for sulfur oxidation. It has recently been shown that the halophilic Marichromatium species will use glycine betaine as an osmotic adaptation to a high-salt environment [64]. A feature search of the genome within PATRIC showed that this genome contains several genes for glycine betaine uptake and metabolism, including a betaine aldehyde dehydrogenase (EC 1.2.1.8), a glycine betaine transporter OpuD, a secondary glycine betaine transporter BetU, and a glycine betaine ABC transporter (consisting of the substrate-binding protein OtaC, permease protein OtaB, and the ATP-binding protein OtaA). This suggests that this species is well adapted to the high salt environment of the NE Salt Marsh.
3.2.3. Sulfurospirillum
Sulfurospirillum are sulfur-reducing, nitrogen-fixing bacteria that have been found to be important for hydrogen and sulfur-based syntrophic interactions in several marine environments [47,65,66,67]. Sulfur-reducing bacteria like Sulfurospirillum produce sulfide that can be toxic to plants and other oxygenic photosynthetic species like algae or cyanobacteria. They are typically found to work in balance with sulfur-oxidizing or sulfur-producing species [47,66]. The genome fragments we obtained from metagenomic binning were only partially complete (22 to 93 percent completeness) with varying levels of contamination present (6% to 61%). This made an accurate species annotation impossible; however, strains UBA2217, SL2-2, and UBA12182, were found to be the closest relatives based on metagenomic analysis. All three were assembled genomes from metagenomic samples from an oil production facility and contaminated mine tailings [68] or a sludge bioreactor to treat contaminated water [69].
Sulfurospirillum are the only organohalide-respiring Epsilonproteobacteria described so far, and they grow on many toxic compounds, allowing them to thrive in polluted habitats [65]. Electron acceptors for anaerobic respiration used by all Sulfurospirillum species so far are nitrate, fumarate, and sulfur (including polysulfide). Interestingly, none of the species reduce sulfate in their energy metabolism, so the species are involved in their environment as sulfur reducers. Although several marine species have been found, only one alkaliphilic species has been isolated, Sulfurospirillum alkalitolerans [70]. This species was isolated from a lab bioreactor that was used to remove H2S from waste gasses. The species shows a pH range from 7.1–9.7 and salt tolerance up to 1.5 M NaCl. From our enrichment analysis, it appeared that Sulfurospirillum was the dominant sulfur reducer in the high-saline, alkaline environment of these NE Salt Marshes. Since both Prosthecochloris and Sulfurospirillum previously have been found to be involved in syntrophic interactions with other species [61,67], it is possible that the sulfur-producing Prosthecochloris sp. SM2 forms a similar syntrophy, and close contact interactions, with the Sulfurospirillum in our enrichment cultures, although further physiological and biochemical analysis will be required to investigate this further.
3.3. The Salt Marsh Sulfur Cycle
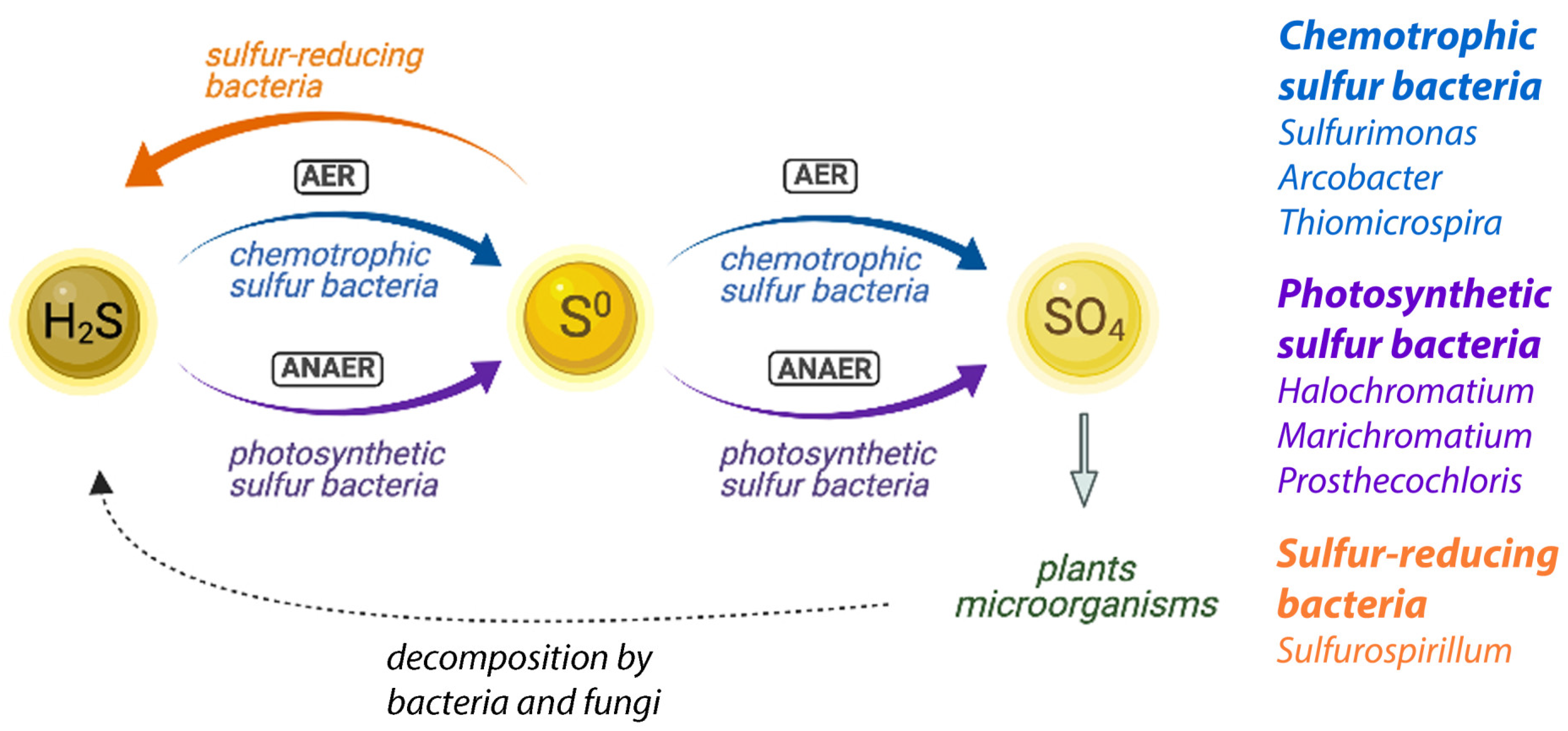
The sulfur cycle and its microbial components have been extensively studied in microbial terrestrial and marine ecosystems [8,9,71]. A simplified model is summarized in Figure 5, where we highlight the different bacterial groups assumed to be involved in the context of the NE Salt Marsh ecosystem.
It has been shown previously that marine microbial benthic ecosystems are dominated by essentially a few functional groups of microbes [8,9], which makes them an ideal model system to study the cycling of sulfur and organic matter. Sulfur is an essential element in all organisms; however, due to the large variety of the possible oxidation states of sulfur, between −2 (as in sulfide) and +6 (as in sulfate), different sulfur species can be useful as electron acceptors or electron donors in a variety of different processes. The processes of sulfur cycling are predominantly mediated by bacteria, and their role in the sulfur cycle is therefore crucially important to maintain an ecosystem.
In simplified terms, the microbial ecosystems consist of cyanobacteria and algae that perform aerobic photosynthesis, which causes oxygen depletion and provides growth substrates for other species. As a consequence of lysis, excretion, and fermentation, organic compounds are produced that are partially or completely degraded by a variety of bacteria and fungi. Some of those species are sulfate-reducing bacteria that produce sulfide, through the reduction of sulfates. Sulfur-reducing bacteria, like Sulfurospirillum, will contribute to the sulfide (H2S) production by further reduction of sulfur (or polysulfides) (orange pathway in Figure 5). This sulfide is reoxidized to sulfate by purple photosynthetic bacteria and colorless sulfur bacteria (purple and blue pathways in Figure 5). Sulfide is inhibitory to oxygenic phototrophs, like cyanobacteria and algae, and its removal by the anaerobic purple and colorless sulfur bacteria is crucial to balance the sulfur cycling in an ecosystem. In addition, these sulfur bacteria produce reducing equivalents for CO2 reduction. Green sulfur bacteria are obligatory anaerobes and grow best under low light conditions, and if they are present in these systems, they are typically found underneath the purple sulfur bacteria [72,73]. They contribute to the anaerobic sulfide oxidation pathway (purple pathway in Figure 5). Purple nonsulfur bacteria, like Rubribacterium, also contribute by anoxygenic photosynthesis and typically use hydrogen as an electron donor, but they can sometimes also use sulfide (at a lower concentration than the photosynthetic sulfur bacteria). We identified several representatives of each of these groups in our microbial analysis of the NE Salt Marshes, and it is clear that a combined, consorted action of all these groups is needed to maintain a balance of nutrient cycling in the ecosystem (Figure 5).
4. Conclusions
The various metabolic capacities of the NE Salt Marsh microbial community are responsible for the biogeochemical cycling of important chemical elements like sulfur, nitrogen, and carbon. From studies of coastal salt marshes, marine microbial sediments, and also ancient stromatolites, it appears that these microbial ecosystems have been resilient for billions of years [9,71]. It is therefore important to study and attempt to preserve these locally important ecosystems. If nothing else, they provide an important glimpse into the biochemical balancing of the planet, now and in the past. The NE Salt Marsh microbiome consists of alkalophilic and halophilic species that appear to be well adapted to this specialized environment. The microbial ecosystem appears to be well balanced as far as sulfur and other nutrients; nevertheless, urban development, agricultural runoff, and the growth of invasive plant species are some of the current threats to maintaining these vital ecosystems. Restoration and conservation efforts of native ecosystems for the most part focus on the insect and plant community; however, a recent study by Lynum et al. [74], specifically on passive salt marsh restoration, showed that the underlying microbial community’s conservation is crucial and paramount to any preservation of marsh vegetation. Although the microbial community can respond quickly in a positive direction, the process of microbial restoration can take several years, before any return to native marsh vegetation is visible [74]. With the current threats to this inland salt marsh system in Nebraska, it is important to understand the underlying microbial composition and complex biochemistry. This current study establishes a good baseline for further studies on how microbiological diversity, nutrient cycling, and bacterial ecology impact this locally important watershed.
Author Contributions
Conceptualization, J.A.K.; methodology, S.R.A. and J.A.K.; software, S.D.; validation, S.R.A. and J.A.K.; formal analysis, S.R.A., S.D. and J.A.K.; investigation, S.R.A., S.D. and J.A.K.; resources, J.A.K.; data curation, S.R.A. and J.A.K.; writing—original draft preparation, J.A.K.; writing—review and editing, S.R.A., S.D. and J.A.K.; visualization, S.D.; supervision, J.A.K.; funding acquisition, J.A.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was sponsored by the Wilson Enhancement Fund for Applied Research in Science at Bellevue University.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The 16S rRNA gene amplicon data sets and WGS have been deposited at DDBJ/ENA/GenBank under Project PRJNA714941. The 16S rRNA gene amplicon data sets can be accessed with SRA Accession Numbers SRR13980338 (SM2A), SRR13982897 (SM2B), SRR13981552 (SM2C), and SRR13981842 (SM2E).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gibbens, S. The remnants of a vast prehistoric sea lie hidden in Nebraska’s endangered marshes. In National Geographic; National Geographic Society: Washington, DC, USA, 2020. [Google Scholar]
- Johnsgard, P.A. The Nature of Nebraska: Ecology and Biodiversity; The University of Nebraska Press: Lincoln, NE, USA, 2001. [Google Scholar]
- Spomer, S.; Higley, L. Population status and distribution of the Salt Creek Tiger Beetle, Cicindela nevadica lincolniana Casey (Coleoptera: Cicindelidae). J. Kans. Entomol. Soc. 1993, 66, 392–398. [Google Scholar]
- Brosius, T.R.; Higley, L.G. Behavioral niche partitioning in a sympatric tiger beetle assemblage and implications for the endangered Salt Creek tiger beetle. PeerJ 2013, 1, e169. [Google Scholar] [CrossRef] [Green Version]
- Ungar, I.; Hogan, W.; McClelland, M. Plant communities of saline soils at Lincoln, Nebraska. Am. Midl. Nat. 1969, 82, 564–577. [Google Scholar] [CrossRef]
- Panella, M. Nebraska’s At-Risk Species Wildlife; Nebraska Game & Parks Commission: Lincoln, NE, USA, 2012; pp. 146–147.
- Malmstrom, T. Saline Wetlands Conservation Partnership 2018 Progress Report; Lincoln Parks and Recreation Department: Lincoln, NE, USA, 2019. Available online: https://www.lincoln.ne.gov/files/sharedassets/public/parks-amp-rec/saline-wetlands/progressrpt2018.pdf (accessed on 13 May 2021).
- Van Gemerden, H. Microbial mats: A joint venture. Mar. Geol. 1993, 113, 3–25. [Google Scholar] [CrossRef]
- Pfennig, N. The phototrophic bacteria and their role in the sulfur cycle. Plant Soil 1975, 43, 1–16. [Google Scholar] [CrossRef]
- Bowen, J.; Crump, B.; Deegan, L.; Hobbie, J.E. Salt marsh sediment bacteria: Their distribution and response to external nutrient inputs. ISME J. 2009, 3, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucl. Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal database project: Data and tools for high throughput rRNA analysis. Nucl. Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Weaver, P.F.; Wall, J.D.; Gest, H. Characterization of Rhodopseudomonas capsulata. Arch. Microbiol. 1975, 105, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucl. Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2014, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rosselló-Móra, R.; Glöckner, F.O.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2015, 32, 929–931. [Google Scholar] [CrossRef]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.J.B. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucl. Acids Res 2019, 47, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spellerberg, I.F.; Peter, J.F. A tribute to Claude Shannon (1916–2001) and a plea for more rigorous use of species richness, species diversity and the ‘Shannon-Wiener’ Index. Glob. Ecol. Biogeogr. 2003, 12, 177–179. [Google Scholar] [CrossRef] [Green Version]
- Tuomisto, H. A diversity of beta diversities: Straightening up a concept gone awry. Part 1. Defining beta diversity as a function of alpha and gamma diversity. Ecography 2010, 33, 2–22. [Google Scholar] [CrossRef]
- Hasle, G.R.; Syvertsen, E.E.; Steidinger, K.A.; Tangen, K. Marine diatoms. In Identifying Marine Diatoms and Dinoflagellates; Tomas, C.R., Ed.; Academic Press: Cambridge, MA, USA, 1996; pp. 5–385. ISBN 978-0-08-053441-1. [Google Scholar]
- Fourtanier, E.; Kociolek, J.P. Catalogue of the diatom genera. Diatom Res. 1999, 14, 1–190. [Google Scholar] [CrossRef]
- Stal, L.J.; Gemerden, H.; Krumbein, W.E. Structure and development of a benthic marine microbial mat. FEMS Microbiol. Lett. 1985, 31, 111–125. [Google Scholar] [CrossRef]
- Bolhuis, H.; Stal, L.J. Analysis of bacterial and archaeal diversity in coastal microbial mats using massive parallel 16S rRNA gene tag sequencing. ISME J. 2011, 5, 1701–1712. [Google Scholar] [CrossRef]
- Boldareva, E.N.; Moskalenko, A.A.; Makhneva, Z.K.; Tourova, T.P.; Kolganova, T.V.; Gorlenko, V.M. Rubribacterium polymorphum gen. nov., sp. nov., a novel alkaliphilic nonsulfur purple bacterium from an Eastern Siberian soda lake. Microbiology 2009, 78, 732–740. [Google Scholar] [CrossRef]
- Imhoff, J.F.; Soliman, G.S.H.; Trüper, H.G. The Wadi Natrun: Chemical composition and microbial mass development in alkaline brines of eutrophic desert lakes. Geomicrobiol. J. 1979, 1, 219–234. [Google Scholar] [CrossRef]
- Kompantseva, E.I.; Bryantseva, I.A.; Komova, A.V.; Namsaraev, B.B. The structure of phototrophic communities of soda lakes of the Southeastern Transbaikal Region. Microbiology 2007, 76, 211–219. [Google Scholar] [CrossRef]
- Kompantseva, E.I.; Komova, A.V.; Krauzova, V.I.; Kolganova, T.V.; Panteleeva, E.E. Purple nonsulfur bacteria in weakly and moderately mineralized soda lakes of the Southern Transbaikal region and Northeastern Mongolia. Microbiology 2009, 78, 246–254. [Google Scholar] [CrossRef]
- Milford, A.D.; Achenbach, L.A.; Jung, D.O.; Madigan, M.T. Rhodobaca bogoriensis gen. nov. and sp. nov. alcaliphilic purple nonsulfur bacterium from African Rift valley soda lakes. Arch. Microbiol. 2000, 174, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Boldareva, E.N.; Akimov, V.N.; Boychenko, V.A.; Stadnichuk, I.N.; Moskalenko, A.A.; Makhneva, Z.K.; Gorlenko, V.M. Rhodobaca barguzinensis sp. nov., a new alkaliphilic purple sulfur bacterium isolated from a soda lake of the Barguzin valley (Buryat Republic, Eastern Siberia). Microbiology 2008, 77, 206–218. [Google Scholar] [CrossRef]
- Holmström, C.; Kjelleberg, S. Marine Pseudoalteromonas species are associated with higher organisms and produce biologically active extracellular agents. FEMS Microbiol. Ecol. 1999, 30, 285–293. [Google Scholar] [CrossRef]
- Harris, J.K.; Kelley, S.T.; Pace, N.R. New perspective on uncultured bacterial phylogenetic division OP11. Appl. Environ. Microbiol. 2004, 70, 845–849. [Google Scholar] [CrossRef] [Green Version]
- Nelson, W.; Stegen, J. The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Front. Microbiol. 2015, 6, 713. [Google Scholar] [CrossRef] [Green Version]
- Wrighton, K.C.; Thomas, B.C.; Sharon, I.; Miller, C.S.; Castelle, C.J.; VerBerkmoes, N.C.; Wilkins, M.J.; Hettich, R.L.; Lipton, M.S.; Williams, K.H.; et al. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 2012, 337, 1661–1665. [Google Scholar] [CrossRef] [Green Version]
- Bowman, J.P.; McCammon, S.A.; Lewis, T.; Skerratt, J.H.; Brown, J.L.; Nichols, D.S.; McMeekin, T.A. Psychroflexus torquis gen. nov., sp. nov., a psychrophilic species from Antarctic sea ice, and reclassification of Flavobacterium gondwanense (Dobson et al. 1993) as Psychroflexus gondwanense gen. nov., comb. nov. Microbiology 1998, 144, 1601–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, J.; Kang, J.Y.; Jahng, K.Y. Psychroflexus salarius sp. nov., isolated from Gomso salt pan. Int. J. Syst. Evol. Microbiol. 2014, 64, 3467–3472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.H.; Kang, S.J.; Jung, Y.T.; Oh, T.K. Psychroflexus salinarum sp. nov., isolated from a marine solar saltern. Int. J. Syst. Evol. Microbiol. 2009, 59, 2404–2407. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.G.; Cui, X.L.; Wang, Y.X.; Tang, S.K.; Zhang, Y.Q.; Li, W.J.; Liu, J.H.; Peng, Q.; Xu, L.H. Psychroflexus sediminis sp. nov., a mesophilic bacterium isolated from salt lake sediment in China. Int. J. Syst. Evol. Microbiol. 2009, 59, 569–573. [Google Scholar] [CrossRef]
- Seiler, H.; Bleicher, A.; Busse, H.J.; Hüfner, J.; Scherer, S. Psychroflexus halocasei sp. nov., isolated from a microbial consortium on a cheese. Int. J. Syst. Evol. Microbiol. 2012, 62, 1850–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventosa, A. Unusual micro-organisms from unusual habitats: Hypersaline environments. In Prokaryotic Diversity: Mechanisms and Significance; Cambridge University Press: Cambridge, UK, 2006; pp. 223–254. [Google Scholar]
- Galisteo, C.; Sánchez-Porro, C.; de la Haba, R.R.; López-Hermoso, C.; Fernández, A.B.; Farias, M.E.; Ventosa, A. Characterization of Salinivibrio socompensis sp. nov., a new halophilic bacterium isolated from the high-altitude hypersaline lake Socompa, Argentina. Microorganisms 2019, 7, 241. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Perner, M. The globally widespread genus Sulfurimonas: Versatile energy metabolisms and adaptations to redox clines. Front. Microbiol. 2015, 6, 989. [Google Scholar] [CrossRef] [Green Version]
- SamKamaleson, A.; Gonsalves, M.-J. Role of sulfur-oxidizing bacteria on the ecology in tropical mangrove sediments. Reg. Stud. Mar. Sci. 2019, 28, 100574. [Google Scholar]
- Aviles, A.F.; Kyndt, J.A. Sequencing of coastal lagoon samples from the Piñones Lagoon, Puerto Rico, reveals important role of bacterial sulfur metabolism in the lagoon ecosystem. Microbiol. Res. Announc. 2021, 10, e00172-21. [Google Scholar]
- Donachie, S.P.; Bowman, J.P.; On, S.L.W.; Alam, M. Arcobacter halophilus sp. nov., the first obligate halophile in the genus Arcobacter. Int. J. Syst. Evol. Microbiol. 2005, 55, 1271–1277. [Google Scholar] [CrossRef]
- McClung, C.R.; Patriquin, D.G.; Davis, R.E. Campylobacter nitrofigilis sp. nov., a nitrogen-fixing bacterium associated with roots of Spartina alterniflora Loisel. Int. J. Syst. Bacteriol. 1983, 33, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Fera, M.T.; Maugeri, T.L.; Gugliandolo, C.; Beninati, C.; Giannone, M.; La Camera, E.; Carbone, M. Detection of Arcobacter spp. in the coastal environment of the Mediterranean Sea. Appl. Environ. Microbiol. 2004, 70, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Wirsen, C.O.; Sievert, S.M.; Cavanaugh, C.M.; Molyneaux, S.J.; Ahmad, A.; Taylor, L.T.; DeLong, E.F.; Taylor, C.D. Characterization of an autotrophic sulfide-oxidizing marine Arcobacter sp. that produces filamentous sulfur. Appl. Environ. Microbiol. 2002, 68, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirsen, C. Is Life Thriving Deep Beneath the Seafloor? Woods Hole Oceanographic Institution: Falmouth, MA, USA, 2004; Available online: https://www.whoi.edu/oceanus/feature/is-life-thriving-deep-beneath-the-seafloor/ (accessed on 1 March 2021).
- Boden, R.; Scott, K.M.; Williams, J.; Russel, S.; Antonenen, K.; Rae, A.W.; Hutt, L.P. An evaluation of Thiomicrospira, Hydrogenovibrio and Thioalkalimicrobium: Reclassification of four species of Thiomicrospira to each Thiomicrorhabdus gen. nov. and Hydrogenovibrio, and reclassification of all four species of Thioalkalimicrobium to Thiomicrospira. Int. J. Syst. Evol. Microbiol. 2017, 67, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, D.Y.; Lysenko, A.M.; Mityushina, L.L.; Tourova, T.P.; Jones, B.E.; Rainey, F.A.; Robertson, L.A.; Kuenen, G.J. Thioalkalimicrobium aerophilum gen. nov., sp. nov. and Thioalkalimicrobium sibericum sp. nov., and Thioalkalivibrio versutus gen. nov., sp. nov., Thioalkalivibrio nitratis sp.nov., novel and Thioalkalivibrio denitrificancs sp. nov., novel obligately alkaliphilic and obligately chemolithoautotrophic sulfur-oxidizing bacteria from soda lakes. Int. J. Syst. Evol. Microbiol. 2001, 51, 565–580. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, D.Y.; Gorlenko, V.M.; Tourova, T.P.; Tsapin, A.I.; Nealson, K.H.; Kuenen, G.J. Thioalkalimicrobium cyclicum sp. nov. and Thioalkalivibrio janaschiijannaschii sp. nov., novel species of haloalkaliphilic, obligately chemolithoautotrophic sulfur-oxidizing bacteria from hypersaline alkaline mono lake (California). Int. J. Syst. Evol. Microbiol. 2002, 52, 913–920. [Google Scholar] [PubMed] [Green Version]
- Sorokin, D.Y.; Foti, M.; Pinkart, H.C.; Muyzer, G. Sulfur-Oxidizing bacteria in soap lake (Washington state), a meromictic, haloalkaline lake with an unprecedented high sulfide content. Appl. Environ. Microbiol. 2007, 73, 451–455. [Google Scholar] [CrossRef] [Green Version]
- Imhoff, J.F.; Süling, J.; Petri, R. Phylogenetic relationships among the Chromatiaceae, their taxonomic reclassification and description of the new genera Allochromatium, Halochromatium, Isochromatium, Marichromatium, Thiococcus, Thiohalocapsa and Thermochromatium. Int. J. Syst. Bacteriol. 1998, 48, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Hunter, C.N.; Daldal, F.; Thurnauer, M.C.; Beatty, J.T. The Purple Phototropic Bacteria; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar]
- Imhoff, J.F. Biology of green sulfur bacteria. In Encyclopedia of Life Sciences; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2014. [Google Scholar] [CrossRef]
- Thiel, V.; Drautz-Moses, D.I.; Purbojati, R.W.; Schuster, S.C.; Lindemann, S.; Bryant, D.A. Genome sequence of Prosthecochloris sp. strain HL-130-GSB from the phylum Chlorobi. Genome Announc. 2017, 5, e00538-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyndt, J.A.; Van Beeumen, J.J.; Meyer, T.E. Simultaneous genome sequencing of Prosthecochloris ethylica and Desulfuromonas acetoxidans within a syntrophic mixture reveals unique pili and protein interactions. Microorganisms 2020, 8, 1939. [Google Scholar] [CrossRef]
- Muller, J.; Overmann, J. Close interspecies interactions between prokaryotes from sulfureous environments. Front. Microbiol. 2011, 2, 146. [Google Scholar] [CrossRef] [Green Version]
- Freed, S.; Robertson, S.; Meyer, T.; Kyndt, J. Draft whole-genome sequence of the green sulfur photosynthetic bacterium Chlorobaculum sp. strain 24CR, isolated from the Carmel River. Microbiol. Resour. Announc. 2019, 8, e00116-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imhoff, J.F.; Rahn, T.; Künzel, S.; Keller, A.; Neulinger, S.C. Osmotic adaptation and compatible solute biosynthesis of phototrophic bacteria as revealed from genome analyses. Microorganisms 2020, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Goris, T.; Diekert, G. The genus sulfurospirillum. In Organohalide-Respiring Bacteria; Adrian, L., Löffler, F., Eds.; Springer: Heidelberg/Berlin, Germany, 2016. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Zhen, Y.; Mi, T.; He, H.; Yu, Z. Microbial diversity and community structure of sulfate-reducing and sulfur-oxidizing bacteria in sediment cores from the East China Sea. Front. Microbiol. 2017, 8, 2133. [Google Scholar] [CrossRef]
- Kruse, S.; Goris, T.; Westermann, M.; Adrian, L.; Diekert, G. Hydrogen production by Sulfurospirillum species enables syntrophic interactions of Epsilonproteobacteria. Nat. Commun. 2018, 9, 4872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Buttet, G.F.; Murray, A.M.; Goris, T.; Burion, M.; Jin, B.; Rolle, M.; Holliger, C.; Maillard, J. Coexistence of two distinct Sulfurospirillum populations respiring tetrachloroethene-genomic and kinetic considerations. FEMS Microbiol. Ecol. 2018, 94. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Tourova, T.P.; Muyzer, G. Isolation and characterization of two novel alkalitolerant sulfidogens from a Thiopaq bioreactor, Desulfonatronum alkalitolerans sp. nov., and Sulfurospirillum alkalitolerans sp. nov. Extremophiles 2013, 17, 535–543. [Google Scholar] [CrossRef]
- Margulis, L.; Barghoorn, E.S.; Ashendorf, D.; Banjeree, S.; Chase, D.; Francis, S.; Giovannoni, S.; Stolz, J. The microbial community in the layered sediments at Laguna Figueroa, Baja California, Mexico: Does it have Precambrian analogues? Precambr. Res. 1980, 11, 93–123. [Google Scholar] [CrossRef]
- Nicholson, J.A.; Stolz, J.F.; Pierson, B.K. Structure of a microbial mat at Great Sippewissett Marsh, Cape Cod, Massachusetts. FEMS Microbiol. Ecol. 1987, 45, 343–346. [Google Scholar] [CrossRef]
- Pierson, B.K.; Oesterle, A.; Murphy, G.L. Pigments, light penetration and photosynthetic activity in the multilayered microbial mats of Great Sippewissett Salt Marsh, Massachusetts. FEMS Microbiol. Ecol. 1987, 45, 365–376. [Google Scholar] [CrossRef]
- Lynum, C.A.; Bulseco, A.N.; Dunphy, C.M.; Osborne, S.M.; Vineis, J.H.; Bowen, J.L. Microbial community response to a passive salt marsh restoration. Estuaries Coasts 2020, 43, 1439–1455. [Google Scholar] [CrossRef]
Figure 1.
(A) Map of the sampling locations for this study. Samples were taken at the valley of Salt Creek and Little Salt Creek near Lincoln, Nebraska. The sampling area within the salt marsh is marked in red. All maps were generated using Google Maps (B) Overview of the NE Salt Marsh sampling locations: SM2B, SM2C, SM2A, and SM2E.
Figure 1.
(A) Map of the sampling locations for this study. Samples were taken at the valley of Salt Creek and Little Salt Creek near Lincoln, Nebraska. The sampling area within the salt marsh is marked in red. All maps were generated using Google Maps (B) Overview of the NE Salt Marsh sampling locations: SM2B, SM2C, SM2A, and SM2E.

Figure 2.
A dendrogram showing the hierarchical clustering of the metagenomes from the three sampling locations, based on genus-level classifications of 16S rRNA gene amplicon samples. Only families with >3% of the sequencing reads are represented in the figure legend.
Figure 2.
A dendrogram showing the hierarchical clustering of the metagenomes from the three sampling locations, based on genus-level classifications of 16S rRNA gene amplicon samples. Only families with >3% of the sequencing reads are represented in the figure legend.

Figure 3.
Whole-genome-based phylogenetic tree of all sequenced Prosthecochloris species. The phylogenetic tree was generated using the CodonTree method within PATRIC [16], which used PGFams as homology groups. The support values for the phylogenetic tree were generated using 100 rounds of the “Rapid bootstrapping” option of RaxML [19]. Chlorobaculum sp. 24CR was used as an outgroup [63]. The branch length tree scale is defined as the mean number of substitutions per site, which is the average across both nucleotide and amino acid changes. The NE Salt Marsh Prosthecochloris sp. SM2 is shown in red.
Figure 3.
Whole-genome-based phylogenetic tree of all sequenced Prosthecochloris species. The phylogenetic tree was generated using the CodonTree method within PATRIC [16], which used PGFams as homology groups. The support values for the phylogenetic tree were generated using 100 rounds of the “Rapid bootstrapping” option of RaxML [19]. Chlorobaculum sp. 24CR was used as an outgroup [63]. The branch length tree scale is defined as the mean number of substitutions per site, which is the average across both nucleotide and amino acid changes. The NE Salt Marsh Prosthecochloris sp. SM2 is shown in red.

Figure 4.
Whole genome-based phylogenetic tree of sequenced Chromatiaceae species. The phylogenetic tree was generated using the CodonTree method within PATRIC [16], which used PGFams as homology groups. The support values for the phylogenetic tree were generated using 100 rounds of the “Rapid bootstrapping” option of RaxML [19]. Halochromatium sp. A10 was used as an outgroup. The branch length tree scale is defined as the mean number of substitutions per site, which is the average across both nucleotide and amino acid changes. The NE Salt Marsh Marichromatium sp. SM2 is shown in red.
Figure 4.
Whole genome-based phylogenetic tree of sequenced Chromatiaceae species. The phylogenetic tree was generated using the CodonTree method within PATRIC [16], which used PGFams as homology groups. The support values for the phylogenetic tree were generated using 100 rounds of the “Rapid bootstrapping” option of RaxML [19]. Halochromatium sp. A10 was used as an outgroup. The branch length tree scale is defined as the mean number of substitutions per site, which is the average across both nucleotide and amino acid changes. The NE Salt Marsh Marichromatium sp. SM2 is shown in red.

Figure 5.
Simplified overview of the proposed sulfur cycling in microbial ecosystems. Bacterial genera identified in the NE Salt Marsh samples that are assumed to be involved are indicated for each group. AER: aerobic reactions; ANAER: anaerobic reactions. Image created using BioRender.com.
Figure 5.
Simplified overview of the proposed sulfur cycling in microbial ecosystems. Bacterial genera identified in the NE Salt Marsh samples that are assumed to be involved are indicated for each group. AER: aerobic reactions; ANAER: anaerobic reactions. Image created using BioRender.com.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Relative abundance at the genus level of the most abundant genera in each of the four samples. The abundance is given as a percentage of the total sequencing reads for each sample, as calculated by the 16S Metagenomics analysis in BaseSpace (Illumina). Abundances over 2% are indicated in bold and discussed in the text; n.d.: not detected.
Table 1.
Relative abundance at the genus level of the most abundant genera in each of the four samples. The abundance is given as a percentage of the total sequencing reads for each sample, as calculated by the 16S Metagenomics analysis in BaseSpace (Illumina). Abundances over 2% are indicated in bold and discussed in the text; n.d.: not detected.
| Classification | SM2A | SM2B | SM2C | SM2E |
|---|---|---|---|---|
| GpI/GpIX | 3.9 | 13.9 | 12.3 | 2.3 |
| Bacillariophyta | 17.1 | 3.4 | 6.4 | 16.8 |
| Rubribacterium | <2 | 4.8 | 5.0 | <2 |
| Pseudoalteromonas | 14.1 | n.d. | n.d. | <2 |
| Parcubacteria | 7.3 | n.d. | <2 | <1 |
| Psychroflexus | 6.3 | <1 | <1 | <2 |
| Sulfurimonas | <2 | <2 | <2 | 16.3 |
| Arcobacter | <2 | n.d. | n.d. | 5 |
| Thiomicrospira | <2 | <2 | <1 | 3.5 |
| Halochromatium | <1 | <2 | <2 | 2.1 |
| Salinivibrio | 2.1 | n.d. | <1 | <1 |
| Unclassified | 12.4 | 31.4 | 27.3 | 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Athen, S.R.; Dubey, S.; Kyndt, J.A. The Eastern Nebraska Salt Marsh Microbiome Is Well Adapted to an Alkaline and Extreme Saline Environment. Life 2021, 11, 446. https://0-doi-org.brum.beds.ac.uk/10.3390/life11050446
AMA Style
Athen SR, Dubey S, Kyndt JA. The Eastern Nebraska Salt Marsh Microbiome Is Well Adapted to an Alkaline and Extreme Saline Environment. Life. 2021; 11(5):446. https://0-doi-org.brum.beds.ac.uk/10.3390/life11050446
Chicago/Turabian StyleAthen, Sierra R., Shivangi Dubey, and John A. Kyndt. 2021. "The Eastern Nebraska Salt Marsh Microbiome Is Well Adapted to an Alkaline and Extreme Saline Environment" Life 11, no. 5: 446. https://0-doi-org.brum.beds.ac.uk/10.3390/life11050446
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.