
Human Amniotic Mesenchymal Stem Cells and Fibroblasts Accelerate Wound Repair of Cystic Fibrosis Epithelium

 , , , ,
, , , ,  and
and
Abstract
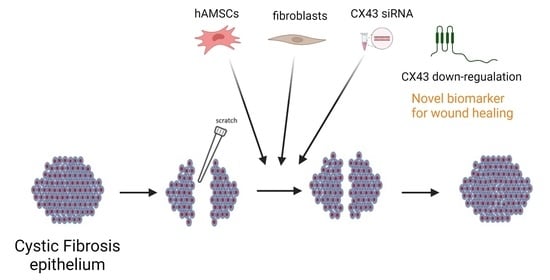
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. hAMSC Cultures and Phenotype
2.2. Cell Cultures
2.3. Co-Cultures onto 24-Well Plates
2.4. Wound Repair Assay in 24-Well Plates
2.5. CM-DiI Labelling of hAMSCs
2.6. Irradiation of CFBE Cells
2.7. Cell Proliferation Assays
2.8. Wound Repair on Irradiated Monolayers (60 mm Dishes)
2.9. Statistical Analysis
3. Results
3.1. Phenotypic Characteristics of hAMSCs Are Maintained until p5
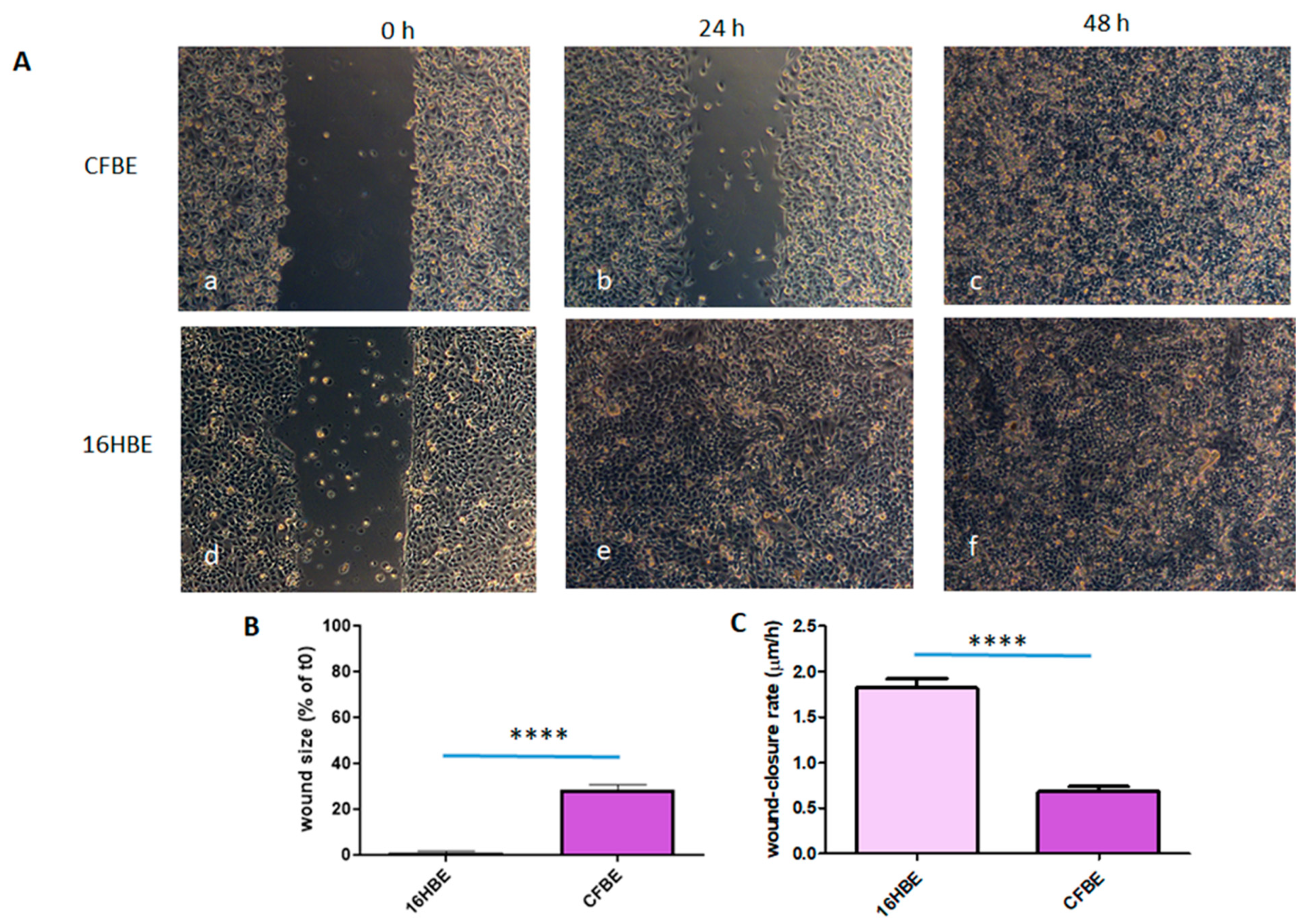
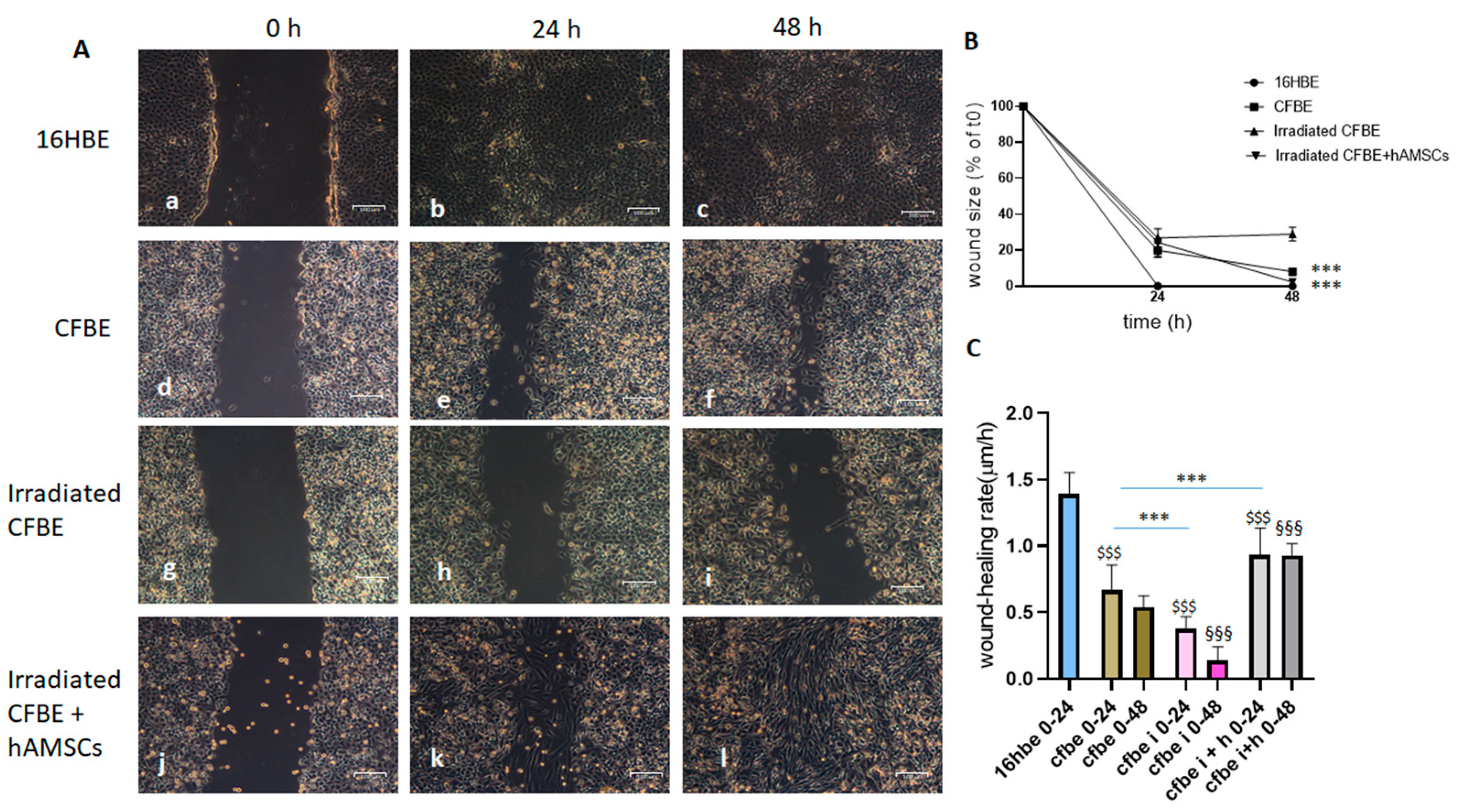
3.2. Wound Closure Is Delayed in CFBE Cells
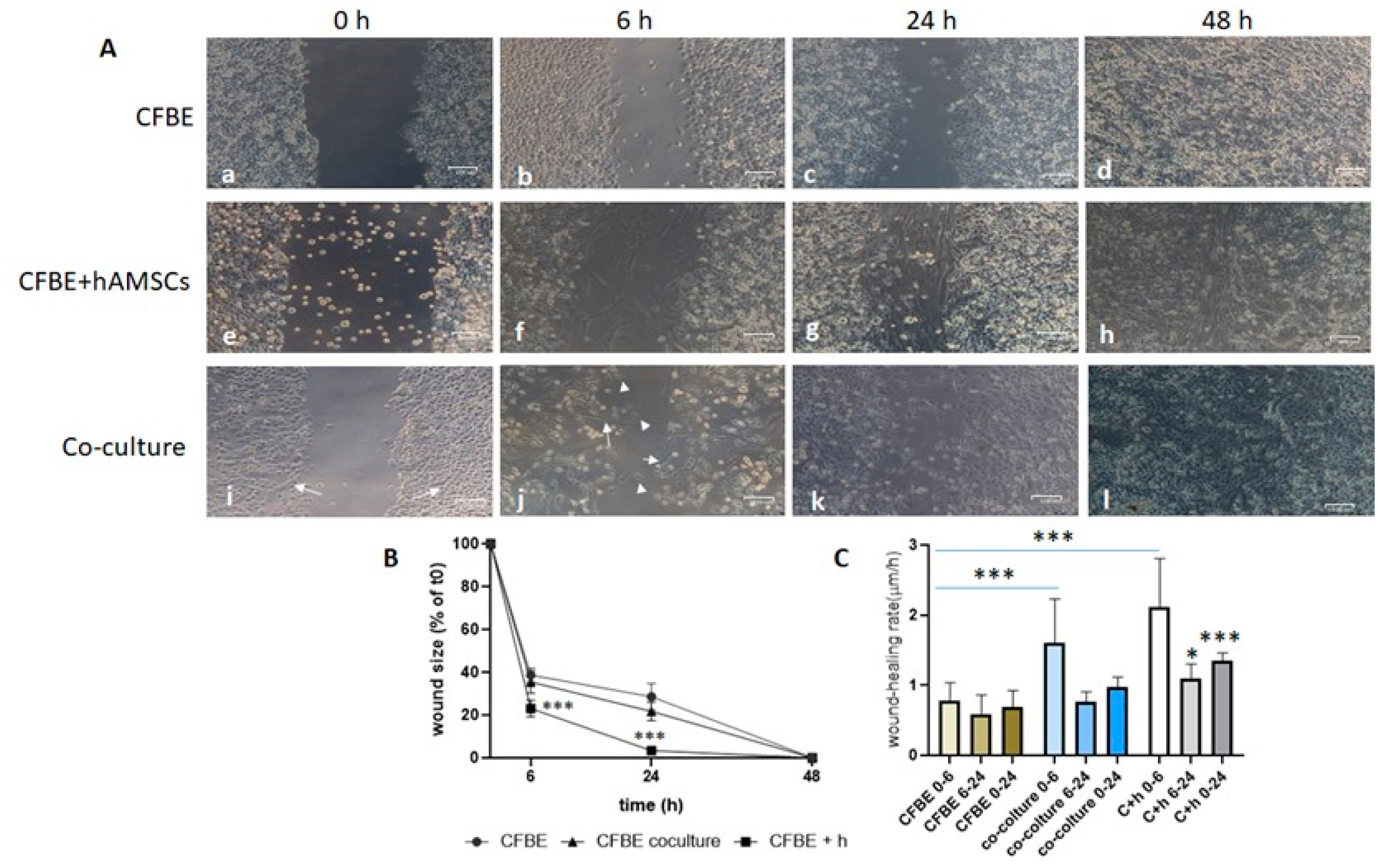
3.3. hAMSCs Accelerate Wound Closure of CF Monolayers
3.4. Block of Cell Proliferation Retards CFBE Wound Closure
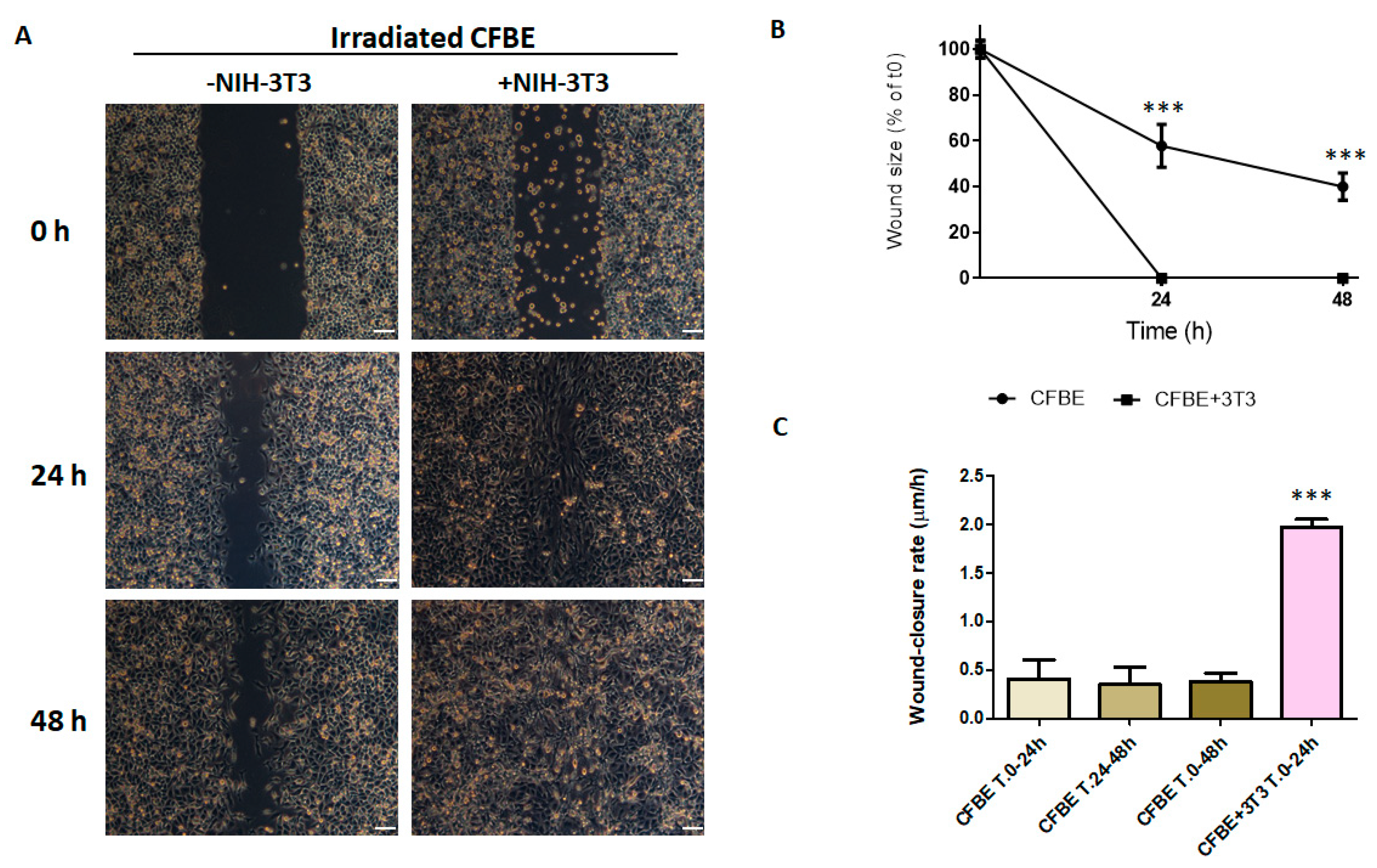
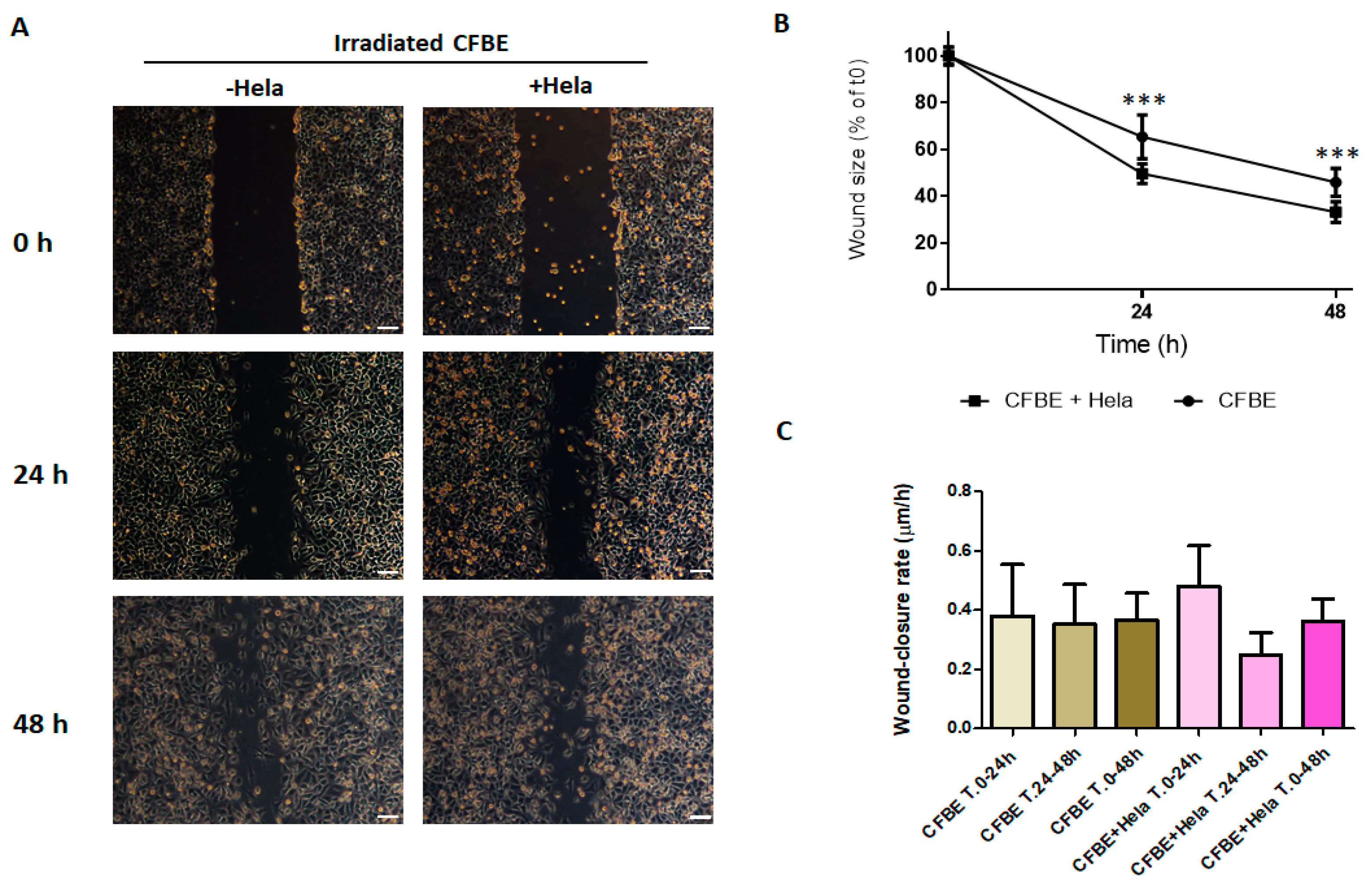
3.5. NIH-3T3 Cells, but Not HeLa Cells, Accelerate Wound Closure
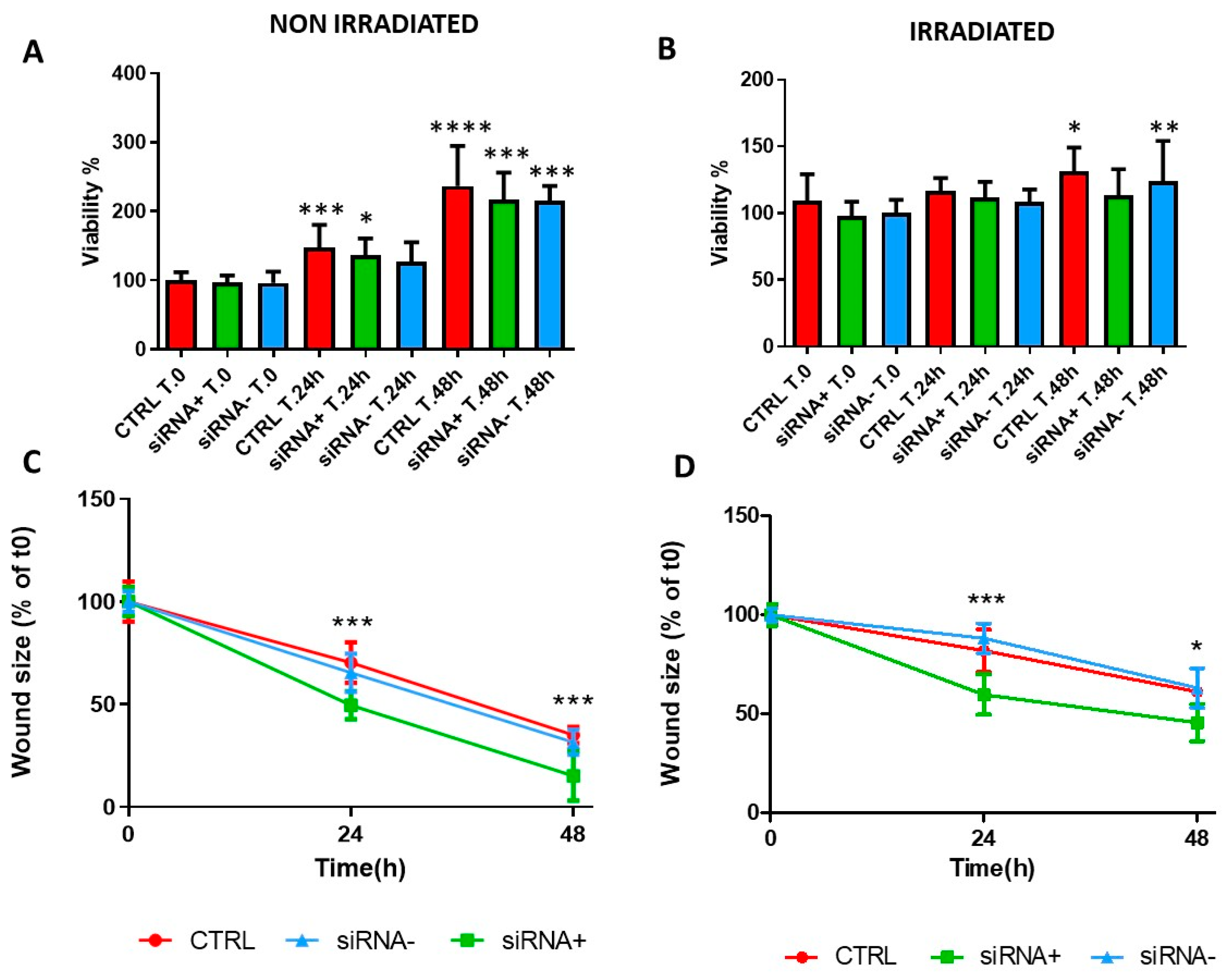
3.6. CX43 siRNA Accelerates CFBE Wound Closure without Affecting Cell Proliferation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Welsh, M.J.; Smith, A.E. Molecolar mechanism of CFTR chloride channel dysfuction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- Matsui, H.; Grubb, B.R.; Tarran, R.; Randell, S.H.; Gatzy, J.T.; Davis, C.W.; Boucher, R.C. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airway disease. Cell 1998, 95, 1005–1015. [Google Scholar] [CrossRef] [Green Version]
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef]
- Hubeau, C.; Lorenzato, M.; Couetil, J.P.; Hubert, D.; Dusser, D.; Puchelle, E.; Gaillard, D. Quantitative analysis of inflammatory cells infiltrating the cystic fibrosis airway mucosa. Clin. Exp. Immunol. 2001, 124, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Voynow, J.A.; Fischer, B.M.; Roberts, B.C.; Proia, A.D. Basal-like cells constitute the proliferating cell population in cystic fibrosis airways. Am. J. Respir. Crit. Care Med. 2005, 172, 1013–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, M.W.; Kylander, J.E.; Yankaskas, J.R.; Boucher, R.C. Cell proliferation in bronchial epithelium and submucosal glands of cystic fibrosis patients. Am. J. Respir. Cell Mol. Biol. 1995, 12, 605–612. [Google Scholar] [CrossRef]
- Adam, D.; Roux-Delrieu, J.; Luczka, E.; Bonnomet, A.; Lesage, J.; Merol, J.C.; Polette, M.; Abely, M.; Coraux, C. Cystic fibrosis airway epithelium remodelling: Involvement of inflammation. J. Pathol. 2015, 235, 408–419. [Google Scholar] [CrossRef]
- Piorunek, T.; Marszalek, A.; Biczysko, W.; Gozdzik, J.; Cofta, S.; Seget, M. Correlation between the stage of cystic fibrosis and the level of morphological changes in adult patients. J. Physiol. Pharmacol. 2008, 59 (Suppl. S6), 565–572. [Google Scholar]
- Tiddens, H.A.; Koopman, L.P.; Lambert, R.K.; Elliott, W.M.; Hop, W.C.; van der Mark, T.W.; de Boer, W.J.; de Jongste, J.C. Cartilaginous airway wall dimensions and airway resistance in cystic fibrosis lungs. Eur. Respir. J. 2000, 15, 735–742. [Google Scholar] [CrossRef] [Green Version]
- Dovey, M.; Wisseman, C.L.; Roggli, V.L.; Roomans, G.M.; Shelburne, J.D.; Spock, A. Ultrastructural morphology of the lung in cystic fibrosis. J. Submicrosc. Cytol. Pathol. 1989, 21, 521–534. [Google Scholar]
- Durieu, I.; Peyrol, S.; Gindre, D.; Bellon, G.; Durand, D.V.; Pacheco, Y. Subepithelial fibrosis and degradation of the bronchial extracellular matrix in cystic fibrosis. Am. J. Respir. Crit. Care Med. 1998, 158, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.D.; Quaresma, M.C.; Pankonien, I. What Role Does CFTR Play in Development, Differentiation, Regeneration and Cancer? Int. J. Mol. Sci. 2020, 21, 3133. [Google Scholar] [CrossRef] [PubMed]
- Rejman, J.; Colombo, C.; Conese, M. Engraftment of bone marrow-derived stem cells to the lung in a model of acute respiratory infection by Pseudomonas aeruginosa. Mol. Ther. 2009, 17, 1257–1265. [Google Scholar] [CrossRef]
- de Bentzmann, S.; Polette, M.; Zahm, J.-M.; Hinnrasky, J.K.C.; Bajolet, O.; Klossek, J.-M.; Filloux, A.; Lazdunski, A.; Puchelle, E. Pseudomonas aeruginosa virulence factors delay airway epithelial wound repair by altering the actin cytoskeleton and inducing overactivation of epithelial matrix metalloproteinase-2. Lab. Investig. 2000, 80, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffin, M.; Bilodeau, C.; Maille, E.; LaFayette, S.L.; McKay, G.A.; Trinh, N.T.; Beaudoin, T.; Desrosiers, M.Y.; Rousseau, S.; Nguyen, D.; et al. Quorum-sensing inhibition abrogates the deleterious impact of Pseudomonas aeruginosa on airway epithelial repair. FASEB J. 2016, 30, 3011–3025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Criq, V.; Villeret, B.; Bastaert, F.; Kheir, S.; Hatton, A.; Cazes, A.; Xing, Z.; Sermet-Gaudelus, I.; Garcia-Verdugo, I.; Edelman, A.; et al. Pseudomonas aeruginosa LasB protease impairs innate immunity in mice and humans by targeting a lung epithelial cystic fibrosis transmembrane regulator-IL-6-antimicrobial-repair pathway. Thorax 2018, 73, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Maheshwari, A.; Chandra, A. Biomarkers for wound healing and their evaluation. J. Wound Care 2016, 25, 46–55. [Google Scholar] [CrossRef]
- Lindley, L.E.; Stojadinovic, O.; Pastar, I.; Tomic-Canic, M. Biology and Biomarkers for Wound Healing. Plast. Reconstr. Surg. 2016, 138, 18S–28S. [Google Scholar] [CrossRef]
- Trinh, N.T.; Prive, A.; Maille, E.; Noel, J.; Brochiero, E. EGF and K+ channel activity control normal and cystic fibrosis bronchial epithelia repair. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L866–L880. [Google Scholar] [CrossRef] [Green Version]
- Maille, E.; Trinh, N.T.; Prive, A.; Bilodeau, C.; Bissonnette, E.; Grandvaux, N.; Brochiero, E. Regulation of normal and cystic fibrosis airway epithelial repair processes by TNF-alpha after injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, L945–L955. [Google Scholar] [CrossRef] [Green Version]
- Trinh, N.T.; Bardou, O.; Prive, A.; Maille, E.; Adam, D.; Lingee, S.; Ferraro, P.; Desrosiers, M.Y.; Coraux, C.; Brochiero, E. Improvement of defective cystic fibrosis airway epithelial wound repair after CFTR rescue. Eur. Respir. J. 2012, 40, 1390–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, R.; Umer, H.M.; Björkqvist, M.; Roomans, G.M. ENaC, iNOS, mucins expression and wound healing in cystic fibrosis airway epithelial and submucosal cells. Cell Biol. Int. Rep. 2014, 21, 25–38. [Google Scholar]
- Adam, D.; Bilodeau, C.; Sognigbe, L.; Maille, E.; Ruffin, M.; Brochiero, E. CFTR rescue with VX-809 and VX-770 favors the repair of primary airway epithelial cell cultures from patients with class II mutations in the presence of Pseudomonas aeruginosa exoproducts. J. Cyst. Fibros. 2018, 17, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, M.C.; Pankonien, I.; Clarke, L.A.; Sousa, L.S.; Silva, I.A.L.; Railean, V.; Dousova, T.; Fuxe, J.; Amaral, M.D. Mutant CFTR Drives TWIST1 mediated epithelial-mesenchymal transition. Cell Death Dis. 2020, 11, 920. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.; Pankonien, I.; Simoes, F.B.; Chanson, M.; Amaral, M.D. Impact of KLF4 on Cell Proliferation and Epithelial Differentiation in the Context of Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 6717. [Google Scholar] [CrossRef]
- Schiller, K.R.; Maniak, P.J.; O’Grady, S.M. Cystic fibrosis transmembrane conductance regulator is involved in airway epithelial wound repair. Am. J. Physiol. Cell Physiol. 2010, 299, C912–C921. [Google Scholar] [CrossRef] [Green Version]
- Itokazu, Y.; Pagano, R.E.; Schroeder, A.S.; O’Grady, S.M.; Limper, A.H.; Marks, D.L. Reduced GM1 ganglioside in CFTR-deficient human airway cells results in decreased beta1-integrin signaling and delayed wound repair. Am. J. Physiol. Cell Physiol. 2014, 306, C819–C830. [Google Scholar] [CrossRef] [Green Version]
- Crespin, S.; Bacchetta, M.; Bou Saab, J.; Tantilipikorn, P.; Bellec, J.; Dudez, T.; Nguyen, T.H.; Kwak, B.R.; Lacroix, J.S.; Huang, S.; et al. Cx26 regulates proliferation of repairing basal airway epithelial cells. Int. J. Biochem. Cell Biol. 2014, 52, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Zoso, A.; Sofoluwe, A.; Bacchetta, M.; Chanson, M. Transcriptomic profile of cystic fibrosis airway epithelial cells undergoing repair. Sci. Data 2019, 6, 240. [Google Scholar] [CrossRef] [Green Version]
- Quaresma, M.C.; Botelho, H.M.; Pankonien, I.; Rodrigues, C.S.; Pinto, M.C.; Costa, P.R.; Duarte, A.; Amaral, M.D. Exploring YAP1-centered networks linking dysfunctional CFTR to epithelial-mesenchymal transition. Life Sci. Alliance 2022, 5, e202101326. [Google Scholar] [CrossRef]
- Mercier, J.; Calmel, C.; Mesinele, J.; Sutanto, E.; Merabtene, F.; Longchampt, E.; Sage, E.; Kicic, A.; Boelle, P.Y.; Corvol, H.; et al. SLC6A14 Impacts Cystic Fibrosis Lung Disease Severity via mTOR and Epithelial Repair Modulation. Front. Mol. Biosci. 2022, 9, 850261. [Google Scholar] [CrossRef] [PubMed]
- Paracchini, V.; Carbone, A.; Colombo, F.; Castellani, S.; Mazzucchelli, S.; Gioia, S.D.; Degiorgio, D.; Seia, M.; Porretti, L.; Colombo, C.; et al. Amniotic mesenchymal stem cells: A new source for hepatocyte-like cells and induction of CFTR expression by coculture with cystic fibrosis airway epithelial cells. J. Biomed. Biotechnol. 2012, 2012, 575471. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Castellani, S.; Favia, M.; Diana, A.; Paracchini, V.; Di Gioia, S.; Seia, M.; Casavola, V.; Colombo, C.; Conese, M. Correction of defective CFTR/ENaC function and tightness of cystic fibrosis airway epithelium by amniotic mesenchymal stromal (stem) cells. J. Cell Mol. Med. 2014, 18, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Zefferino, R.; Beccia, E.; Casavola, V.; Castellani, S.; Di Gioia, S.; Giannone, V.; Seia, M.; Angiolillo, A.; Colombo, C.; et al. Gap Junctions Are Involved in the Rescue of CFTR-Dependent Chloride Efflux by Amniotic Mesenchymal Stem Cells in Coculture with Cystic Fibrosis CFBE41o-Cells. Stem. Cells Int. 2018, 2018, 1203717. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, M.; Kannan, S. Fibroblasts and mesenchymal stem cells: Two sides of the same coin? J. Cell Physiol. 2018, 233, 9099–9109. [Google Scholar] [CrossRef]
- Zupan, J. Mesenchymal Stem/Stromal Cells and Fibroblasts: Their Roles in Tissue Injury and Regeneration, and Age-Related Degeneration. In Fibroblasts—Advances in Inflammation, Autoimmunity and Cancer; Bertoncelj, M.F., Lakota, K., Eds.; IntechOpen: London, UK, 2021; pp. 1–25. [Google Scholar]
- Buechler, M.B.; Turley, S.J. A short field guide to fibroblast function in immunity. Semin. Immunol. 2018, 35, 48–58. [Google Scholar] [CrossRef]
- Ichim, T.E.; O’Heeron, P.; Kesari, S. Fibroblasts as a practical alternative to mesenchymal stem cells. J. Transl. Med. 2018, 16, 212. [Google Scholar] [CrossRef] [Green Version]
- Sutton, M.T.; Fletcher, D.; Episalla, N.; Auster, L.; Kaur, S.; Gwin, M.C.; Folz, M.; Velasquez, D.; Roy, V.; van Heeckeren, R.; et al. Mesenchymal Stem Cell Soluble Mediators and Cystic Fibrosis. J. Stem. Cell Res. Ther. 2017, 7, 400. [Google Scholar] [CrossRef] [Green Version]
- Sutton, M.T.; Fletcher, D.; Ghosh, S.K.; Weinberg, A.; van Heeckeren, R.; Kaur, S.; Sadeghi, Z.; Hijaz, A.; Reese, J.; Lazarus, H.M.; et al. Antimicrobial Properties of Mesenchymal Stem Cells: Therapeutic Potential for Cystic Fibrosis Infection, and Treatment. Stem. Cells Int. 2016, 2016, 5303048. [Google Scholar] [CrossRef] [Green Version]
- Matteo, M.; Beccia, E.; Carbone, A.; Castellani, S.; Milillo, L.; Lauritano, D.; Di Gioia, S.; Angiolillo, A.; Conese, M. Effect of Mother’s Age and Pathology on Functional Behavior of Amniotic Mesenchymal Stromal Cells—Hints for Bone Regeneration. Appl. Sci. 2019, 9, 3471. [Google Scholar] [CrossRef] [Green Version]
- Gruenert, D.C.; Willems, M.; Cassiman, J.J.; Frizzell, R.A. Established cell lines used in cystic fibrosis research. J. Cyst. Fibros. 2004, 3, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- HeLa. Available online: http://www.atcc.org/products/ccl-2 (accessed on 16 April 2022).
- Trapani, A.; Guerra, L.; Corbo, F.; Castellani, S.; Sanna, E.; Capobianco, L.; Monteduro, A.G.; Manno, D.E.; Mandracchia, D.; Di Gioia, S.; et al. Cyto/Biocompatibility of Dopamine Combined with the Antioxidant Grape Seed-Derived Polyphenol Compounds in Solid Lipid Nanoparticles. Molecules 2021, 26, 916. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Lo, C.W. Expression of a connexin 43/beta-galactosidase fusion protein inhibits gap junctional communication in NIH3T3 cells. J. Cell Biol. 1995, 130, 419–429. [Google Scholar] [CrossRef]
- Mesnil, M.; Krutovskikh, V.; Piccoli, C.; Elfgang, C.; Traub, O.; Willecke, K.; Yamasaki, H. Negative growth control of HeLa cells by connexin genes: Connexin species specificity. Cancer Res. 1995, 55, 629–639. [Google Scholar] [PubMed]
- Elfgang, C.; Eckert, R.; Lichtenberg-Frate, H.; Butterweck, A.; Traub, O.; Klein, R.A.; Hulser, D.F.; Willecke, K. Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J. Cell Biol. 1995, 129, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Conese, M.; Beccia, E.; Castellani, S.; Di Gioia, S.; Colombo, C.; Angiolillo, A.; Carbone, A. The long and winding road: Stem cells for cystic fibrosis. Expert. Opin. Biol. Ther. 2018, 18, 281–292. [Google Scholar] [CrossRef]
- Bonfield, T.L.; Lennon, D.; Ghosh, S.K.; DiMarino, A.M.; Weinberg, A.; Caplan, A.I. Cell based therapy aides in infection and inflammation resolution in the murine model of cystic fibrosis lung disease. Stem. Cell Discov. 2013, 3, 139–153. [Google Scholar] [CrossRef] [Green Version]
- Zulueta, A.; Colombo, M.; Peli, V.; Falleni, M.; Tosi, D.; Ricciardi, M.; Baisi, A.; Bulfamante, G.; Chiaramonte, R.; Caretti, A. Lung mesenchymal stem cells-derived extracellular vesicles attenuate the inflammatory profile of Cystic Fibrosis epithelial cells. Cell Signal 2018, 51, 110–118. [Google Scholar] [CrossRef]
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, L715–L731. [Google Scholar] [CrossRef] [Green Version]
- Coraux, C.; Martinella-Catusse, C.; Nawrocki-Raby, B.; Hajj, R.; Burlet, H.; Escotte, S.; Laplace, V.; Birembaut, P.; Puchelle, E. Differential expression of matrix metalloproteinases and interleukin-8 during regeneration of human airway epithelium in vivo. J. Pathol. 2005, 206, 160–169. [Google Scholar] [CrossRef]
- Hajj, R.; Lesimple, P.; Nawrocki-Raby, B.; Birembaut, P.; Puchelle, E.; Coraux, C. Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. J. Pathol. 2007, 211, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T. Connexins: Junctional and non-junctional modulators of proliferation. Cell Tissue Res. 2015, 360, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M. Introduction: Connexins, pannexins and their channels as gatekeepers of organ physiology. Cell Mol. Life Sci. 2015, 72, 2775–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.; Tan, T.; Chan, C.; Laxton, V.; Chan, Y.W.; Liu, T.; Wong, W.T.; Tse, G. The Role of Connexins in Wound Healing and Repair: Novel Therapeutic Approaches. Front. Physiol. 2016, 7, 596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanson, M.; Derouette, J.P.; Roth, I.; Foglia, B.; Scerri, I.; Dudez, T.; Kwak, B.R. Gap junctional communication in tissue inflammation and repair. Biochim. Biophys. Acta 2005, 1711, 197–207. [Google Scholar] [CrossRef]
- Chanson, M.; Watanabe, M.; O’Shaughnessy, E.M.; Zoso, A.; Martin, P.E. Connexin Communication Compartments and Wound Repair in Epithelial Tissue. Int. J. Mol. Sci. 2018, 19, 1354. [Google Scholar] [CrossRef]
- Conese, M.; Di Gioia, S. Pathophysiology of Lung Disease and Wound Repair in Cystic Fibrosis. Pathophysiology 2021, 28, 155–188. [Google Scholar] [CrossRef]
- Pankonien, I.; Quaresma, M.C.; Rodrigues, C.S.; Amaral, M.D. CFTR, Cell Junctions and the Cytoskeleton. Int. J. Mol. Sci. 2022, 23, 2688. [Google Scholar] [CrossRef]
- Faniku, C.; O’Shaughnessy, E.; Lorraine, C.; Johnstone, S.R.; Graham, A.; Greenhough, S.; Martin, P.E.M. The Connexin Mimetic Peptide Gap27 and Cx43-Knockdown Reveal Differential Roles for Connexin43 in Wound Closure Events in Skin Model Systems. Int. J. Mol. Sci. 2018, 19, 604. [Google Scholar] [CrossRef] [Green Version]
- Goliger, J.A.; Paul, D.L. Wounding alters epidermal connexin expression and gap junction-mediated intercellular communication. Mol. Biol. Cell 1995, 6, 1491–1501. [Google Scholar] [CrossRef] [Green Version]
- Kretz, M.; Euwens, C.; Hombach, S.; Eckardt, D.; Teubner, B.; Traub, O.; Willecke, K.; Ott, T. Altered connexin expression and wound healing in the epidermis of connexin-deficient mice. J. Cell Sci. 2003, 116, 3443–3452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polusani, S.R.; Kalmykov, E.A.; Chandrasekhar, A.; Zucker, S.N.; Nicholson, B.J. Cell coupling mediated by connexin 26 selectively contributes to reduced adhesivity and increased migration. J. Cell Sci. 2016, 129, 4399–4410. [Google Scholar] [PubMed] [Green Version]
- Ugurlu, B.; Karaoz, E. Comparison of similar cells: Mesenchymal stromal cells and fibroblasts. Acta Histochem. 2020, 122, 151634. [Google Scholar] [CrossRef]
- Alt, E.; Yan, Y.; Gehmert, S.; Song, Y.H.; Altman, A.; Gehmert, S.; Vykoukal, D.; Bai, X. Fibroblasts share mesenchymal phenotypes with stem cells, but lack their differentiation and colony-forming potential. Biol. Cell 2011, 103, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Khatun, M.; Sorjamaa, A.; Kangasniemi, M.; Sutinen, M.; Salo, T.; Liakka, A.; Lehenkari, P.; Tapanainen, J.S.; Vuolteenaho, O.; Chen, J.C.; et al. Niche matters: The comparison between bone marrow stem cells and endometrial stem cells and stromal fibroblasts reveal distinct migration and cytokine profiles in response to inflammatory stimulus. PLoS ONE 2017, 12, e0175986. [Google Scholar] [CrossRef] [PubMed]
- Berical, A.; Lee, R.E.; Randell, S.H.; Hawkins, F. Challenges Facing Airway Epithelial Cell-Based Therapy for Cystic Fibrosis. Front. Pharmacol. 2019, 10, 74. [Google Scholar] [CrossRef]
- Partridge, L.; Deelen, J.; Slagboom, P.E. Facing up to the global challenges of ageing. Nature 2018, 561, 45–56. [Google Scholar] [CrossRef]
- Camernik, K.; Mihelic, A.; Mihalic, R.; Haring, G.; Herman, S.; Marolt Presen, D.; Janez, A.; Trebse, R.; Marc, J.; Zupan, J. Comprehensive analysis of skeletal muscle- and bone-derived mesenchymal stem/stromal cells in patients with osteoarthritis and femoral neck fracture. Stem Cell Res. Ther. 2020, 11, 146. [Google Scholar] [CrossRef]
- Conese, M.; Tirelli, A.S.; Alicandro, G.; Di Gioia, S.; Carbone, A.; Castellani, S.; Colombo, C. Biomarkers of Inflammation and Remodelling in Cystic Fibrosis. Clin. Immunol. Endocr. Metab. Drugs 2016, 3, 92–108. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beccia, E.; Daniello, V.; Laselva, O.; Leccese, G.; Mangiacotti, M.; Di Gioia, S.; La Bella, G.; Guerra, L.; Matteo, M.; Angiolillo, A.; et al. Human Amniotic Mesenchymal Stem Cells and Fibroblasts Accelerate Wound Repair of Cystic Fibrosis Epithelium. Life 2022, 12, 756. https://0-doi-org.brum.beds.ac.uk/10.3390/life12050756
Beccia E, Daniello V, Laselva O, Leccese G, Mangiacotti M, Di Gioia S, La Bella G, Guerra L, Matteo M, Angiolillo A, et al. Human Amniotic Mesenchymal Stem Cells and Fibroblasts Accelerate Wound Repair of Cystic Fibrosis Epithelium. Life. 2022; 12(5):756. https://0-doi-org.brum.beds.ac.uk/10.3390/life12050756
Chicago/Turabian StyleBeccia, Elisa, Valeria Daniello, Onofrio Laselva, Giorgia Leccese, Michele Mangiacotti, Sante Di Gioia, Gianfranco La Bella, Lorenzo Guerra, Maria Matteo, Antonella Angiolillo, and et al. 2022. "Human Amniotic Mesenchymal Stem Cells and Fibroblasts Accelerate Wound Repair of Cystic Fibrosis Epithelium" Life 12, no. 5: 756. https://0-doi-org.brum.beds.ac.uk/10.3390/life12050756