
Revisiting DNA Sequence-Dependent Deformability in High-Resolution Structures: Effects of Flanking Base Pairs on Dinucleotide Morphology and Global Chain Configuration


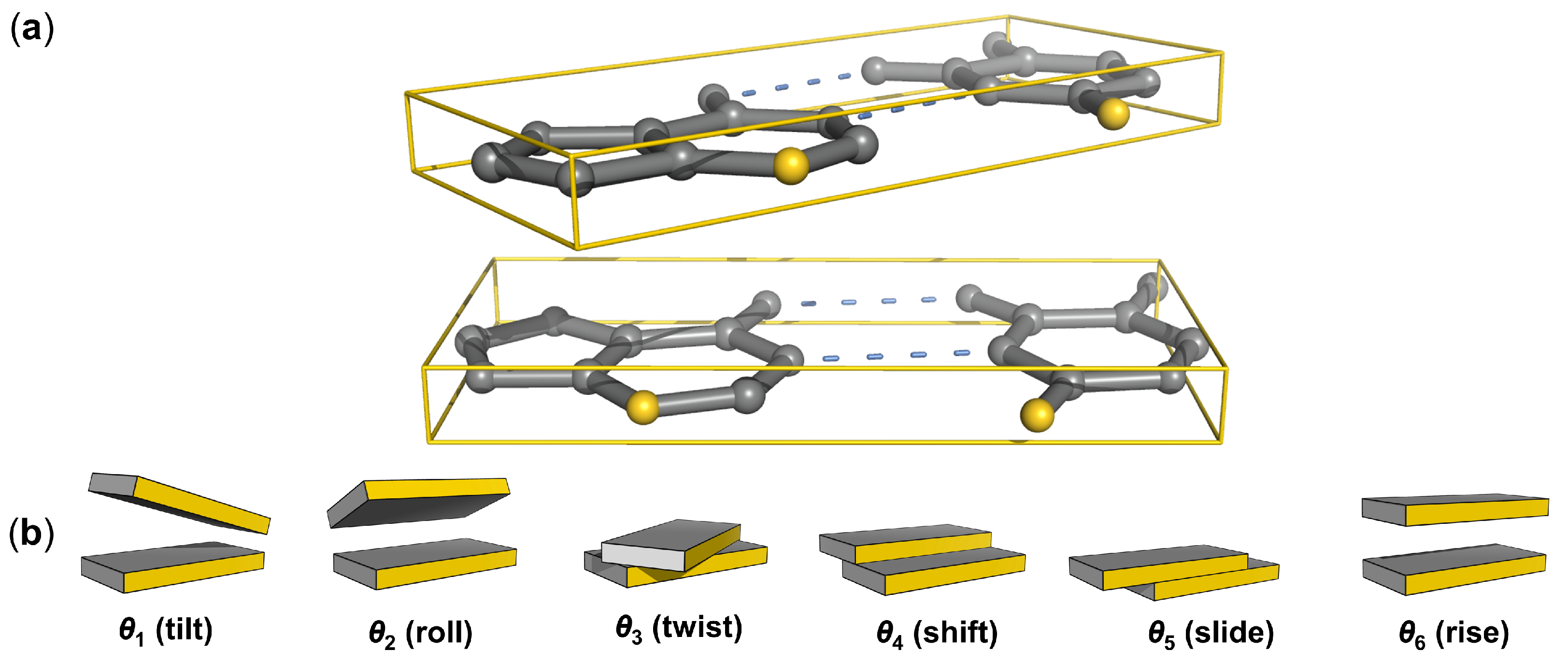{kind=link}
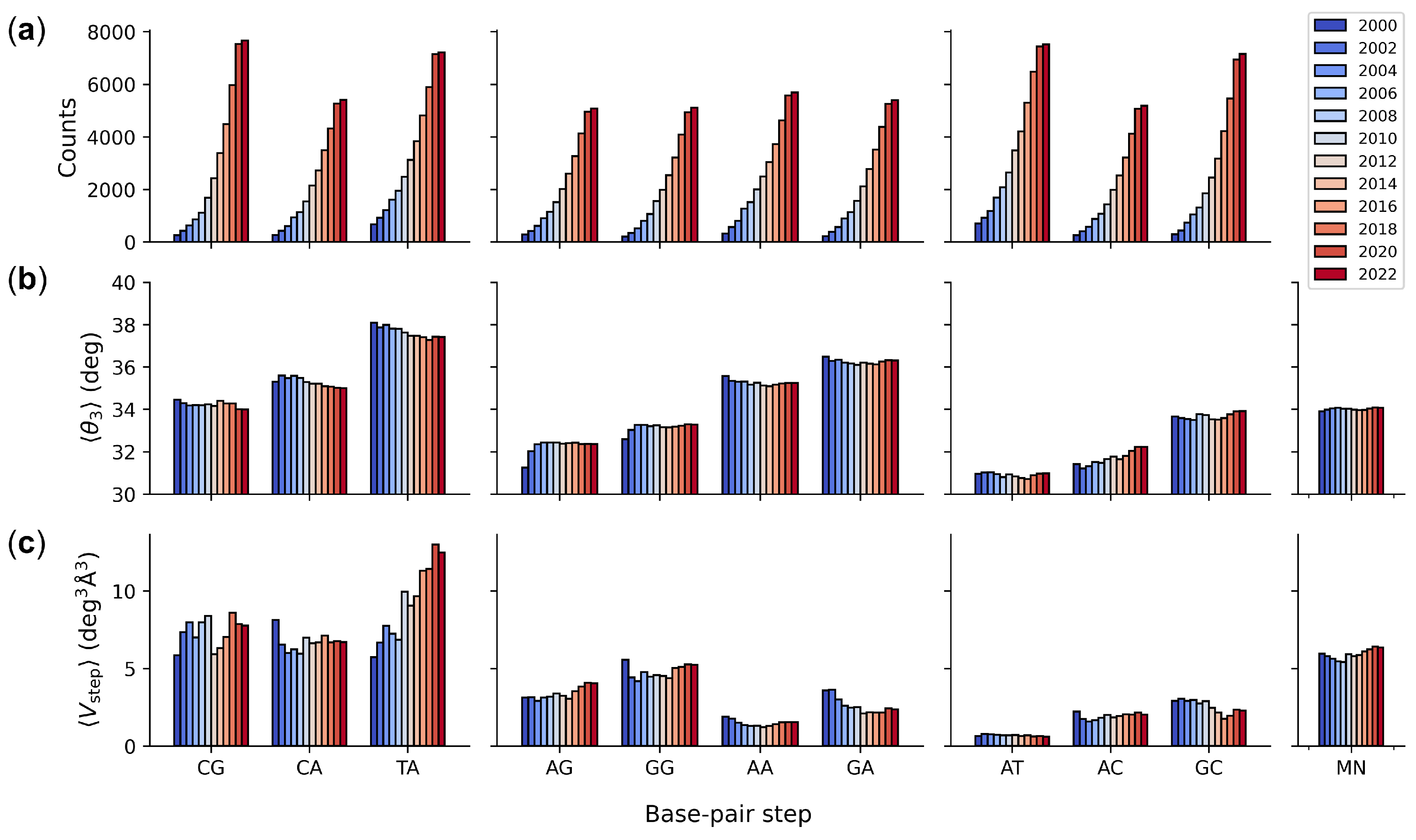{kind=link}
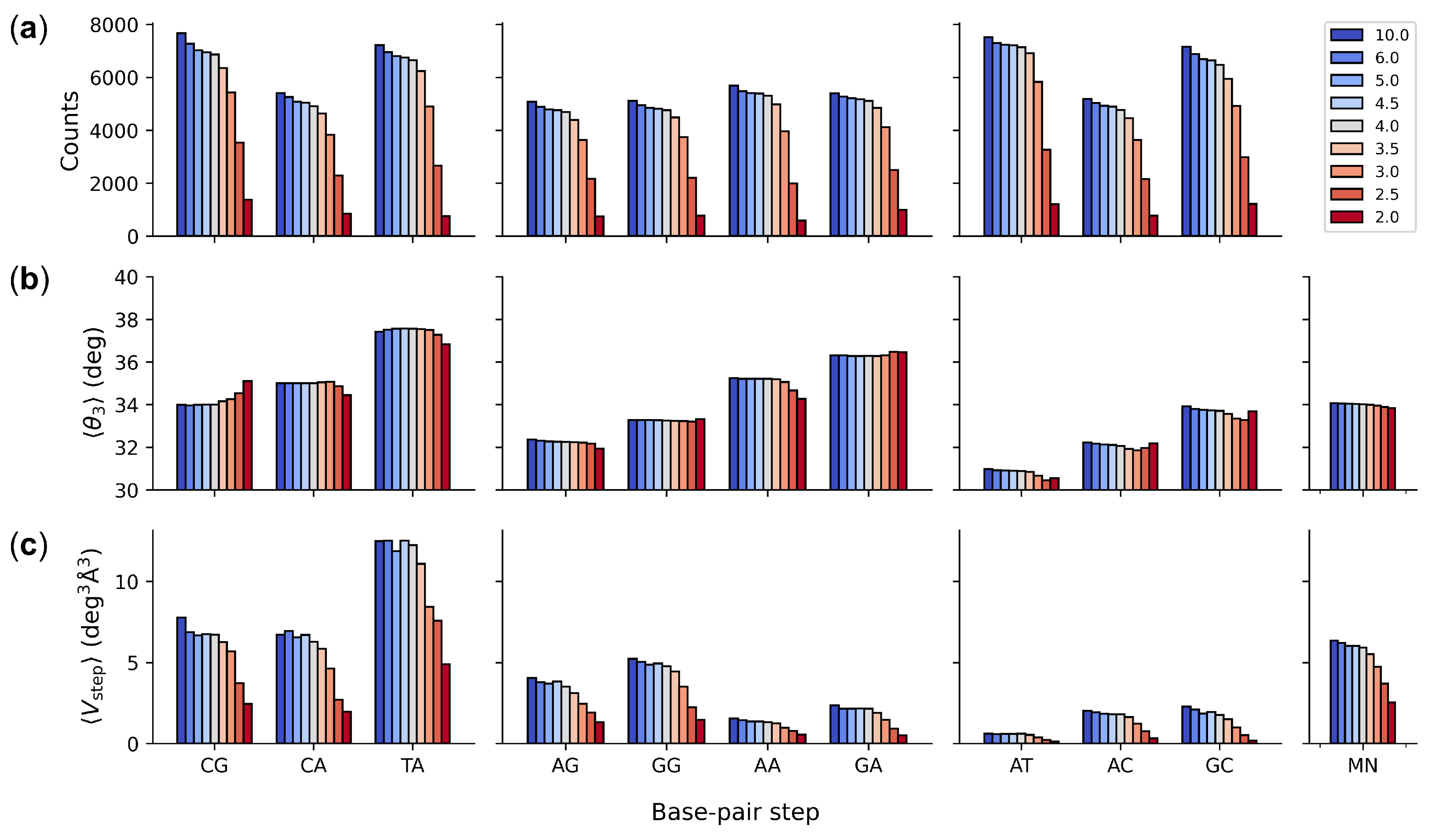{kind=link}
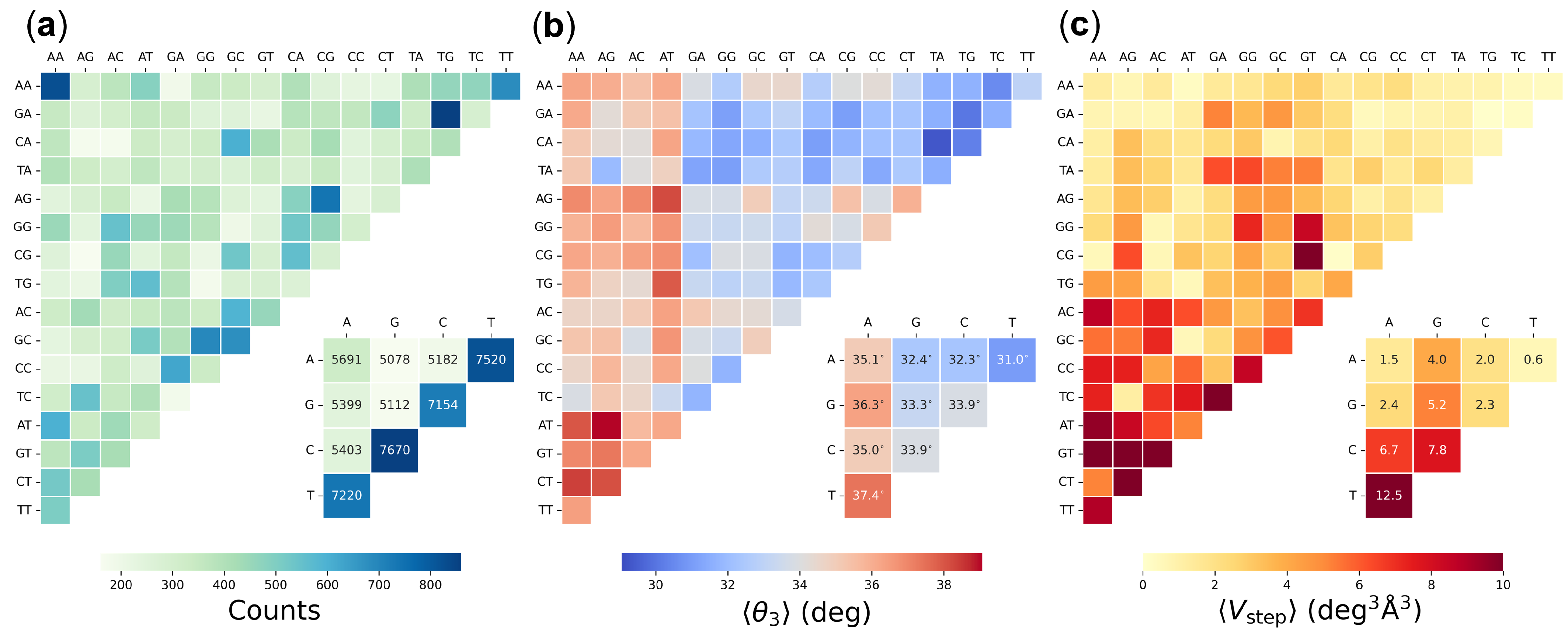{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Configurational States
2.3. Knowledge-Based Potentials
2.4. Energy Optimization
2.5. Ring-Closure Propensities
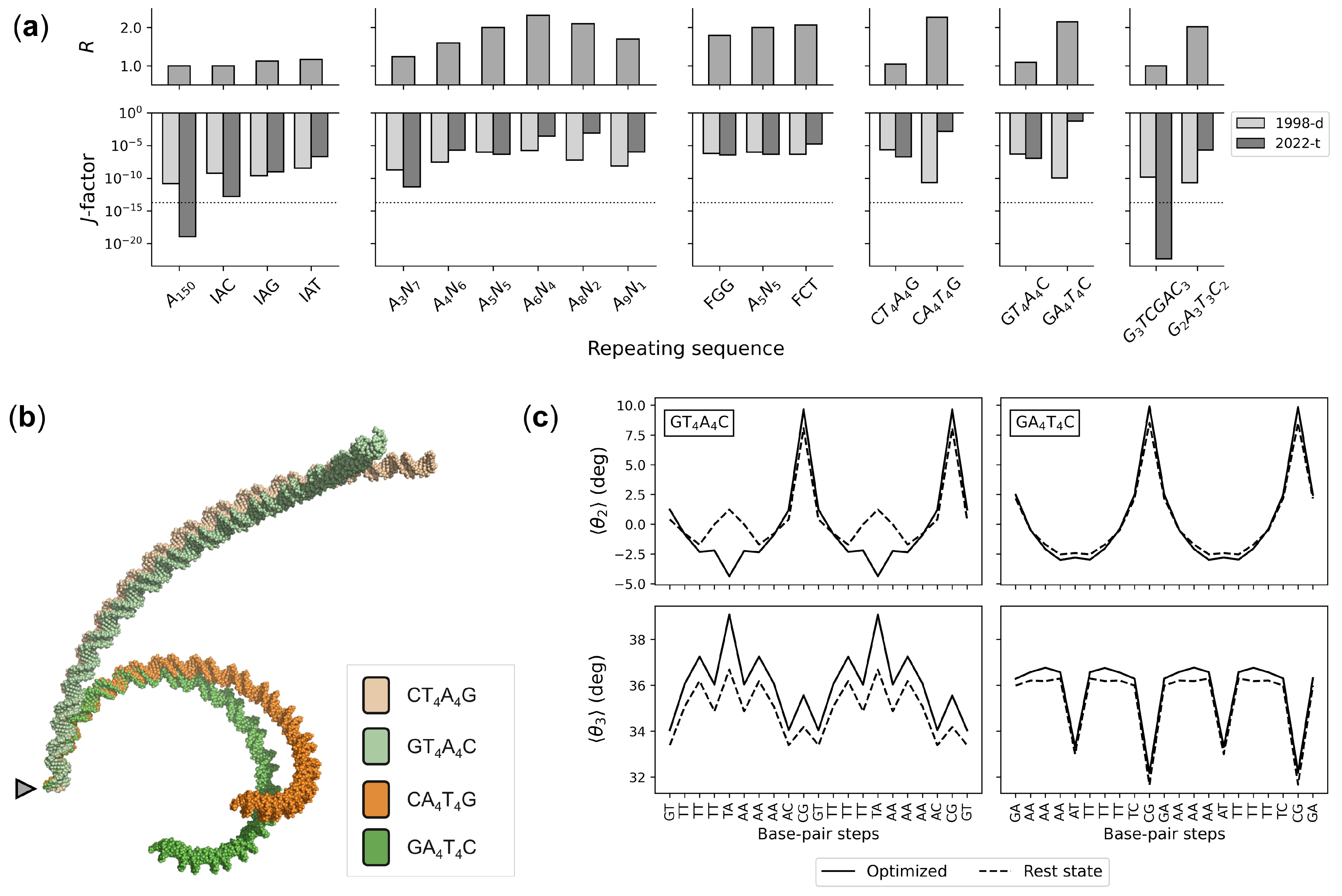
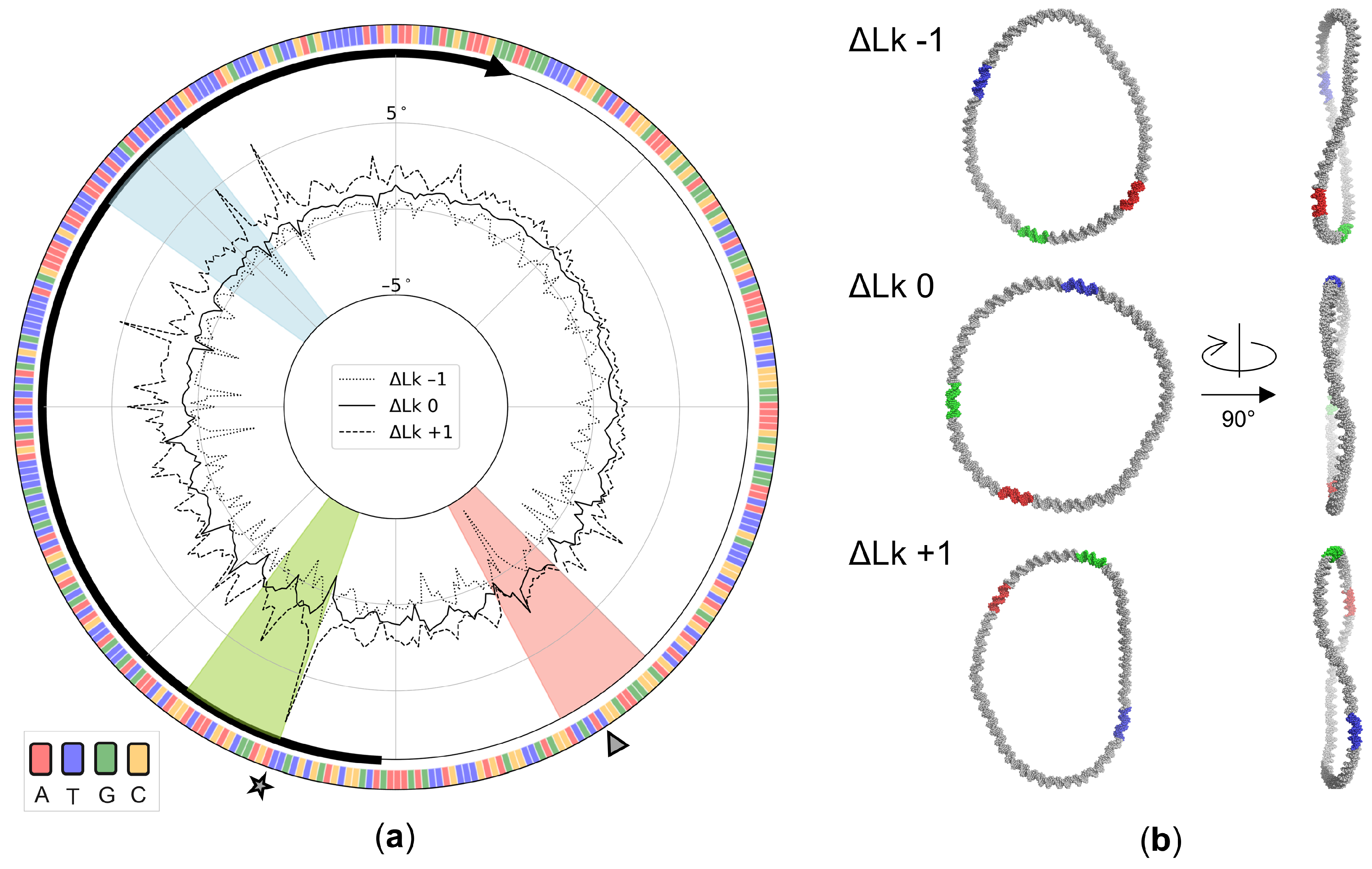
3. Results
3.1. Base-Pair Steps within High-Resolution Structures
3.2. Effects of Sequence Context on Base-Pair Structure and Deformability
3.3. Sequence-Dependent DNA Curvature
3.4. Sequence-Dependent Twist Uptake in DNA Minicircles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Watson, J.D.; Crick, F.H.C. Molecular structure of nucleic acids. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.E.; Gosling, R.G. Molecular configuration in sodium thymonucleate. Nature 1953, 171, 740–741. [Google Scholar] [CrossRef] [PubMed]
- Marvin, D.A.; Spencer, M.; Wilkins, M.H.F.; Hamilton, L.D. A new configuration of deoxyribonucleic acid. Nature 1958, 182, 387–388. [Google Scholar] [CrossRef] [PubMed]
- Arnott, S.; Dover, S.D.; Wonacott, A.J. Least-squares refinement of the crystal and molecular structures of DNA and RNA from X-ray data and standard bond lengths and angles. Acta Crystallogr. 1969, B25, 2192–2206. [Google Scholar] [CrossRef]
- Lu, X.J.; Olson, W.K. 3DNA: A software package for the analysis, rebuilding, and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef] [Green Version]
- Watson, J.D.; Crick, F.H.C. Genetical implications of the structure of deoxyribonucleic acid. Nature 1953, 171, 964–967. [Google Scholar] [CrossRef]
- Calladine, C.R. Mechanics of sequence-dependent stacking of bases in B-DNA. J. Mol. Biol. 1982, 161, 343–352. [Google Scholar] [CrossRef]
- Gorin, A.A.; Zhurkin, V.B.; Olson, W.K. B-DNA twisting correlates with base-pair morphology. J. Mol. Biol. 1995, 247, 34–48. [Google Scholar] [CrossRef]
- Lu, X.J.; Shakked, Z.; Olson, W.K. A-form conformational motifs in ligand-bound DNA structures. J. Mol. Biol. 2000, 300, 819–840. [Google Scholar] [CrossRef]
- Olson, W.K.; Zhurkin, V.B. Working the kinks out of nucleosomal DNA. Curr. Opin. Struct. Biol. 2011, 21, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Selsing, E.; Wells, R.D.; Alden, C.J.; Arnott, S. Bent DNA: Visualization of a base-paired and stacked A-B conformational junction. J. Biol. Chem. 1979, 254, 5417–5422. [Google Scholar] [CrossRef]
- Marky, N.L.; Olson, W.K. Spatial translational motions of base pairs in DNA molecules: Application of the extended matrix generator method. Biopolymers 1994, 34, 121–142. [Google Scholar] [CrossRef] [PubMed]
- Olson, W.K.; Gorin, A.A.; Lu, X.J.; Hock, L.M.; Zhurkin, V.B. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc. Natl. Acad. Sci. USA 1998, 95, 11163–11168. [Google Scholar] [CrossRef] [Green Version]
- Lankas, F.; Sponer, J.; Langowski, J.; Cheatham, T.E., 3rd. DNA basepair step deformability inferred from molecular dynamics simulations. Biophys. J. 2003, 85, 2872–2883. [Google Scholar] [CrossRef] [Green Version]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satchwell, S.C.; Drew, H.R.; Travers, A.A. Sequence periodicities in chicken nucleosome core DNA. J. Mol. Biol. 1986, 191, 659–675. [Google Scholar] [CrossRef]
- Chua, E.Y.D.; Vasudevan, D.; Davey, G.E.; Wu, B.; Davey, C.A. The mechanics behind DNA sequence-dependent properties of the nucleosome. Nucleic Acids Res. 2012, 40, 6338–6352. [Google Scholar] [CrossRef] [Green Version]
- Burkhoff, A.M.; Tullius, T.D. The unusual conformation adopted by the adenine tracts in kinetoplast DNA. Cell 1987, 48, 935–943. [Google Scholar] [CrossRef]
- Price, M.A.; Tullius, T.D. How the structure of an adenine tract depends on sequence context: A new model for the structure of TnAn DNA sequences. Biochemistry 1993, 32, 127–136. [Google Scholar] [CrossRef]
- Koo, H.S.; Crothers, D.M. Calibration of DNA curvature and a unified description of sequence-directed bending. Proc. Natl. Acad. Sci. USA 1988, 85, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Brukner, I.; Sanchez, R.; Suck, D.; Pongor, S. Sequence-dependent bending propensity of DNA as revealed by DNase I: Parameters for trinucleotides. EMBO J. 1995, 14, 1812–1818. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.J.; Dauncey, M.P.; Hunter, C.A. Sequence-dependent DNA structure: Tetranucleotide conformational maps. J. Mol. Biol. 2000, 295, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Beveridge, D.L.; Barreiro, G.; Byun, K.S.; Case, D.A.; Cheatham, T.E., 3rd; Dixit, S.B.; Giudice, E.; Lankas, F.; Lavery, R.; Maddocks, J.H.; et al. Molecular dynamics simulations of the 136 unique tetranucleotide sequences of DNA oligonucleotides. I. Research design and results on d(CpG) steps. Biophys. J. 2004, 87, 3799–3813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, S.B.; Beveridge, D.L.; Case, D.A.; Cheatham, T.E., 3rd; Giudice, E.; Lankas, F.; Lavery, R.; Maddocks, J.H.; Osman, R.; Sklenar, H.; et al. Molecular dynamics simulations of the 136 unique tetranucleotide sequences of DNA oligonucleotides. II: Sequence context effects on the dynamical structures of the 10 unique dinucleotide steps. Biophys. J. 2005, 89, 3721–3740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, S.; Kono, H.; Takenaka, S.; Go, N.; Sarai, A. Sequence-dependent DNA deformability studied using molecular dynamics simulations. Nucleic Acids Res. 2007, 35, 6063–6074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavery, R.; Zakrzewska, K.; Beveridge, D.; Bishop, T.C.; Case, D.A.; Cheatham, T.E., 3rd; Dixit, S.; Jayaram, B.; Lankas, F.; Laughton, C.; et al. A systematic molecular dynamics study of nearest-neighbor effects on base pair and base pair step conformations and fluctuations in B-DNA. Nucleic Acids Res. 2010, 38, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Pasi, M.; Maddocks, J.H.; Beveridge, D.; Bishop, T.C.; Case, D.A.; Cheatham, T., 3rd; Dans, P.D.; Jayaram, B.; Lankas, F.; Laughton, C.; et al. μABC: A systematic microsecond molecular dynamics study of tetranucleotide sequence effects in B-DNA. Nucleic Acids Res. 2014, 42, 12272–12283. [Google Scholar] [CrossRef]
- Balaceanu, A.; Buitrago, D.; Walther, J.; Hospital, A.; Dans, P.D.; Orozco, M. Modulation of the helical properties of DNA: Next-to-nearest neighbour effects and beyond. Nucleic Acids Res. 2019, 47, 4418–4430. [Google Scholar] [CrossRef] [Green Version]
- Hagerman, P.J. Sequence dependence of the curvature of DNA: A test of the phasing hypothesis. Biochemistry 1985, 24, 7033–7037. [Google Scholar] [CrossRef]
- Hagerman, P.J. Sequence-directed curvature of DNA. Nature 1986, 321, 449–450. [Google Scholar] [CrossRef]
- Koo, H.S.; Wu, H.M.; Crothers, D.M. DNA bending at adenine·thymine tracts. Nature 1986, 320, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Fogg, J.M.; Judge, A.K.; Stricker, E.; Chan, H.L.; Zechiedrich, L. Supercoiling and looping promote DNA base accessibility and coordination among distant sites. Nat. Commun. 2021, 12, 5683. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Schaeffer, D.; Liao, Y.; Kinch, L.N.; Pei, J.; Shi, S.; Kim, B.H.; Grishin, N.V. ECOD: An evolutionary classification of protein domains. PLoS Comput. Biol. 2014, 10, e1003926. [Google Scholar] [CrossRef]
- Schaeffer, R.D.; Liao, Y.; Cheng, H.; Grishin, N.V. ECOD: New developments in the evolutionary classification of domains. Nucleic Acids Res. 2017, 45, D296–D302. [Google Scholar] [CrossRef] [Green Version]
- Dickerson, R.E.; Bansal, M.; Calladine, C.R.; Diekmann, S.; Hunter, W.N.; Kennard, O.; von Kitzing, E.; Lavery, R.; Nelson, H.C.M.; Olson, W.K.; et al. Definitions and nomenclature of nucleic acid structure parameters. Nucleic Acids Res. 1989, 17, 1797–1803. [Google Scholar] [CrossRef] [Green Version]
- Coleman, B.D.; Olson, W.K.; Swigon, D. Theory of sequence-dependent DNA elasticity. J. Chem. Phys. 2003, 118, 7127–7140. [Google Scholar] [CrossRef]
- Olson, W.K.; Bansal, M.; Burley, S.K.; Dickerson, R.E.; Gerstein, M.; Harvey, S.C.; Heinemann, U.; Lu, X.J.; Neidle, S.; Shakked, Z.; et al. A standard reference frame for the description of nucleic acid base-pair geometry. J. Mol. Biol. 2001, 313, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Olson, W.K.; Colasanti, A.V.; Lu, X.J.; Zhurkin, V.B. Watson–Crick base pairs: Character and recognition. In Wiley Encyclopedia of Chemical Biology; John Wiley & Sons, Inc.: New York, NY, USA, 2008. [Google Scholar] [CrossRef]
- Olson, W.K.; Esguerra, M.; Xin, Y.; Lu, X.J. New information content in RNA base pairing deduced from quantitative analysis of high-resolution structures. Methods 2009, 47, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Peckham, H.E.; Olson, W.K. Nucleic-acid structural deformability deduced from anisotropic displacement parameters. Biopolymers 2011, 95, 254–269. [Google Scholar] [CrossRef]
- Clauvelin, N.; Olson, W.K. Synergy between protein positioning and DNA elasticity: Energy minimization of protein-decorated DNA minicircles. J. Phys. Chem. B 2021, 125, 2277–2287. [Google Scholar] [CrossRef] [PubMed]
- Young, R.T.; Clauvelin, N.; Olson, W.K. emDNA— A tool for modeling protein-decorated DNA loops and minicircles at the base-pair step level. J. Mol. Biol. 2022, 167558. [Google Scholar] [CrossRef] [PubMed]
- Britton, L.; Olson, W.K.; Tobias, I. Two perspectives on the twist of DNA. J. Chem. Phys. 2009, 131, 245101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clauvelin, N.; Tobias, I.; Olson, W.K. Characterization of the geometry and topology of DNA pictured as a discrete collection of atoms. J. Chem. Theor. Comp. 2012, 8, 1092–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G.S. The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides. Q. Rev. Biophys. 1978, 11, 179–246. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C.; Trifonov, E.N. The ten helical twist angles of B-DNA. Nucleic Acids Res. 1982, 10, 1097–1104. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.; Klug, A. Helical periodicity of DNA determined by enzyme digestion. Nature 1980, 286, 573–578. [Google Scholar] [CrossRef]
- Wang, J.C. Helical repeat of DNA in solution. Proc. Natl. Acad. Sci. USA 1979, 76, 200–203. [Google Scholar] [CrossRef] [Green Version]
- Peck, L.J.; Wang, J.C. Sequence dependence of the helical repeat of DNA in solution. Nature 1981, 292, 375–378. [Google Scholar] [CrossRef]
- Marini, J.C.; Levene, S.D.; Crothers, D.M.; Englund, P.T. Bent helical structure in kinetoplast DNA. Proc. Natl. Acad. Sci. USA 1983, 80, 7664–7678, Correction ibid 1983, 80, 7678. [Google Scholar] [CrossRef] [Green Version]
- Tolstorukov, M.Y.; Colasanti, A.V.; McCandlish, D.; Olson, W.K.; Zhurkin, V.B. A novel roll-and-slide mechanism of DNA folding in chromatin: Implications for nucleosome positioning. J. Mol. Biol. 2007, 371, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifonov, E.N.; Sussman, J. The pitch of chromatin DNA is reflected in its nucleotide sequence. Proc. Natl. Acad. Sci. USA 1980, 77, 3816–3820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulanovsky, L.E.; Trifonov, E.N. Estimation of wedge components in curved DNA. Nature 1987, 326, 720–722. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, P.J. Evidence for the existence of stable curvature of DNA in solution. Proc. Natl. Acad. Sci. USA 1984, 81, 4632–4636. [Google Scholar] [CrossRef] [Green Version]
- Zhurkin, V.B. Sequence-dependent bending of DNA and phasing of nucleosomes. J. Biomol. Struct. Dyn. 1985, 2, 785–804. [Google Scholar] [CrossRef]
- Stefl, R.; Wu, H.; Ravindranathan, S.; Sklenár, V.; Feigon, J. DNA A-tract bending in three dimensions: Solving the dA4T4 vs. dT4A4 conundrum. Proc. Natl. Acad. Sci. USA 2004, 101, 1177–1182. [Google Scholar] [CrossRef] [Green Version]
- Irobalieva, R.N.; Fogg, J.M.; Catanese, D.J.; Sutthibutpong, T.; Chen, M.; Barker, A.K.; Ludtke, S.J.; Harris, S.A.; Schmid, M.F.; Chiu, W.; et al. Structural diversity of supercoiled DNA. Nat. Commun. 2015, 12, 8440. [Google Scholar] [CrossRef]
- Gray, H.B., Jr.; Lu, T. The BAL 31 nucleases (EC 3.1.11). Methods Mol. Biol. 1993, 16, 231–251. [Google Scholar]
- Chaudhry, M.A.; Weinfeld, M. Induction of double-strand breaks by S1 nuclease, mung bean nuclease and nuclease P1 in DNA containing abasic sites and nicks. Nucleic Acids Res. 1995, 23, 3805–3809. [Google Scholar] [CrossRef] [Green Version]
- Grindley, N.D.F.; Whiteson, K.L.; Rice, P.A. Mechanisms of site-specific recombination. Annu. Rev. Biochem. 2006, 75, 567–605. [Google Scholar] [CrossRef] [Green Version]
- Olson, W.K. Simulating DNA at low resolution. Curr. Opin. Struct. Biol. 1996, 6, 242–256. [Google Scholar] [CrossRef]
- Černý, J.; Božíková, P.; Svoboda, J.; Schneider, B. A unified dinucleotide alphabet describing both RNA and DNA structures. Nucleic Acids Res. 2020, 48, 6367–6381. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Irobalieva, R.N.; Chiu, W.; Schmid, M.F.; Fogg, J.M.; Zechiedrich, L.; Pettitt, B.M. Influence of DNA sequence on the structure of minicircles under torsional stress. Nucleic Acids Res. 2017, 45, 7633–7642. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, R.T.; Czapla, L.; Wefers, Z.O.; Cohen, B.M.; Olson, W.K. Revisiting DNA Sequence-Dependent Deformability in High-Resolution Structures: Effects of Flanking Base Pairs on Dinucleotide Morphology and Global Chain Configuration. Life 2022, 12, 759. https://0-doi-org.brum.beds.ac.uk/10.3390/life12050759
Young RT, Czapla L, Wefers ZO, Cohen BM, Olson WK. Revisiting DNA Sequence-Dependent Deformability in High-Resolution Structures: Effects of Flanking Base Pairs on Dinucleotide Morphology and Global Chain Configuration. Life. 2022; 12(5):759. https://0-doi-org.brum.beds.ac.uk/10.3390/life12050759
Chicago/Turabian StyleYoung, Robert T., Luke Czapla, Zoe O. Wefers, Benjamin M. Cohen, and Wilma K. Olson. 2022. "Revisiting DNA Sequence-Dependent Deformability in High-Resolution Structures: Effects of Flanking Base Pairs on Dinucleotide Morphology and Global Chain Configuration" Life 12, no. 5: 759. https://0-doi-org.brum.beds.ac.uk/10.3390/life12050759