
Translational Medicine in Uremic Vascular Calcification: Scavenging ROS Attenuates p-Cresyl Sulfate-Activated Caspase-1, NLRP3 Inflammasome and Eicosanoid Inflammation in Human Arterial Smooth Muscle Cells
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
2.1. CKD Patients Had an Increase in Circulating Levels of PCS and Medial Arterial Calcification
2.2. CKD Arteries Exhibited More Serious Elastic Lamina (EL) Disruption in Parallel with UVC Severity
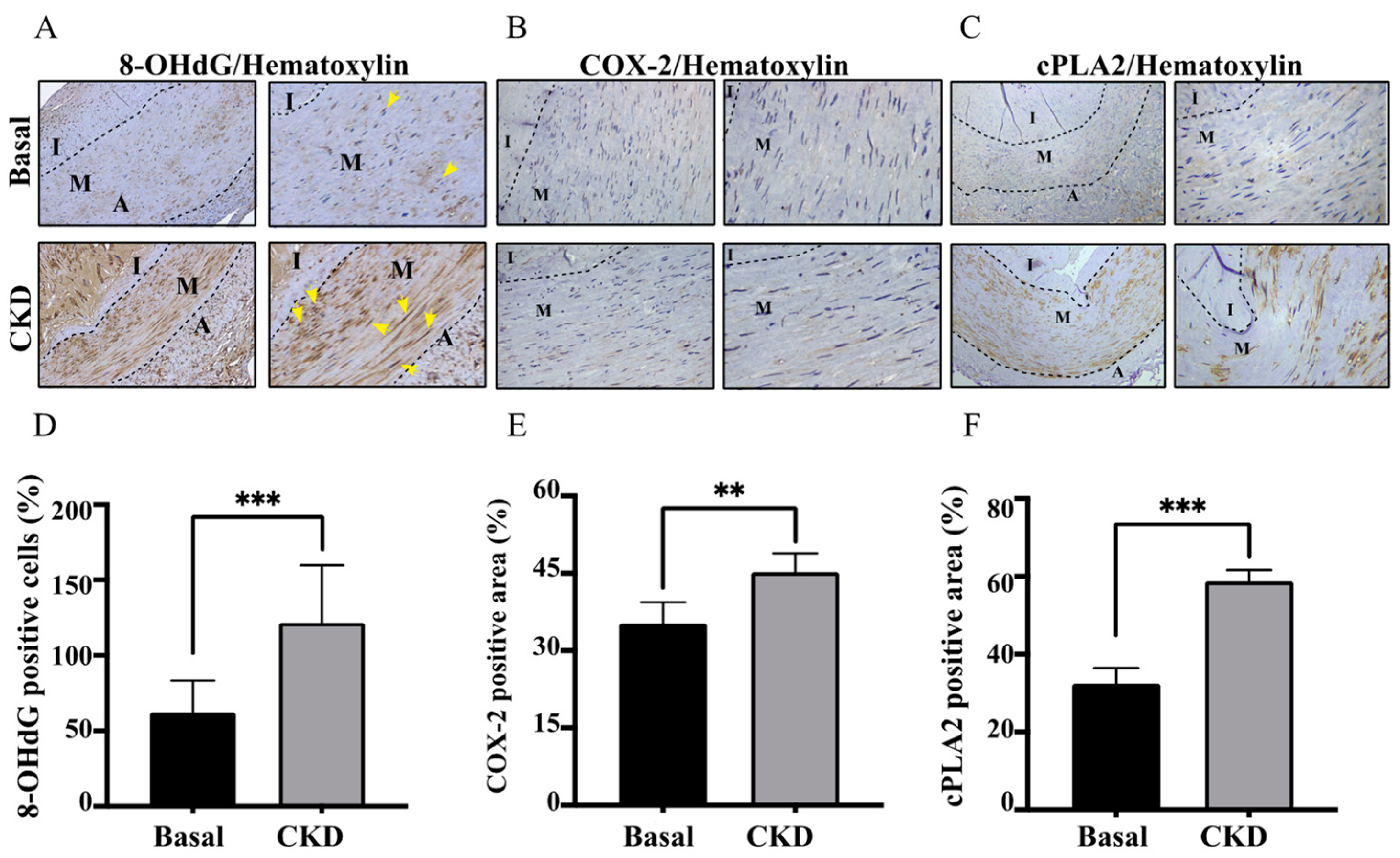
2.3. Concordant with UVC Severity, CKD Arteries Manifested Enhanced Expressions of COX-2/cPLA-2 and 8-OHdG, Indicative of Eicosanoid Inflammation and Oxidative Injury
2.4. Higher Productions of Caspase-1, IL-1β and NLRP3 Inflammasome in CKD-UVC Arteries and PCS-Stimulated HASMC
2.5. ROS Scavenger Attenuates PCS-Triggered Coupling Expressions of COX-2/cPLA-2 in HASMC Model
2.6. ROS Scavenger Attenuates PCS-Triggered Expressions of Pro-Caspase-1 and NLRP3 in HASMC Model
3. Discussion
3.1. Pro-Oxidant PCS-Induced Internal EL Disruption as an Emerging Key Player of UVC Progression
3.2. Higher Circulating Levels of PCS Is in Parallel with UVC Severity, Oxidative EL Injury, Coupling Expressions of COX-2/cPLA-2, and ROS Scavenger Inhibits PCS-Induced Eicosanoid Inflammation
3.3. CKD-UVC Arteries Exert Higher Expressions of Caspase-1, IL-1β and NLRP3 Inflammasome, and ROS Scavenger Inhibits PCS-Activated Caspase-1 and NLRP3 Inflammasome
4. Materials and Methods
4.1. Patients and Arterial Specimens
4.2. Reagents for Immunohistochemistry (IHC) Staining and Cell Models
4.3. Cell and Treatments
4.4. RNA Isolation and Quantitative Real-Time PCR
4.5. Measurement of Circulating PCS Levels
4.6. Statistics
5. Conclusions
6. Patients
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Falconi, C.A.; da Cruz Junho, C.V.; Fogaça-Ruiz, F.; Vernier, I.C.S.; da Cunha, R.S.; Stinghen, A.E.M.; Carneiro-Ramos, M.S. Uremic toxins: An alarming danger concerning the cardiovascular system. Front. Physiol. 2021, 12, 686249. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Dounousi, E.; Salmas, M.; Eleftheriadis, T.; Liakopoulos, V. Vascular calcification in chronic kidney disease: The role of vitamin K-dependent matrix Gla protein. Front. Med. 2020, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-F.; Liu, S.-H.; Lu, K.-C.; Ka, S.-M.; Hsieh, C.-Y.; Ho, C.-T.; Lin, W.-N.; Wen, L.-L.; Liou, J.-C.; Chang, S.-W.; et al. Uremic Vascular Calcification Is Correlated With Oxidative Elastic Lamina Injury, Contractile Smooth Muscle Cell Loss, Osteogenesis, and Apoptosis: The Human Pathobiological Evidence. Front. Med. 2020, 7, 78. [Google Scholar] [CrossRef]
- Lekawanvijit, S.; Kompa, A.R.; Krum, H. Protein-bound uremic toxins: A long overlooked culprit in cardiorenal syndrome. Am. J. Physiol. Ren. Physiol. 2016, 311, F52–F62. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.F.; Hsieh, C.Y.; Liou, J.C.; Liu, S.H.; Hung, C.F.; Lu, K.C.; Lin, C.C.; Wu, C.C.; Ka, S.M.; Wen, L.L.; et al. Scavenging Intracellular ROS Attenuates p-Cresyl Sulfate-Triggered Osteogenesis through MAPK Signaling Pathway and NF-κB Activation in Human Arterial Smooth Muscle Cells. Toxins 2020, 12, 472. [Google Scholar] [CrossRef]
- Chang, J.F.; Liang, S.S.; Thanasekaran, P.; Chang, H.W.; Wen, L.L.; Chen, C.H.; Liou, J.C.; Yeh, J.C.; Liu, S.H.; Dai, H.M.; et al. Translational Medicine in Pulmonary-Renal Crosstalk: Therapeutic Targeting of p-Cresyl Sulfate Triggered Nonspecific ROS and Chemoattractants in Dyspneic Patients with Uremic Lung Injury. J. Clin. Med. 2018, 7, 266. [Google Scholar] [CrossRef]
- Boini, K.M.; Hussain, T.; Li, P.-L.; Koka, S.S. Trimethylamine-N-oxide instigates NLRP3 inflammasome activation and endothelial dysfunction. Cell. Physiol. Biochem. 2017, 44, 152–162. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Yang, P.; Liu, X.; Lu, L.; Chen, Y.; Zhong, X.; Li, Z.; Liu, H.; Ou, C. Trimethylamine-N-Oxide promotes vascular calcification through activation of NLRP3 (nucleotide-binding domain, Leucine-Rich-Containing family, pyrin Domain-Containing-3) inflammasome and NF-κB (nuclear factor κB) signals. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 751–765. [Google Scholar] [CrossRef]
- Rossi, M.N.; Pascarella, A.; Licursi, V.; Caiello, I.; Taranta, A.; Rega, L.R.; Levtchenko, E.; Emma, F.; De Benedetti, F.; Prencipe, G. NLRP2 regulates proinflammatory and antiapoptotic responses in proximal tubular epithelial cells. Front. Cell Dev. Biol. 2019, 7, 252. [Google Scholar] [CrossRef]
- Chang, J.-F.; Yeh, J.-C.; Ho, C.-T.; Liu, S.-H.; Hsieh, C.-Y.; Wang, T.-M.; Chang, S.-W.; Lee, I.T.; Huang, K.-Y.; Wang, J.-Y.; et al. Targeting ROS and cPLA2/COX2 Expressions Ameliorated Renal Damage in Obese Mice with Endotoxemia. Int. J. Mol. Sci. 2019, 20, 4393. [Google Scholar] [CrossRef]
- Zhao, M.M.; Xu, M.J.; Cai, Y.; Zhao, G.; Guan, Y.; Kong, W.; Tang, C.; Wang, X. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011, 79, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-F.; Chou, Y.-S.; Wu, C.-C.; Chen, P.-C.; Ko, W.-C.; Liou, J.-C.; Hsieh, C.-Y.; Lin, W.-N.; Wen, L.-L.; Chang, S.-W. A Joint Evaluation of Neurohormone Vasopressin-Neurophysin II-Copeptin and Aortic Arch Calcification on Mortality Risks in Hemodialysis Patients. Front. Med. 2020, 7, 102. [Google Scholar] [CrossRef]
- Chang, J.-F.; Hsieh, C.-Y.; Liou, J.-C.; Lu, K.-C.; Zheng, C.-M.; Wu, M.-S.; Chang, S.-W.; Wang, T.-M.; Wu, C.-C. Circulating p-Cresyl Sulfate, Non-Hepatic Alkaline Phosphatase and Risk of Bone Fracture Events in Chronic Kidney Disease-Mineral Bone Disease. Toxins 2021, 13, 479. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-C.; Chang, S.-W.; Hsieh, C.-Y.; Liou, J.-C.; Chang, J.-F.; Wang, T.-M. Fat-Bone Relationship in Chronic Kidney Disease—Mineral Bone Disorders: Adiponectin Is Associated with Skeletal Events among Hemodialysis Patients. Diagnostics 2021, 11, 1254. [Google Scholar] [CrossRef]
- London, G.M. Mechanisms of arterial calcifications and consequences for cardiovascular function. Kidney Int. Suppl. 2013, 3, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Tada, S.; Tarbell, J.M. Internal elastic lamina affects the distribution of macromolecules in the arterial wall: A computational study. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H905–H913. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-F.; Chen, P.-C.; Hsieh, C.-Y.; Liou, J.-C. A Growth Differentiation Factor 15-Based Risk Score Model to Predict Mortality in Hemodialysis Patients. Diagnostics 2021, 11, 286. [Google Scholar] [CrossRef]
- De Vriese, A.S. Should Statins Be Banned from Dialysis? J. Am. Soc. Nephrol. JASN 2017, 28, 1675–1676. [Google Scholar] [CrossRef]
- Mackenzie, I.S.; McEniery, C.M.; Dhakam, Z.; Brown, M.J.; Cockcroft, J.R.; Wilkinson, I.B. Comparison of the effects of antihypertensive agents on central blood pressure and arterial stiffness in isolated systolic hypertension. Hypertension 2009, 54, 409–413. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Goel, A.; Mehta, J.L. LOX-1: Regulation, Signaling and Its Role in Atherosclerosis. Antioxidants 2019, 8, 218. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Taniguchi, M.; Tokumoto, M.; Toyonaga, J.; Fujisaki, K.; Suehiro, T.; Noguchi, H.; Iida, M.; Tsuruya, K.; Kitazono, T. The antioxidant tempol ameliorates arterial medial calcification in uremic rats: Important role of oxidative stress in the pathogenesis of vascular calcification in chronic kidney disease. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2012, 27, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Oxidative stress in uremia: Nature, mechanisms, and potential consequences. Semin. Nephrol. 2004, 24, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Hénaut, L.; Mary, A.; Chillon, J.-M.; Kamel, S.; Massy, Z.A. The Impact of Uremic Toxins on Vascular Smooth Muscle Cell Function. Toxins 2018, 10, 218. [Google Scholar] [CrossRef]
- Hung, K.-C.; Chang, J.-F.; Hsu, Y.-H.; Hsieh, C.-Y.; Wu, M.-S.; Wu, M.-Y.; Chiu, I.-J.; Syu, R.-S.; Wang, T.-M.; Wu, C.-C.; et al. Therapeutic Effect of Calcimimetics on Osteoclast–Osteoblast Crosslink in Chronic Kidney Disease and Mineral Bone Disease. Int. J. Mol. Sci. 2020, 21, 8712. [Google Scholar] [CrossRef]
- Jiang, H.; Yan, Y.; Jiang, W.; Zhou, R. NLRP3 inflammasome: Activation, regulation, and role in diseases. Sci. SInica Vitae 2017, 47, 125–131. [Google Scholar]
- He, Y.; Hara, H.; Núñez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Feng, Y.-S.; Tan, Z.-X.; Wang, M.-M.; Xing, Y.; Dong, F.; Zhang, F. Inhibition of NLRP3 inflammasome: A prospective target for the treatment of ischemic stroke. Front. Cell. Neurosci. 2020, 14, 155. [Google Scholar] [CrossRef]
- Sun, Y.; Johnson, C.; Zhou, J.; Wang, L.; Li, Y.-F.; Lu, Y.; Nanayakkara, G.; Fu, H.; Shao, Y.; Sanchez, C. Uremic toxins are conditional danger-or homeostasis-associated molecular patterns. Front. Biosci. 2018, 23, 348. [Google Scholar]
- Glorieux, G.; Vanholder, R.; Van Biesen, W.; Pletinck, A.; Schepers, E.; Neirynck, N.; Speeckaert, M.; De Bacquer, D.; Verbeke, F. Free p-cresyl sulfate shows the highest association with cardiovascular outcome in chronic kidney disease. Nephrol. Dial. Transplant. 2021, 36, 998–1005. [Google Scholar] [CrossRef]
- Yeh, J.C.; Wu, C.C.; Choy, C.S.; Chang, S.W.; Liou, J.C.; Chen, K.S.; Tung, T.H.; Lin, W.N.; Hsieh, C.Y.; Ho, C.T.; et al. Non-Hepatic Alkaline Phosphatase, hs-CRP and Progression of Vertebral Fracture in Patients with Rheumatoid Arthritis: A Population-Based Longitudinal Study. J. Clin. Med. 2018, 7, 439. [Google Scholar] [CrossRef] [PubMed]
- Schoppet, M.; Shanahan, C.M. Role for alkaline phosphatase as an inducer of vascular calcification in renal failure? Kidney Int. 2008, 73, 989–991. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Controls (n = 12) | CKD (n = 18) | p-Value |
|---|---|---|---|
| Age (years) | 67.3 ± 15.4 | 65.9 ± 12.8 | 0.78 |
| Male, n (%) | 9 (75.0) | 13 (72.2) | 0.87 |
| Diabetes, n (%) | 4 (33.3) | 14 (77.8) | <0.05 |
| Cardiovascular disease, n (%) | 4 (33.3) | 7 (38.9) | 0.77 |
| Heart failure, n (%) | 3 (25.0) | 5 (27.8) | 0.87 |
| Anemia, n (%) | 5 (41.7) | 10 (55.6) | 0.48 |
| Systolic blood pressure (mmHg) | 130.8 ± 34.0 | 143.1 ± 25.7 | 0.27 |
| Diastolic blood pressure (mmHg) | 75.4 ± 16.1 | 82.9 ± 19.7 | 0.43 |
| Blood urea nitrogen (mg/dL) | 18.0 ± 6.1 | 47.2 ± 23.8 | <0.01 |
| Creatinine (mg/dL) | 0.9 ± 0.2 | 3.9 ± 2.4 | <0.01 |
| eGFR (mL/min) b | 80.9 ± 13.8 | 22.6 ± 15.5 | <0.01 |
| Sodium (mmol/L) | 135.1 ± 4.2 | 136.4 ± 5.4 | 0.46 |
| Potassium (mmol/L) | 4.2 ± 0.7 | 3.9 ± 0.5 | 0.22 |
| Glucose (mg/dL) | 151.6 ± 43.9 | 243.9 ± 113.2 | <0.05 |
| Alanine aminotransferase (IU/L) | 29.2 ± 21.0 | 18.5 ± 11.7 | 0.10 |
| Alkaline phosphatase (IU/L) | 125.6 ± 35.4 | 176.4 ± 60.9 | <0.05 |
| Calcium (mg/dL) | 9.0 ± 0.4 | 8.8 ± 0.9 | 0.62 |
| Phosphate (mg/dL) | 2.9 ± 0.4 | 5.0 ± 1.2 | <0.01 |
| Calcium-phosphate product c | 26.1 ± 2.8 | 43.4 ± 9.7 | <0.01 |
| p-cresyl sulfate | 1.2 ± 0.9 | 15.2 ± 8.1 | <0.01 |
| Medial arterial calcification area (%) d | 2.1 ± 1.0 | 4.2 ± 1.9 | <0.01 |
| p-Cresyl Sulfate (ug/mL) | Indoxyl Sulfate (ug/mL) | p-Value | |
|---|---|---|---|
| Creatinine (mg/dL) | 0.50 | 0.52 | <0.01 |
| eGFR (ml/min) | 0.71 | 0.67 | <0.01 |
| Blood urea nitrogen (mg/dL) | 0.42 | 0.41 | <0.05 |
| Calcium-phosphate product | 0.76 | 0.70 | <0.01 |
| Medial arterial calcification area (%) | 0.42 | 0.41 | <0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, J.-F.; Kuo, H.-L.; Liu, S.-H.; Hsieh, C.-Y.; Hsu, C.-P.; Hung, K.-C.; Wang, T.-M.; Wu, C.-C.; Lu, K.-C.; Lin, W.-N.; et al. Translational Medicine in Uremic Vascular Calcification: Scavenging ROS Attenuates p-Cresyl Sulfate-Activated Caspase-1, NLRP3 Inflammasome and Eicosanoid Inflammation in Human Arterial Smooth Muscle Cells. Life 2022, 12, 769. https://0-doi-org.brum.beds.ac.uk/10.3390/life12050769
Chang J-F, Kuo H-L, Liu S-H, Hsieh C-Y, Hsu C-P, Hung K-C, Wang T-M, Wu C-C, Lu K-C, Lin W-N, et al. Translational Medicine in Uremic Vascular Calcification: Scavenging ROS Attenuates p-Cresyl Sulfate-Activated Caspase-1, NLRP3 Inflammasome and Eicosanoid Inflammation in Human Arterial Smooth Muscle Cells. Life. 2022; 12(5):769. https://0-doi-org.brum.beds.ac.uk/10.3390/life12050769
Chicago/Turabian StyleChang, Jia-Feng, Hsiao-Ling Kuo, Shih-Hao Liu, Chih-Yu Hsieh, Chih-Ping Hsu, Kuo-Chin Hung, Ting-Ming Wang, Chang-Chin Wu, Kuo-Cheng Lu, Wei-Ning Lin, and et al. 2022. "Translational Medicine in Uremic Vascular Calcification: Scavenging ROS Attenuates p-Cresyl Sulfate-Activated Caspase-1, NLRP3 Inflammasome and Eicosanoid Inflammation in Human Arterial Smooth Muscle Cells" Life 12, no. 5: 769. https://0-doi-org.brum.beds.ac.uk/10.3390/life12050769