On the Contribution of Protein Spatial Organization to the Physicochemical Interconnection between Proteins and Their Cognate mRNAs

Abstract
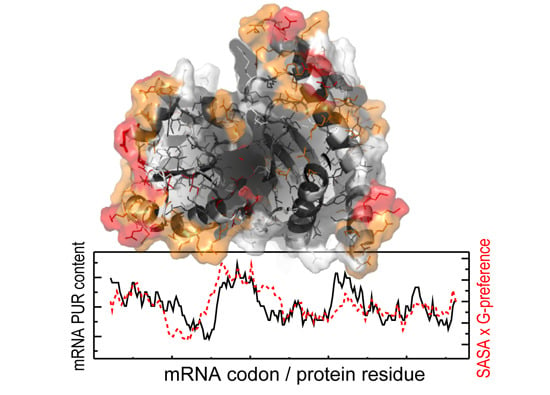
:
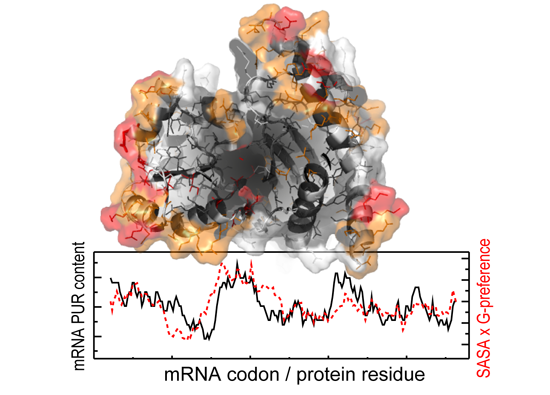{kind=link}
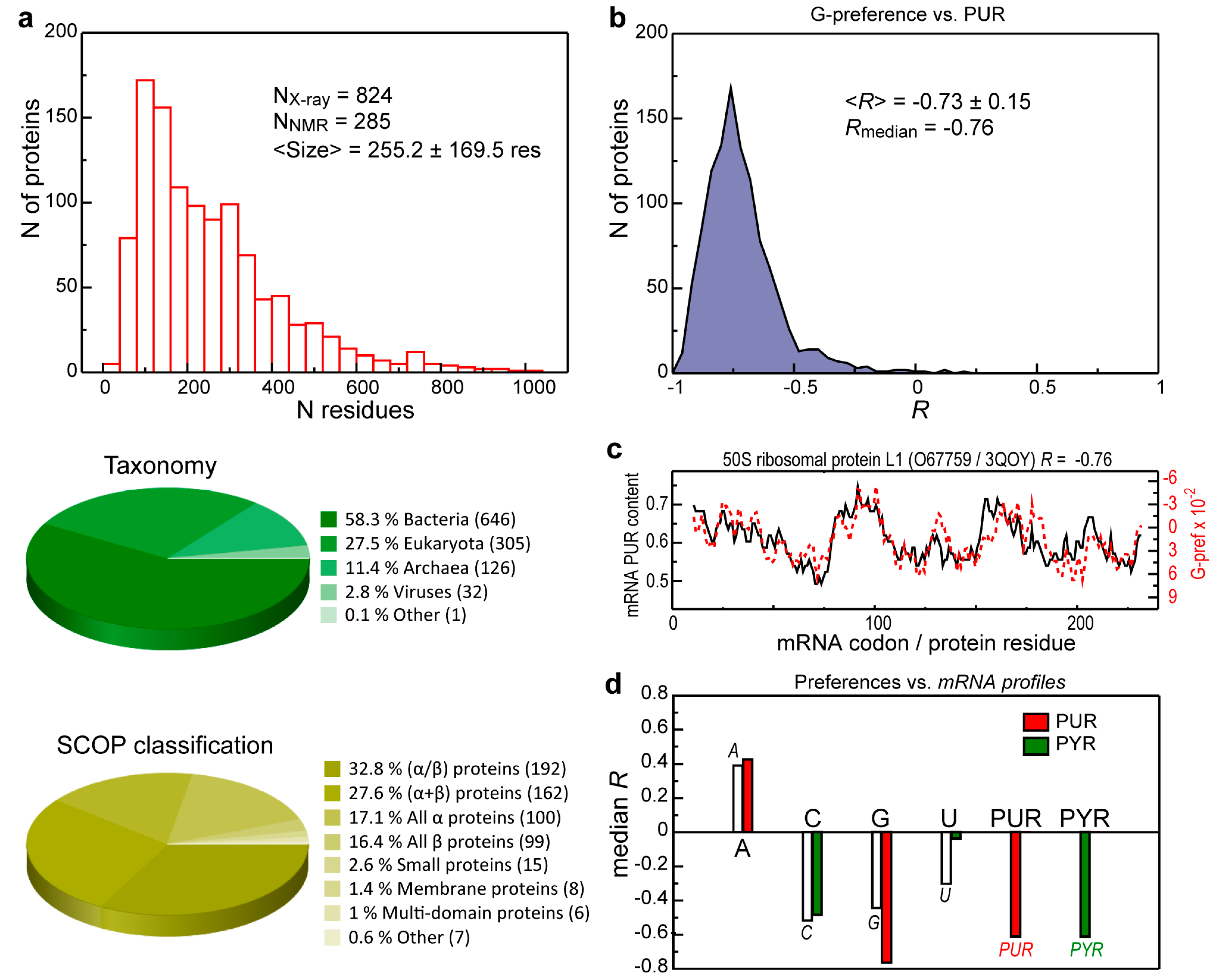{kind=link}
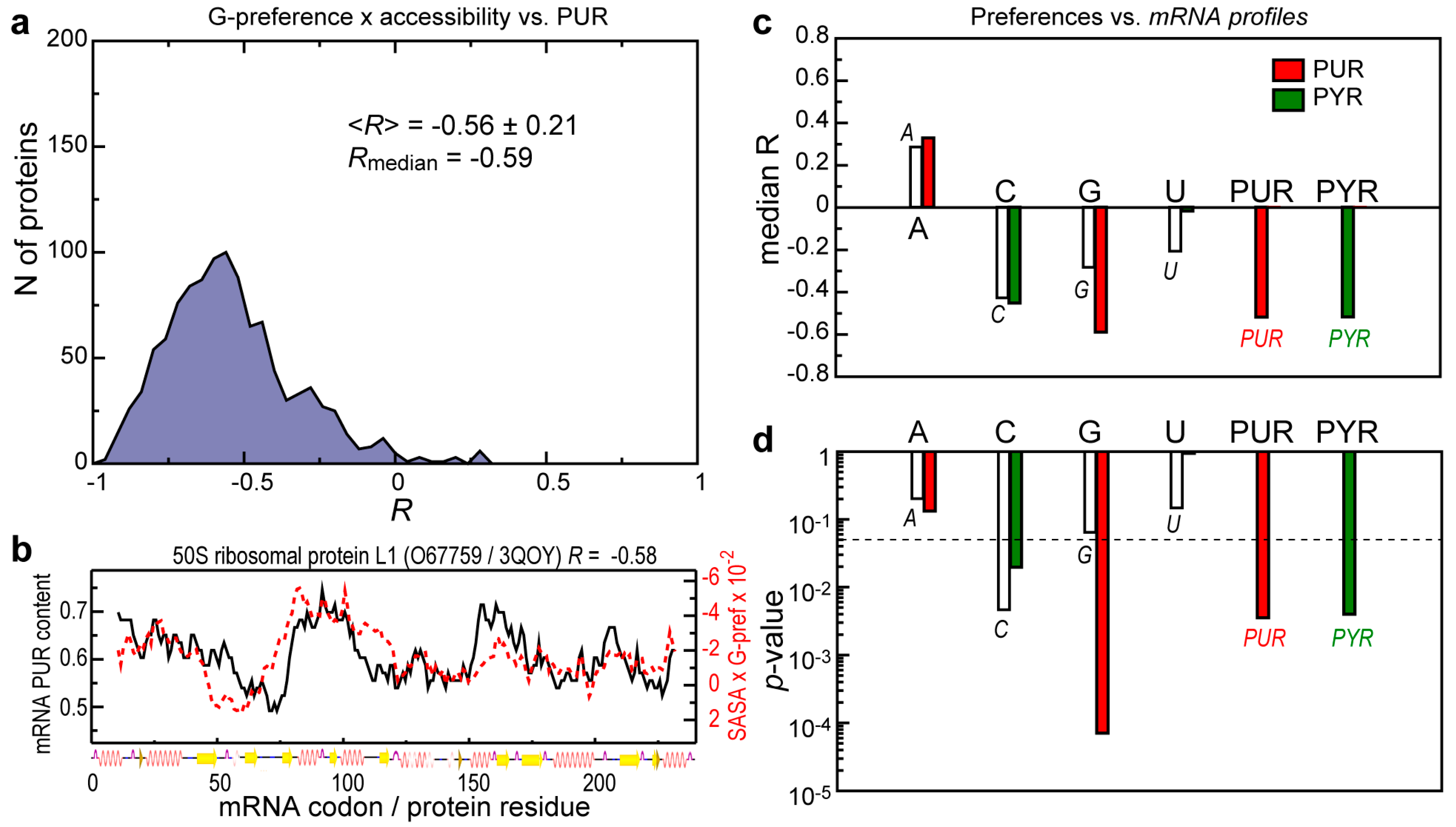{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Set of Protein 3D Structures
2.2. Calculations of Protein/mRNA Sequence Correlations
2.3. Analysis of Protein Structures
2.4. Enrichment Analysis
3. Results
3.1. Intrinsic Sequence Correlations in the 3D Set

3.2. Contribution of Protein Residue Accessibilities to Matching with Cognate mRNAs

3.3. Distribution of Nucleobase Interaction Preferences in Folded Protein Structures


4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nirenberg, M.W.; Jones, O.W.; Leder, P.; Clark, F.C.; Sly, S.; Petska, S. On the coding of genetic information. Cold Spring Harb. Symp. Quant. Biol. 1963, 28, 549–557. [Google Scholar] [CrossRef]
- Koonin, E.V.; Novozhilov, A.S. Origin and evolution of the genetic code: The universal enigma. IUBMB Life 2009, 61, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H.C. The origin of the genetic code. J. Mol. Biol. 1968, 38, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Haig, D.; Hurst, L.D. A quantitative measure of error minimization in the genetic-code. J. Mol. Evol. 1991, 33, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Freeland, S.J.; Knight, R.D.; Landweber, L.F.; Hurst, L.D. Early fixation of an optimal genetic code. Mol. Biol. Evol. 2000, 17, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R. On the evolution of the genetic code. Proc. Natl. Acad. Sci. USA 1965, 54, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Yarus, M.; Widmann, J.J.; Knight, R. Rna-amino acid binding: A stereochemical era for the genetic code. J. Mol. Evol. 2009, 69, 406–429. [Google Scholar] [CrossRef] [PubMed]
- Baltz, A.G.; Munschauer, M.; Schwanhausser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mrna-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Konig, J.; Zarnack, K.; Luscombe, N.M.; Ule, J. Protein-RNA interactions: New genomic technologies and perspectives. Nat. Rev. Genet. 2012, 13, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Kyrpides, N.C.; Ouzounis, C.A. Mechanisms of specificity in messenger-RNA degradation—Autoregulation and cognate interactions. J. Theor. Biol. 1993, 163, 373–392. [Google Scholar] [CrossRef] [PubMed]
- Ouzounis, C.A.; Kyrpides, N.C. Reverse interpretation—A hypothetical selection mechanism for adaptive mutagenesis based on autoregulated messenger-RNA stability. J. Theor. Biol. 1994, 167, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.; Copur, S.M.; Ju, J.; Chen, T.M.; Khleif, S.; Voeller, D.M.; Mizunuma, N.; Patel, M.; Maley, G.F.; Maley, F.; et al. Thymidylate synthase protein and p53 mRNA form an in vivo ribonucleoprotein complex. Mol. Cell. Biol. 1999, 19, 1582–1594. [Google Scholar] [PubMed]
- Tai, N.; Schmitz, J.C.; Liu, J.; Lin, X.; Bailly, M.; Chen, T.M.; Chu, E. Translational autoregulation of thymidylate synthase and dihydrofolate reductase. Front. Biosci. 2004, 9, 2521–2526. [Google Scholar] [CrossRef] [PubMed]
- Schuttpelz, M.; Schoning, J.C.; Doose, S.; Neuweiler, H.; Peters, E.; Staiger, D.; Sauer, M. Changes in conformational dynamics of mRNA upon atgrp7 binding studied by fluorescence correlation spectroscopy. J. Am. Chem. Soc. 2008, 130, 9507–9513. [Google Scholar] [CrossRef] [PubMed]
- Anders, G.; Mackowiak, S.D.; Jens, M.; Maaskola, J.; Kuntzagk, A.; Rajewsky, N.; Landthaler, M.; Dieterich, C. Dorina: A database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res. 2012, 40, D180–D186. [Google Scholar] [CrossRef] [PubMed]
- Hlevnjak, M.; Polyansky, A.A.; Zagrovic, B. Sequence signatures of direct complementarity between mRNAs and cognate proteins on multiple levels. Nucleic Acids Res. 2012, 40, 8874–8882. [Google Scholar] [CrossRef] [PubMed]
- Polyansky, A.A.; Zagrovic, B. Evidence of direct complementary interactions between messenger RNAs and their cognate proteins. Nucleic Acids Res. 2013, 41, 8434–8443. [Google Scholar] [CrossRef] [PubMed]
- Polyansky, A.A.; Hlevnjak, M.; Zagrovic, B. Proteome-wide analysis reveals clues of complementary interactions between mRNAs and their cognate proteins as the physicochemical foundation of the genetic code. RNA Biol. 2013, 10, 1248–1254. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R. Evolution of the genetic code. Naturwissenschaften 1973, 60, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Mathew, D.C.; Luthey-Schulten, Z. On the physical basis of the amino acid polar requirement. J. Mol. Evol. 2008, 66, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Noller, H.F. Evolution of protein synthesis from an RNA world. Cold Spring Harb. Perspect. Biol. 2012, 4, U1–U20. [Google Scholar] [CrossRef]
- Tompa, P. Unstructural biology coming of age. Curr. Opin. Struct. Biol. 2011, 21, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Polyansky, A.A.; Hlevnjak, M.; Zagrovic, B. Analogue encoding of physicochemical properties of proteins in their cognate messenger RNAs. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Schrodinger, L.L.C. The Pymol Molecular Graphics System, version 1.2r1; PyMOL: New York, NY, USA, 2010. [Google Scholar]
- Morales, P.; Garneau, L.; Klein, H.; Lavoie, M.F.; Parent, L.; Sauve, R. Contribution of the KCa3.1 channel-calmodulin interactions to the regulation of the KCa3.1 gating process. J. Gen. Physiol. 2013, 142, 37–60. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; Urey, H.C.; Oro, J. Origin of organic compounds on the primitive earth and in meteorites. J. Mol. Evol. 1976, 9, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, E.N.; Kirzhner, A.; Kirzhner, V.M.; Berezovsky, I.N. Distinct stages of protein evolution as suggested by protein sequence analysis. J. Mol. Evol. 2001, 53, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Segre, D.; Lancet, D. Composing life. EMBO Rep. 2000, 1, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, Y.; Kwok, C.K.; Zhang, Y.; Bevilacqua, P.C.; Assmann, S.M. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014, 505, 696–700. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beier, A.; Zagrovic, B.; Polyansky, A.A. On the Contribution of Protein Spatial Organization to the Physicochemical Interconnection between Proteins and Their Cognate mRNAs. Life 2014, 4, 788-799. https://0-doi-org.brum.beds.ac.uk/10.3390/life4040788
Beier A, Zagrovic B, Polyansky AA. On the Contribution of Protein Spatial Organization to the Physicochemical Interconnection between Proteins and Their Cognate mRNAs. Life. 2014; 4(4):788-799. https://0-doi-org.brum.beds.ac.uk/10.3390/life4040788
Chicago/Turabian StyleBeier, Andreas, Bojan Zagrovic, and Anton A. Polyansky. 2014. "On the Contribution of Protein Spatial Organization to the Physicochemical Interconnection between Proteins and Their Cognate mRNAs" Life 4, no. 4: 788-799. https://0-doi-org.brum.beds.ac.uk/10.3390/life4040788