
Does a Barcoding Gap Exist in Prokaryotes? Evidences from Species Delimitation in Cyanobacteria

Abstract
:1. Introduction

2. Experimental Section
2.1. Selection of Sequences
2.2. Testing for the Barcoding Gap
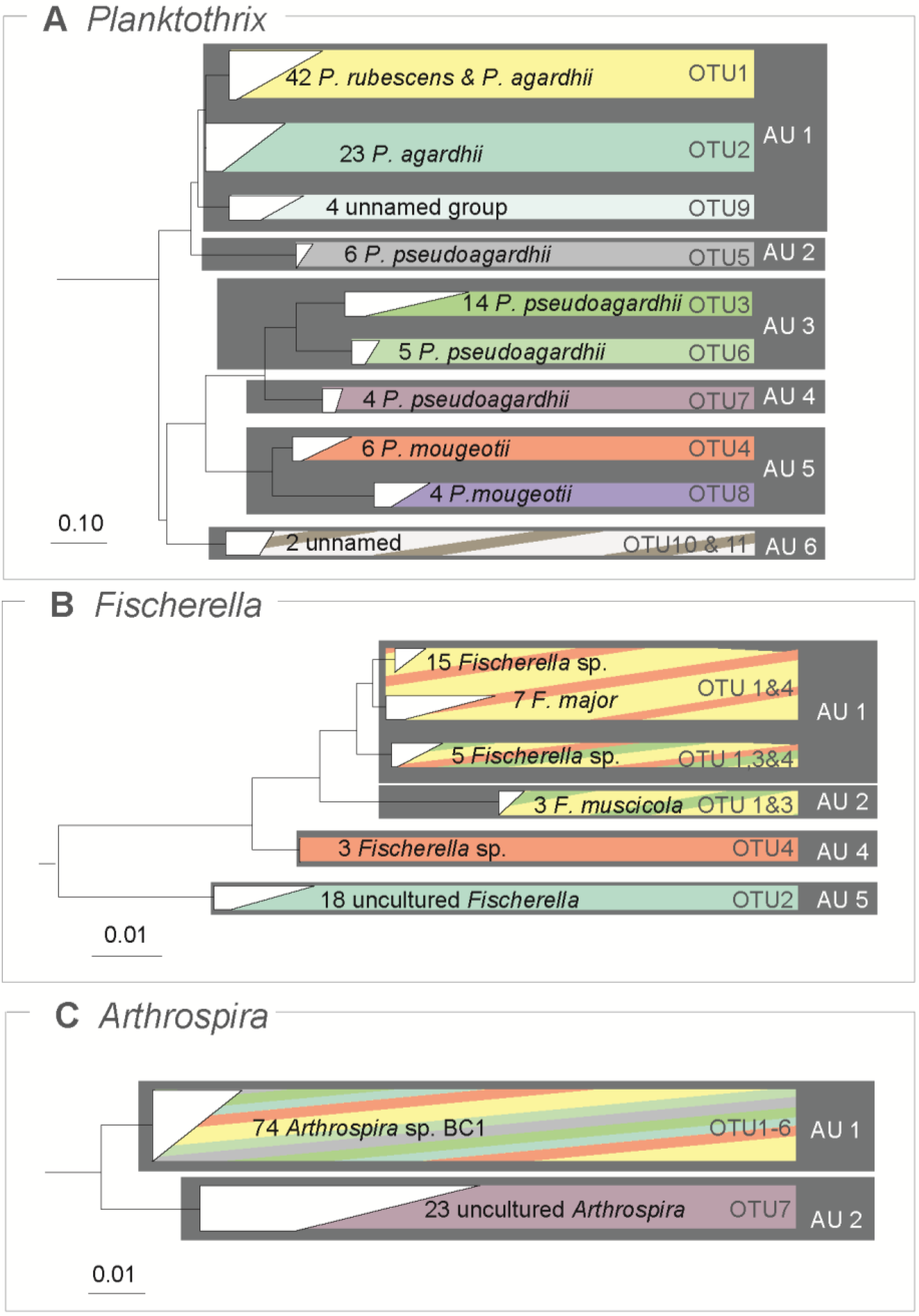
2.3. Traditional OTUs Delimitation
2.4. Assessing for Explanations and Confounding Factors
3. Results
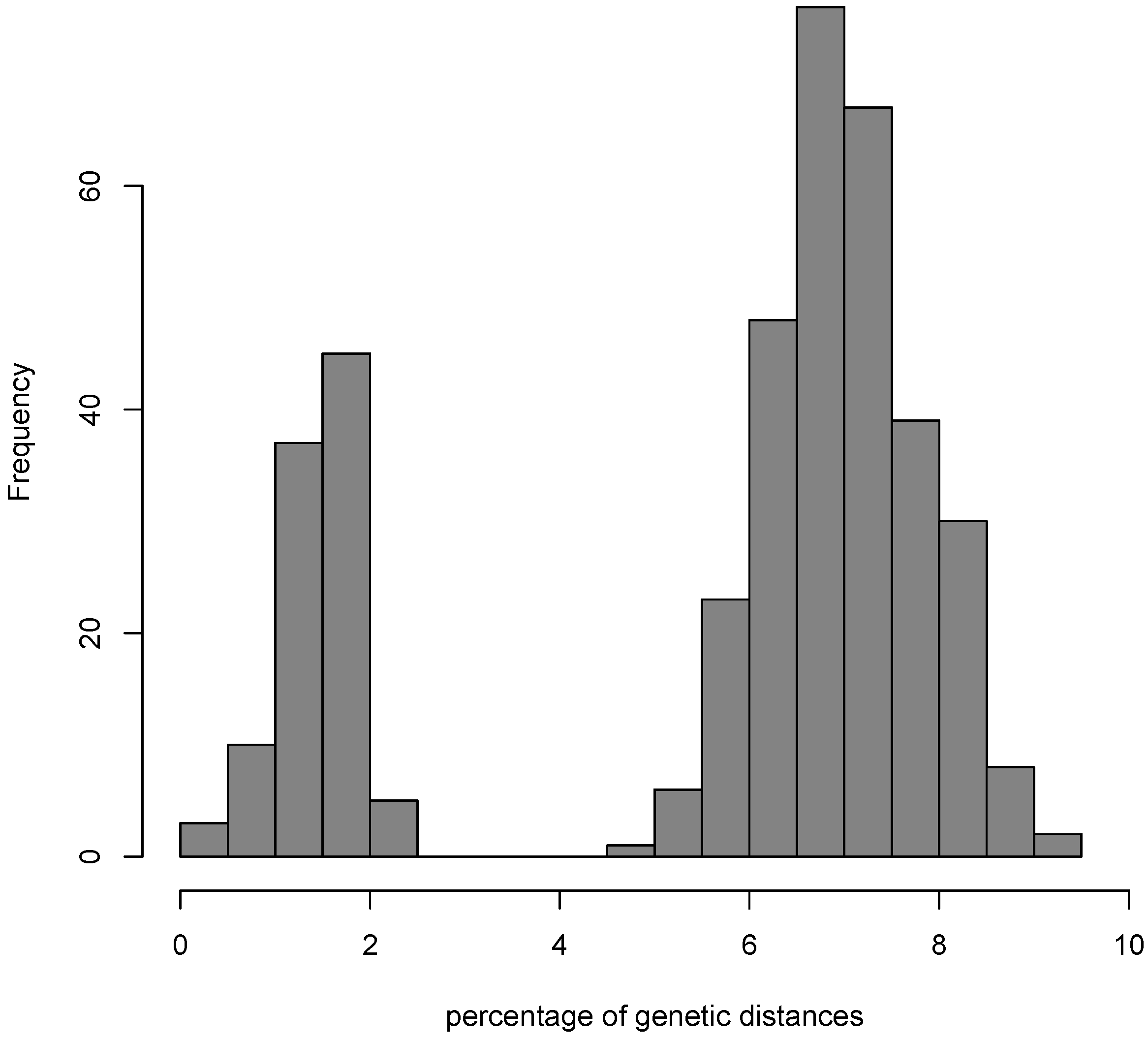
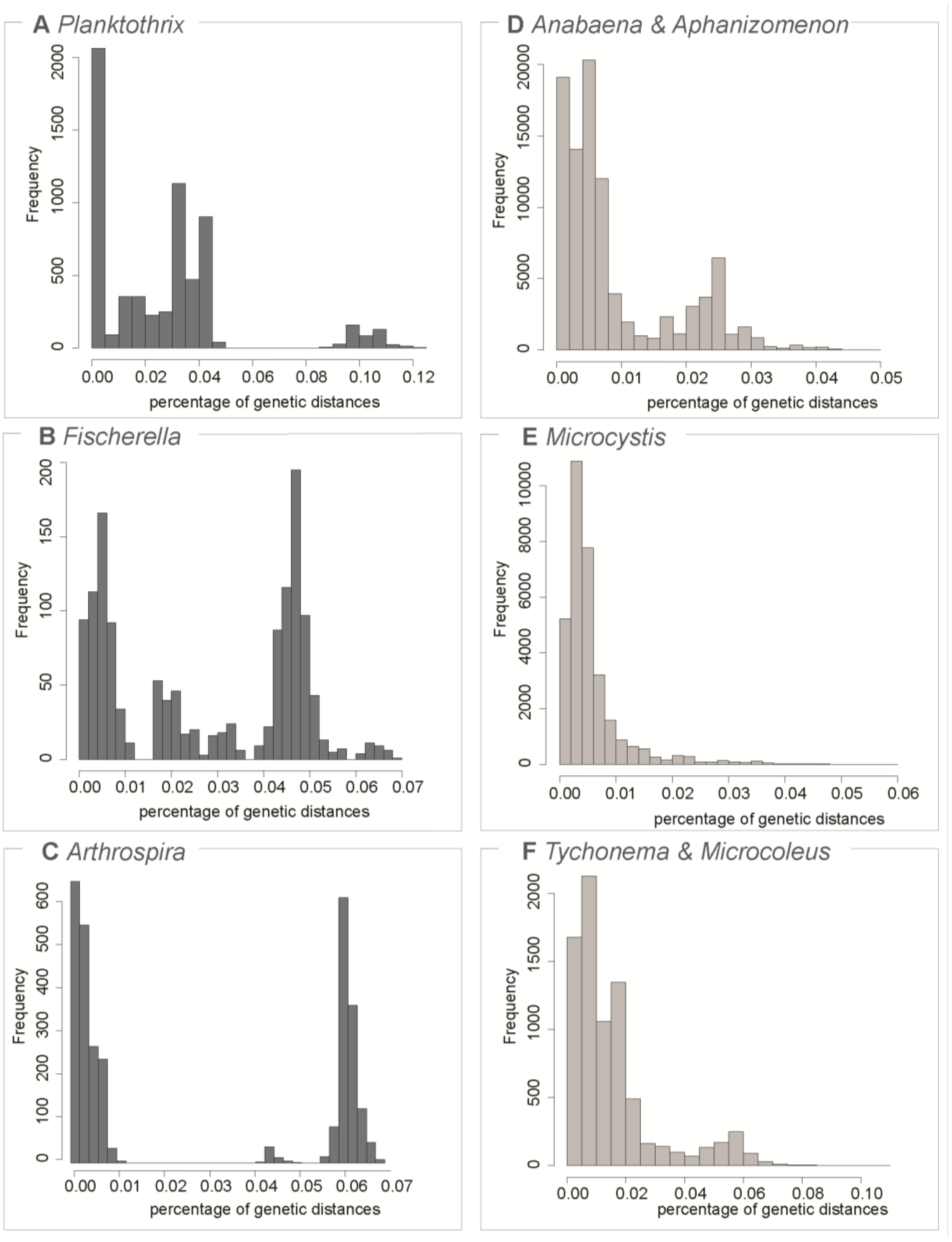
3.1. Testing for the Barcoding Gap
3.2. The Datasets


{kind=link}
{kind=link}
{kind=link}
| Dataset | Number of Sequences | Maximum Genetic Diversity | Barcoding Gap | ABGD Units | OTU 97% | OTU 99% |
|---|---|---|---|---|---|---|
| A. ovalisporum & A. bergii | 34 | 0.03 | yes | 2 (2) | 2 (2) | 6 (6) |
| Anabaena & Aphanizomenon | 508 | 0.05 | no | - | 5 (4) | 33 (28) |
| Arthrospira | 97 | 0.07 | yes | 3 (2) | 3 (3) | 7 (7) |
| Calothrix | 71 | 0.13 | no | - | 8 (5) | 34 (13) |
| Chroococcidiopsis | 88 | 0.11 | no | - | 4 (3) | 25 (9) |
| Cylindrospermopsis | 157 | 0.14 | yes | 2 (2) | 3 (3) | 25 (18) |
| Fischerella | 58 | 0.07 | yes | 6 (4) | 2 (2) | 4 (4) |
| Leptolyngbya & Chamaesiphon | 400 | 0.26 | no | - | 8 (3) | 153 (67) |
| Microcystis | 316 | 0.06 | no | - | 2 (2) | 11 (7) |
| Nodularia | 74 | 0.05 | yes | 2 (2) | 4 (2) | 12 (6) |
| Nostoc | 323 | 0.04 | no | - | 5 (4) | 19 (14) |
| Phormidium | 70 | 0.16 | yes | 5 (4) | 3 (2) | 24 (8) |
| Planktothrix | 108 | 0.12 | yes | 13 (6) | 3 (3) | 10 (9) |
| Prochlorococcus | 1011 | 0.05 | no | - | 3 (2) | 29 (13) |
| Prochlorococcus & Synechococcus | 2448 | 0.17 | no | - | 40 (18) | 286 (99) |
| Tychonema & Microcoleus | 135 | 0.108 | no | - | 1 (1) | 12 (7) |
3.3. Traditional OTUs Delimitation
| (A) Presence of a Barcoding Gap | Estimate | p |
| (intercept) | −14.19 ± 566.51 | 0.898 |
| Number of sequences | −0.03 ± 0.02 | 0.286 |
| Maximum genetic diversity | −22.3 ± 33.3 | 0.504 |
| Number of sequences: Maximum genetic diversity | 0.08 ± 0.14 | 0.578 |
| Shape | 19.66 ± 566.1 | 0.897 |
| (B) ABGD units | Estimate | p |
| (intercept) | 1.35 ± 0.44 (1.13 ± 0.53) | 0.002 (0.035) |
| Number of sequences | 0.00 ± 0.00 (0.00 ± 0.00) | 0.624 (0.979) |
| (C) OTUs 97% | Estimate | p |
| (intercept) | 1.06 ± 0.15 (0.87 ± 0.17) | <0.0001 (<0.0001) |
| Number of sequences | 0.00 ± 0.00 (0.00 ± 0.00) | <0.0001 (<0.0001) |
| (D) OTUs 99% | Estimate | p |
| (intercept) | 2.99 ± 0.05 (0.22 ± 0.07) | <0.0001 (<0.0001) |
| Number of sequences | 0.00 ± 0.00 (0.01 ± 0.00) | <0.0001 (<0.0001) |
3.4. Comparison of ABGD Units and OTUs

4. Discussion
Acknowledgments
Author Contributions
Supplementary Materials
Conflicts of Interest
References
- De Queiroz, K. Species concepts and species delimitation. Syst. Biol. 2007, 56, 879–886. [Google Scholar]
- Nosil, P. Ecological Speciation; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Diekmann, U.; Doebeli, M.; Metz, J.A.J.; Tautz, D. Adaptive Speciation; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Butlin, R.; Bridle, J.; Schluter, D. Speciation and Patterns of Diversity; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Coyne, J.A.; Orr, H.A. Speciation; Sinauer Associates Inc.: Sunderland, MA, USA, 2004. [Google Scholar]
- Koonin, E.V.; Makarova, K.S.; Aravind, L. Horizontal gene transfer in prokaryotes: Quantification and classification. Ann. Rev. Microbiol. 2001, 55, 709–742. [Google Scholar] [CrossRef]
- Wiedenbeck, J.; Cohan, F.M. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol. Rev. 2011, 35, 957–976. [Google Scholar] [PubMed]
- Schleifer, K.H. Classification of Bacteria and Archaea: Past, present and future. Syst. Appl. Microbiol. 2009, 32, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Rosselló-Mora, R.; Amann, R. The species concept for prokaryotes. FEMS Microbiol. Rev. 2001, 25, 39–67. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E. Defining taxonomic ranks. In The Prokaryotes; Springer: New York, NY, USA, 2006; pp. 29–57. [Google Scholar]
- Cadillo-Quiroz, H.; Didelot, X.; Held, N.L.; Herrera, A.; Darling, A.; Reno, M.L.; Krause, D.J.; Whitaker, R.J. Patterns of gene flow define species of thermophilic Archaea. PLoS Biol. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glockner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzeby, J.; Amann, R.; Rossello-Mora, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Vos, M. A species concept for bacteria based on adaptive divergence. Trends Microbiol. 2011, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cohan, F.M.; Aracena, S. Prokaryotic sex: Eukaryote-Like qualities of recombination in an Archaean lineage. Curr. Biol. 2012, 22, 601–602. [Google Scholar] [CrossRef] [PubMed]
- Cohan, F.M. Bacterial species and speciation. Syst. Biol. 2001, 50, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Cohan, F.M. Are Species Cohesive?—A View from Bacteriology. In Population Genetics of Bacteria: A Tribute to Thomas S. Whittam; Walk, S.T., Feng, P.C., Eds.; ASM Press: Washington, DC, USA, 2011. [Google Scholar]
- Cohan, F.M. Species. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 506–511. [Google Scholar]
- Barraclough, T.G.; Balbi, K.J.; Ellis, R.J. Evolving concepts of bacterial species. Evol. Biol. 2012, 39, 148–157. [Google Scholar] [CrossRef]
- Rosselló-Mora, R. Updating prokaryotic taxonomy. J. Bacteriol. 2005, 187, 6255–6257. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Goebel, B. Taxonomic note: A place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Bacteriol. 1994, 44, 846–849. [Google Scholar] [CrossRef]
- Acinas, S.G.; Klepac-Ceraj, V.; Hunt, D.E.; Pharino, C.; Ceraj, I.; Distel, D.L.; Polz, M.F. Fine-scale phylogenetic architecture of a complex bacterial community. Nature 2004, 430, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Handelsman, J. Toward a census of bacteria in soil. PLoS Comput. Biol. 2006, 2. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Ebers, J. Taxonomic parameters revisited: Tarnished gold standards. Microbiol. Today 2006, 33, 152–155. [Google Scholar]
- Yarza, P.; Spröer, C.; Swiderski, J.; Mrotzek, N.; Spring, S.; Tindall, B.J.; Gronow, S.; Pukall, R.; Klenk, H.-P.; Lang, E.; et al. Sequencing orphan species initiative (SOS): Filling the gaps in the 16S rRNA gene sequence database for all species with validly published names. Syst. Appl. Microbiol. 2013, 36, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.; Cywinska, A.; Ball, S.L. Biological identifications through DNA barcodes. Proc. R. Soc. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Barraclough, T.G.; Hughes, M.; Ashford-Hodges, N.; Fujisawa, T. Inferring evolutionarily significant units of bacterial diversity from broad environmental surveys of single-locus data. Biol. Lett. 2009, 5, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Koeppel, A.F.; Wu, M. Surprisingly extensive mixed phylogenetic and ecological signals among bacterial Operational Taxonomic Units. Nucleic Acids Res. 2013, 41, 5175–5188. [Google Scholar] [CrossRef] [PubMed]
- Komárek, J. Cyanobacterial taxonomy: Current problems and prospects for the integration of traditional and molecular approaches. Algae 2006, 21, 349–375. [Google Scholar] [CrossRef]
- Komárek, J. Modern Classification of Cyanobacteria; Wiley: Chichester, UK, 2014; pp. 21–39. [Google Scholar]
- Youssef, N.; Sheik, C.S.; Krumholz, L.R.; Najar, F.Z.; Roe, B.A.; Elshahed, M.S. Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Appl. Environ. Microbiol. 2009, 75, 5227–5236. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Ludwig, W.; Strunk, O.; Westram, R.; Richter, L.; Meier, H.; Yadhukumar; Buchner, A.; Lai, T.; Steppi, S.; Jobb, G.; et al. ARB: A software environment for sequence data. Nucleic Acids Res. 2004, 32, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Komárek, J. Recent changes (2008) in cyanobacteria taxonomy based on a combination of molecular background with phenotype and ecological consequences (genus and species concept). Hydrobiologia 2010, 639, 245–259. [Google Scholar] [CrossRef]
- Zapomělová, E.; Skácelová, O.; Pumann, P.; Kopp, R.; Janeček, E. Biogeographically interesting planktonic Nostocales (Cyanobacteria) in the Czech Republic and their polyphasic evaluation resulting in taxonomic revisions of Anabaena bergii Ostenfeld 1908 (Chrysosporum gen. nov.) and A. tenericaulis Nygaard 1949 (Dolichospermum tenericaule comb. nova). Hydrobiologia 2012, 698, 353–365. [Google Scholar] [CrossRef]
- Honda, D.; Yokota, A.; Sugiyama, J. Detection of Seven Major Evolutionary Lineages in Cyanobacteria Based on the 16S rRNA Gene Sequence Analysis with New Sequences of Five Marine Synechococcus Strains. J. Mol. Evol. 1999, 48, 723–739. [Google Scholar] [CrossRef]
- Robertson, B.R.; Tezuka, N.; Watanabe, M.M. Phylogenetic analyses of Synechococcus strains (cyanobacteria) using sequences of 16S rDNA and part of the phycocyanin operon reveal multiple evolutionary lines and reflect phycobilin content. Int. J. Syst. Evol. Microbiol. 2001, 51, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Callieri, C.; Coci, M.; Corno, G.; Macek, M.; Modenutti, B.; Balseiro, E.; Bertoni, R. Phylogenetic diversity of nonmarine picocyanobacteria. FEMS Microbiol. Ecol. 2013, 85, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Ashelford, K.E.; Chuzhanova, N.A.; Fry, J.C.; Jones, A.J.; Weightman, A.J. At Least 1 in 20 16S rRNA Sequence Records Currently Held in Public Repositories Is Estimated To Contain Substantial Anomalies. Appl. Environ. Microbiol. 2005, 71, 7724–7736. [Google Scholar] [CrossRef] [PubMed]
- Klopfstein, S.; Vilhelmsen, L.; Heraty, J.M.; Sharkey, M.; Ronquist, F. The hymenopteran tree of life: Evidence from protein-coding genes and objectively aligned ribosomal data. PLoS One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef]
- Fontaneto, D.; Flot, J.-F.; Tang, C. Guidelines for DNA taxonomy, with a focus on the meiofauna. Mar. Biodiv. 2014. Submitted for publicaiton. [Google Scholar]
- Tang, C.Q.; Leasi, F.; Obertegger, U.; Kieneke, A.; Barraclough, T.G.; Fontaneto, D. The widely used small subunit 18S rDNA molecule greatly underestimates true diversity in biodiversity surveys of the meiofauna. Proc. Natl. Acad. Sci. USA 2012, 109, 16208–16212. [Google Scholar] [CrossRef] [PubMed]
- Kekkonen, M.; Hebert, P.D. DNA barcode-based delineation of putative species: Efficient start for taxonomic workflows. Mol. Ecol. Resour. 2014, 14, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Castenholz, R.W.; Wilmotte, A.; Herdman, M.; Rippka, R.; Waterbury, J.B.; Iteman, I.; Hoffmann, L. Phylum BX. Cyanobacteria. In Bergey’s Manual® of Systematic Bacteriology; Springer: New York, NY, USA, 2001; pp. 473–599. [Google Scholar]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Young, K.D. The Selective Value of Bacterial Shape. Microbiol. Mol. Biol. Rev. 2006, 70, 660–703. [Google Scholar] [CrossRef] [PubMed]
- Crawley, M.J. The R book; Wiley: Chichester, UK, 2013. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2009. [Google Scholar]
- Cohan, F.M.; Perry, E.B. A systematics for discovering the fundamental units of bacterial diversity. Curr. Biol. 2007, 17, R373–R386. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Baracaldo, P.; Hayes, P.; Blank, C. Morphological and habitat evolution in the Cyanobacteria using a compartmentalization approach. Geobiology 2005, 3, 145–165. [Google Scholar] [CrossRef]
- Schlick-Steiner, B.C.; Steiner, F.M.; Seifert, B.; Stauffer, C.; Christian, E.; Crozier, R.H. Integrative taxonomy: A multisource approach to exploring biodiversity. Annu. Rev. Entomol. 2010, 55, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Birky, C.W., Jr.; Ricci, C.; Melone, G.; Fontaneto, D. Integrating DNA and morphological taxonomy to describe diversity in poorly studied microscopic animals: New species of the genus Abrochtha Bryce, 1910 (Rotifera: Bdelloidea: Philodinavidae). Zool. J. Linn. Soc. 2011, 161, 723–734. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eckert, E.M.; Fontaneto, D.; Coci, M.; Callieri, C. Does a Barcoding Gap Exist in Prokaryotes? Evidences from Species Delimitation in Cyanobacteria. Life 2015, 5, 50-64. https://0-doi-org.brum.beds.ac.uk/10.3390/life5010050
Eckert EM, Fontaneto D, Coci M, Callieri C. Does a Barcoding Gap Exist in Prokaryotes? Evidences from Species Delimitation in Cyanobacteria. Life. 2015; 5(1):50-64. https://0-doi-org.brum.beds.ac.uk/10.3390/life5010050
Chicago/Turabian StyleEckert, Ester M., Diego Fontaneto, Manuela Coci, and Cristiana Callieri. 2015. "Does a Barcoding Gap Exist in Prokaryotes? Evidences from Species Delimitation in Cyanobacteria" Life 5, no. 1: 50-64. https://0-doi-org.brum.beds.ac.uk/10.3390/life5010050