
Rosette-Forming Glioneuronal Tumor of the Fourth Ventricle: A Case of Relapse Treated with Proton Beam Therapy

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
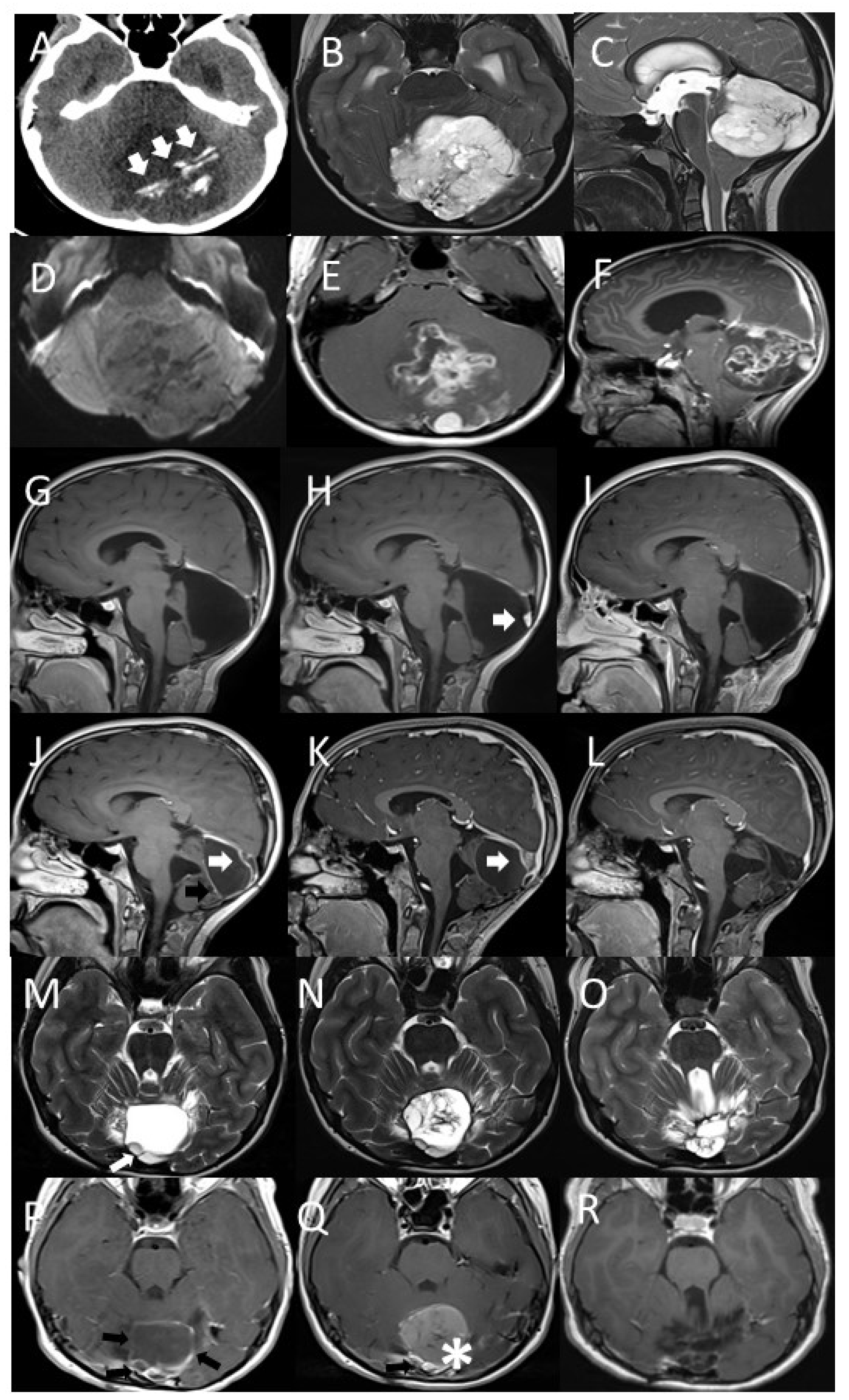
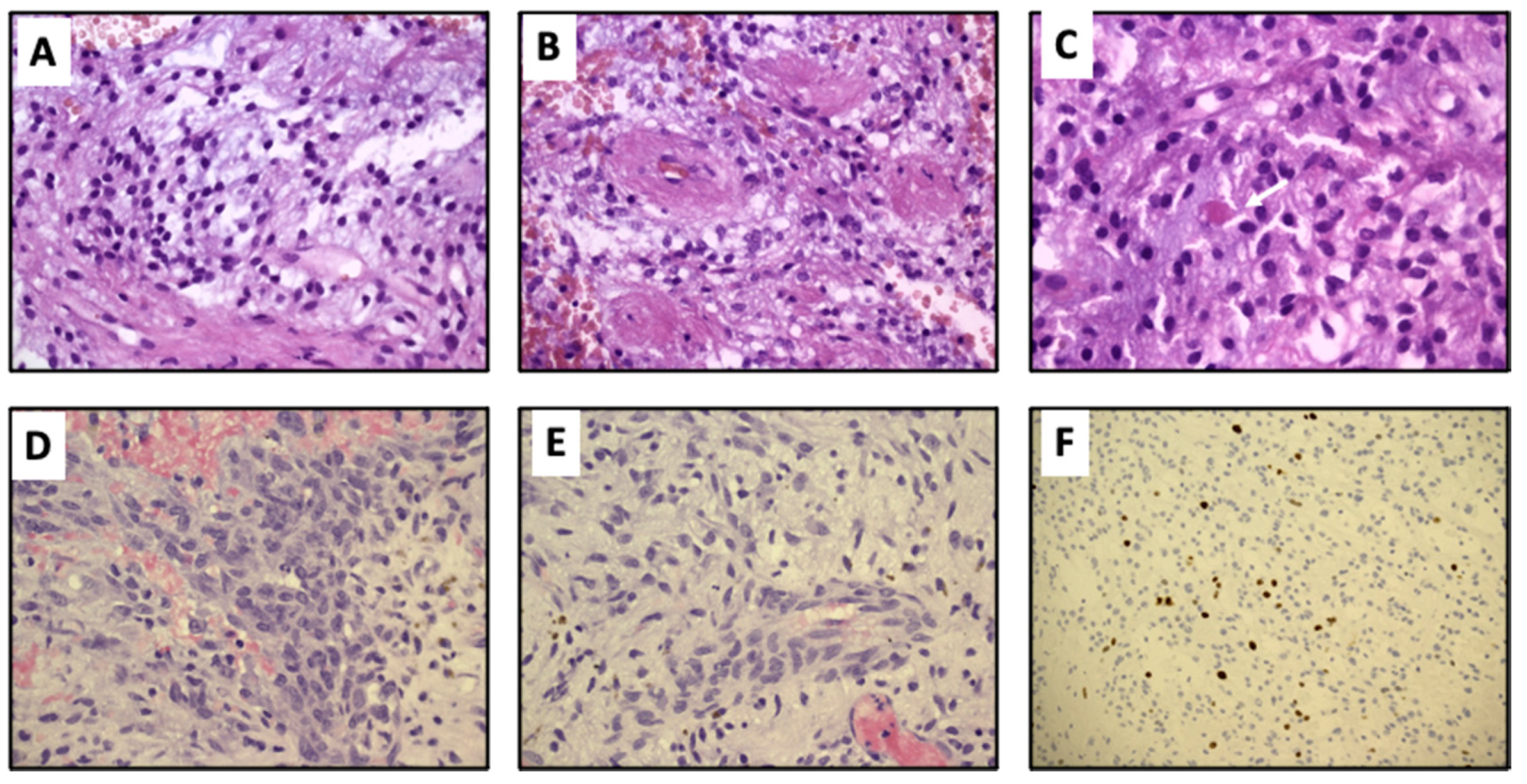
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, T.; Scheithauer, B.W.; Hirose, T. A rosette-forming glioneuronal tumor of the fourth ventricle: Infratentorial form of dysembryoplastic neuroepithelial tumor? Am. J. Surg. Pathol. 2002, 26, 582–591. [Google Scholar] [CrossRef]
- Eaton, B.R.; Esiashvili, N.; Kim, S.; Weyman, E.A.; Thornton, L.T.; Mazewski, C.; MacDonald, T.; Ebb, D.; MacDonald, S.M.; Tarbell, N.J.; et al. Clinical outcomes among children with standard-risk medulloblastoma treated with proton and photon radiation therapy: A comparison of disease control and overall survival. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Eaton, B.R.; Chowdhry, V.K.; Weaver, K.; Liu, L.; Ebb, D.H.; Macdonald, S.M.; Tarbell, N.J.; Yock, T.I. Use of proton therapy for re-irradiation in pediatric intracranial ependymoma. Radiother. Oncol. 2015, 116, 301–308. [Google Scholar] [CrossRef]
- Merchant, T.E.; Hua, C.H.; Shukla, H.; Ying, X.; Nill, S.; Oelfke, U. Proton versus photon radiotherapy for common pediatric brain tumors: Comparison of models of dose characteristics and their relationship to cognitive function. Pediatr. Blood Cancer 2008, 51, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Schlamann, A.; von Bueren, A.; Hagel, C.; Zwiener, I.; Seidel, C.; Kortmann, R.-D.; Muller, K. An individual patient data me-ta-analysis on characteristics and outcome of patients with papillary glioneuronal tumor, rosette glioneuronal tumor with neuropil-like islands and rosette forming glioneuronal tumor of the fourth ventricle. PLoS ONE 2014, 9, e101211. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; Huse, J.T. 2016 World Health Organization Classification of Central Nervous System Tumors. Contin. Lifelong Learn. Neurol. 2017, 23, 1531–1547. [Google Scholar] [CrossRef]
- Thurston, B.; Gunny, R.; Anderson, G.; Paine, S.; Thompson, D.; Jacques, T.; Ternier, J. Fourth ventricle rosette-forming glioneuronal tumour in children: An unusual presentation in an 8-year-old patient, discussion and review of the literature. Child’s Nerv. Syst. 2013, 29, 839–847. [Google Scholar] [CrossRef]
- Hsu, C.; Kwan, G.; Lau, Q.; Bhuta, S. Rosette-forming glioneuronal tumour: Imaging features, histopathological correlation and a comprehensive review of literature. Br. J. Neurosurg. 2012, 26, 668–673. [Google Scholar] [CrossRef]
- Rosenblum, M.K. The 2007 WHO Classification of Nervous System Tumors: Newly recognized members of the mixed glio-neuronal group. Brain Pathol. Zurich Switz. 2007, 17, 308–313. [Google Scholar] [CrossRef]
- Kemp, S.; Achan, A.; Ng, T.; Dexter, M.A.J. Rosette-forming glioneuronal tumour of the lateral ventricle in a patient with neurofibromatosis 1. J. Clin. Neurosci. 2012, 19, 1180–1181. [Google Scholar] [CrossRef] [PubMed]
- Karafin, M.; Jallo, G.I.; Ayars, M.; Eberhart, C.G.; Rodriguez, F.J. Rosette forming glioneuronal tumor in association with Noonan syndrome: Pathobiological implications. Clin. Neuropathol. 2011, 30, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.Y.; Bergstrom, K.; Person, R.; Bavle, A.; Ballester, L.Y.; Scollon, S.; Martinez, R.-R.; Jea, A.; Birchansky, S.; Wheeler, D.A.; et al. Integrated tumor and germline whole-exome sequencing identifies mutations in MAPK and PI3K pathway genes in an adolescent with rosette-forming glioneuronal tumor of the fourth ventricle. Cold Spring Harb. Mol. Case Stud. 2016, 2, a001057. [Google Scholar] [CrossRef]
- Arai, A.; Sasayama, T.; Tamaki, M.; Sakagami, Y.; Enoki, E.; Ohbayashi, C.; Kohmura, E. Rosette-forming glioneuronal tumor of the fourth ventricle--case report. Neurol. Med. Chir. 2010, 50, 224–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacques, T.S.; Eldridge, C.; Patel, A.; Saleem, N.M.; Powell, M.; Kitchen, N.D.; Thom, M.; Revesz, T. Mixed glioneuronal tumour of the fourth ventricle with prominent rosette formation. Neuropathol. Appl. Neurobiol. 2006, 32, 217–220. [Google Scholar] [CrossRef]
- Matsumura, N.; Wang, Y.; Nakazato, Y. Coexpression of glial and neuronal markers in the neurocytic rosettes of ro-sette-forming glioneuronal tumors. Brain Tumor Pathol. 2014, 31, 17–22. [Google Scholar] [CrossRef]
- Allende, D.S.; Prayson, R.A. The Expanding Family of Glioneuronal Tumors. Adv. Anat. Pathol. 2009, 16, 33–39. [Google Scholar] [CrossRef]
- Gessi, M.; Lambert, S.R.; Lauriola, L.; Waha, A.; Collins, V.P.; Pietsch, T. Absence of KIAA1549-BRAF fusion in rosette-forming glioneuronal tumors of the fourth ventricle (RGNT). J. Neurooncol. 2012, 110, 21–25. [Google Scholar] [CrossRef]
- Ellezam, B.; Theeler, B.J.; Luthra, R.; Adesina, A.M.; Aldape, K.D.; Gilbert, M.R. Recurrent PIK3CA mutations in rosette-forming glio-neuronal tumor. Acta Neuropathol. 2012, 123, 285–287. [Google Scholar] [CrossRef]
- Eye, P.G.; Davidson, L.; Malafronte, P.J.; Cantrell, S.; Theeler, B.J. PIK3CA mutation in a mixed dysembryoplastic neu-roepithelial tumor and rosette forming glioneuronal tumor, a case report and literature review. J. Neurol. Sci. 2017, 373, 280–284. [Google Scholar] [CrossRef]
- Sievers, P.; Appay, R.; Schrimpf, D.; Stichel, D.; Reuss, D.E.; Wefers, A.K.; Reinhardt, A.; Coras, R.; Ruf, V.C.; Schmid, S.; et al. Rosette-forming glioneuronal tumors share a distinct DNA methylation profile and mutations in FGFR1, with recurrent co-mutation of PIK3CA and NF1. Acta Neuropathol. 2019, 138, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Nobusawa, S.; Yamazaki, T.; Teranishi, T.; Watanabe, S.; Murayama, K.; Ohba, S.; Okabe, A.; Sakurai, K.; Urano, M.; et al. An epilepsy-associated glioneuronal tumor with mixed morphology harboringFGFR1mutation. Pathol. Int. 2019, 69, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Malignant Transformation of a Rosette-Forming Glioneuronal Tumor with IDH1 Mutation: A Case Report and Literature ReviewPubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/31218281/ (accessed on 1 February 2021).
- Bidinotto, L.T.; Scapulatempo-Neto, C.; Mackay, A.; Caravina de Almeida, G.; Scheithauer, B.W.; Berardinelli, G.N.; Torrieri, R.; Clara, C.A.; Feltrin, L.T.; Viana-Pereira, M.; et al. Molecular Profiling of a Rare Rosette-Forming Glioneuronal Tumor Arising in the Spinal Cord. PLoS ONE 2015, 10, e0137690. [Google Scholar] [CrossRef]
- Cebula, H.; Chibbaro, S.; Santin, M.; Kremer, S.; Chaussemy, D.; Proust, F. Thalamic rosette-forming a glioneuronal tumor in an elderly patient: Case report and literature review. Neurochirurgie 2016, 62, 60–63. [Google Scholar] [CrossRef]
- Haryu, S.; Saito, R.; Kanamori, M.; Sonoda, Y.; Kumabe, T.; Watanabe, M.; Tonga, F.; Tominaga, T. Rosette-forming Glioneuronal Tu-mor: Rare Case Presented with Spontaneous Disappearance of Contrast Enhancement. NMC Case Rep. J. 2015, 2, 65–67. [Google Scholar] [CrossRef] [Green Version]
- Lath, R.; Sharma, P.; Swain, M.; De Padua, M.; Ranjan, A. Rosette-forming glioneuronal tumors: A report of two cases. Neurol. India 2011, 59, 276–80. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.P.; Chakraborty, A.R.; Pelargos, P.E.; Shi, H.H.; Milton, C.K.; Sung, S.; McCoy, T.; Peterson, J.E.; Glenn, C.A. Rosette-forming glioneuronal tumor: An illustrative case and a systematic review. Neuro-Oncol. Adv. 2020, 2, vdaa116. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.; Prudowsky, Z.D.; Shetty, V.; Geller, T.; Elbabaa, S.K.; Guzman, M.; AbdelBaki, M.S. Rosette-Forming Glioneuronal Tumor of the Fourth Ventricle in Children: Case Report and Literature Review. World Neurosurg. 2017, 107, 1045.e9–1045.e16. [Google Scholar] [CrossRef]
- Bitterman, D.S.; MacDonald, S.M.; Yock, T.I.; Tarbell, N.J.; Wright, K.D.; Chi, N.S.; Marcus, K.J.; Kogan, D.A.H. Revisiting the Role of Ra-diation Therapy for Pediatric Low-Grade Glioma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 3335–3339. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.T. Long-term survivors of childhood central nervous system malignancies: The experience of the Childhood Cancer Survivor Study. Eur. J. Paediatr. Neurol. 2010, 14, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhakta, N.; Liu, Q.; Ness, K.K.; Baassiri, M.; Eissa, H.; Yeo, F.; Chemaitilly, W.; Ehrhardt, M.J.; Bass, J.; Bishop, M.W.; et al. The cumulative burden of surviving childhood cancer: An initial report from the St Jude Lifetime Cohort Study (SJLIFE). Lancet 2017, 390, 2569–2582. [Google Scholar] [CrossRef]
- Packer, R.J.; Gurney, J.G.; Punyko, J.A.; Donaldson, S.S.; Inskip, P.D.; Stovall, M.; Yasui, Y.; Mertens, A.C.; Sklar, C.A.; Nicholson, H.S.; et al. Long-term neurologic and neurosensory sequelae in adult survivors of a childhood brain tumor: Childhood cancer survivor study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 3255–3261. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.T.; Reddick, W.E.; Petersen, R.C.; Santucci, A.; Zhang, N.; Srivastava, D.; Ogg, R.J.; Hillenbrand, C.M.; Sabin, N.; Krasin, M.J.; et al. Evaluation of memory impairment in aging adult survivors of childhood acute lymphoblastic leu-kemia treated with cranial radiotherapy. J. Natl. Cancer Inst. 2013, 105, 899–907. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cacchione, A.; Mastronuzzi, A.; Carai, A.; Colafati, G.S.; Diomedi-Camassei, F.; Marrazzo, A.; Carboni, A.; Miele, E.; Pedace, L.; Tartaglia, M.; et al. Rosette-Forming Glioneuronal Tumor of the Fourth Ventricle: A Case of Relapse Treated with Proton Beam Therapy. Diagnostics 2021, 11, 903. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11050903
Cacchione A, Mastronuzzi A, Carai A, Colafati GS, Diomedi-Camassei F, Marrazzo A, Carboni A, Miele E, Pedace L, Tartaglia M, et al. Rosette-Forming Glioneuronal Tumor of the Fourth Ventricle: A Case of Relapse Treated with Proton Beam Therapy. Diagnostics. 2021; 11(5):903. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11050903
Chicago/Turabian StyleCacchione, Antonella, Angela Mastronuzzi, Andrea Carai, Giovanna Stefania Colafati, Francesca Diomedi-Camassei, Antonio Marrazzo, Alessia Carboni, Evelina Miele, Lucia Pedace, Marco Tartaglia, and et al. 2021. "Rosette-Forming Glioneuronal Tumor of the Fourth Ventricle: A Case of Relapse Treated with Proton Beam Therapy" Diagnostics 11, no. 5: 903. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11050903