
Laboratory Diagnosis of Porphyria

 , , , , ,
, , , , , 
Abstract
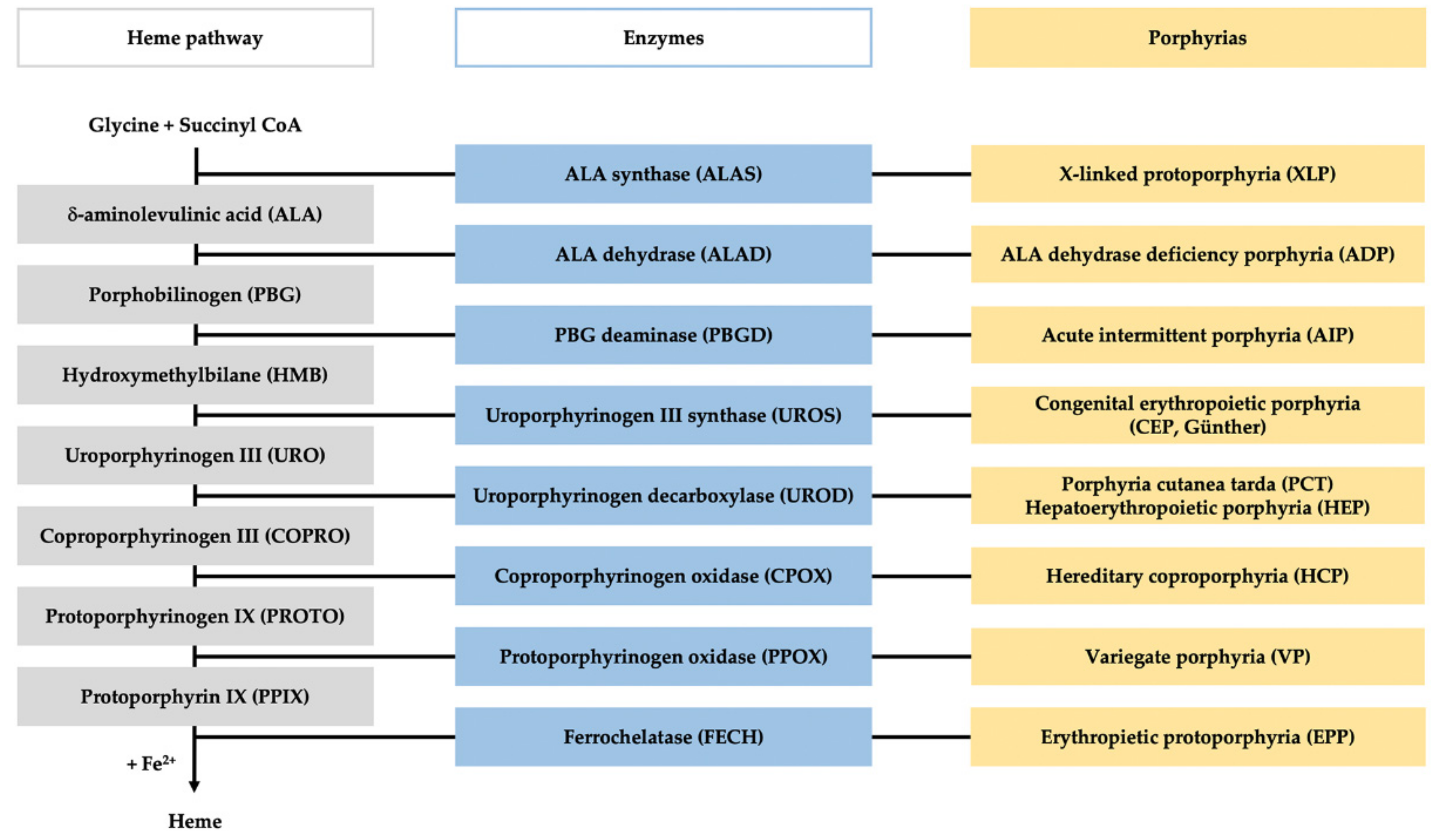
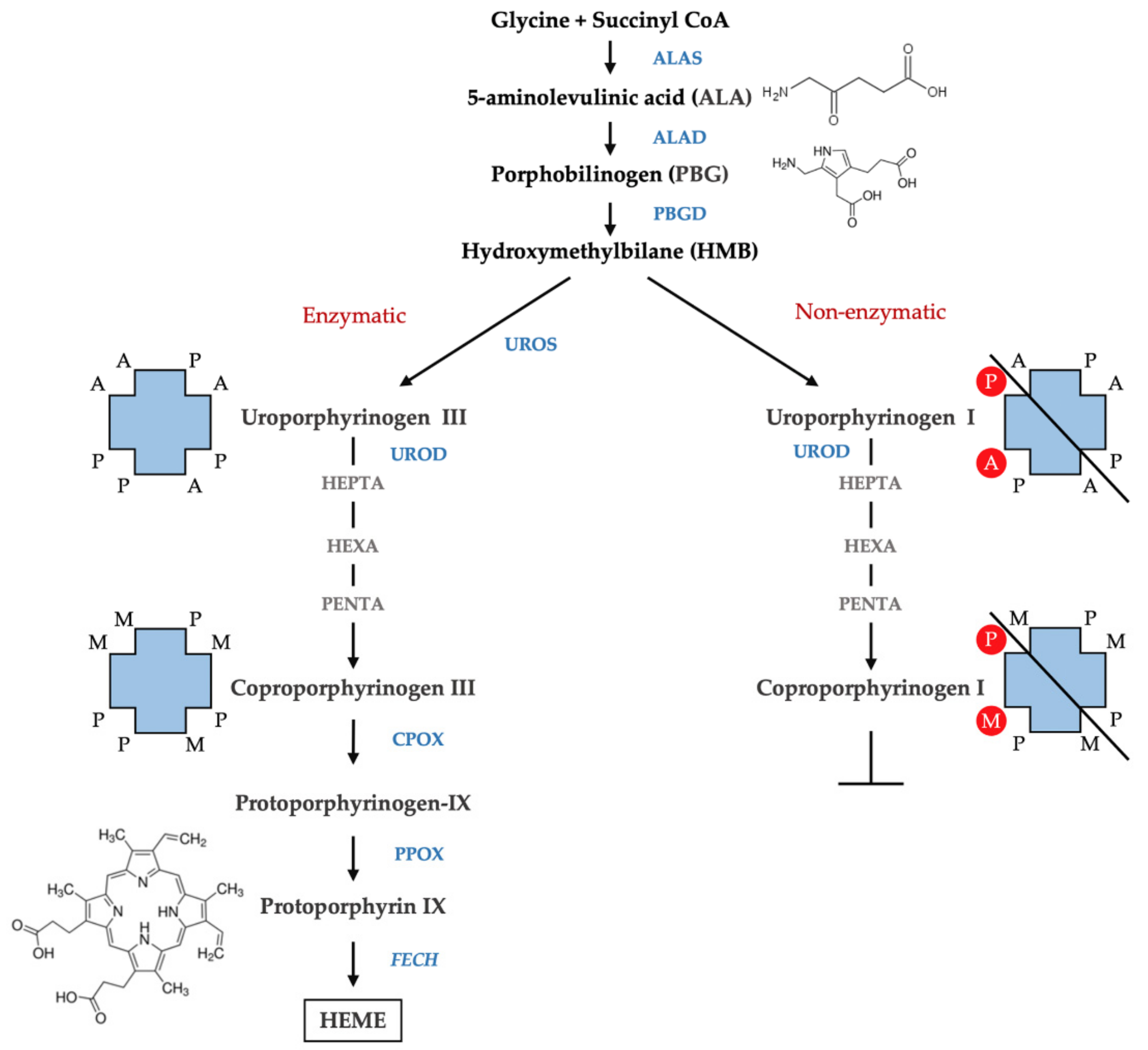
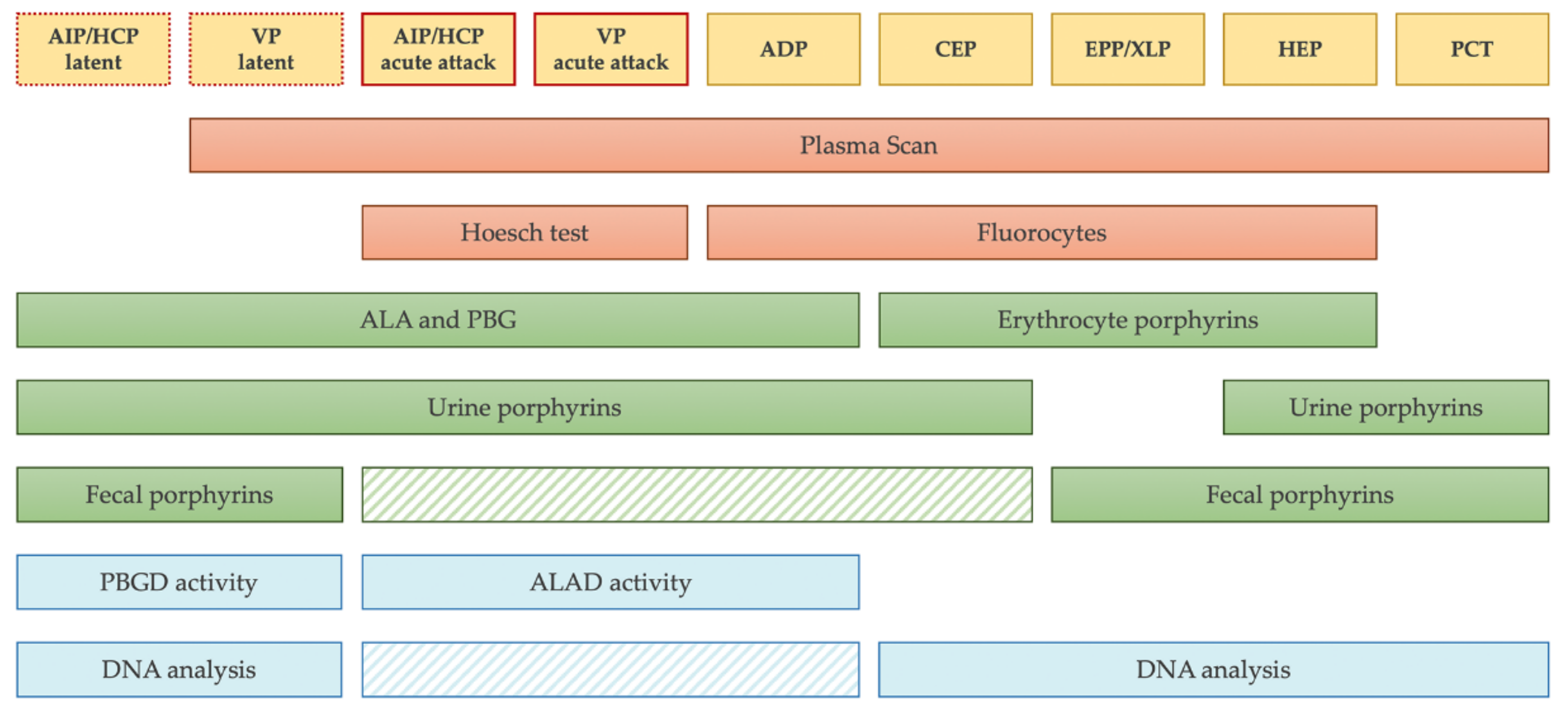
:1. Introduction
2. Qualitative Screening Tests
2.1. Plasma Scan
2.2. Fluorocytes
2.3. Hoesch Test
3. Quantitative Confirmatory Tests
3.1. ALA and PBG Determination
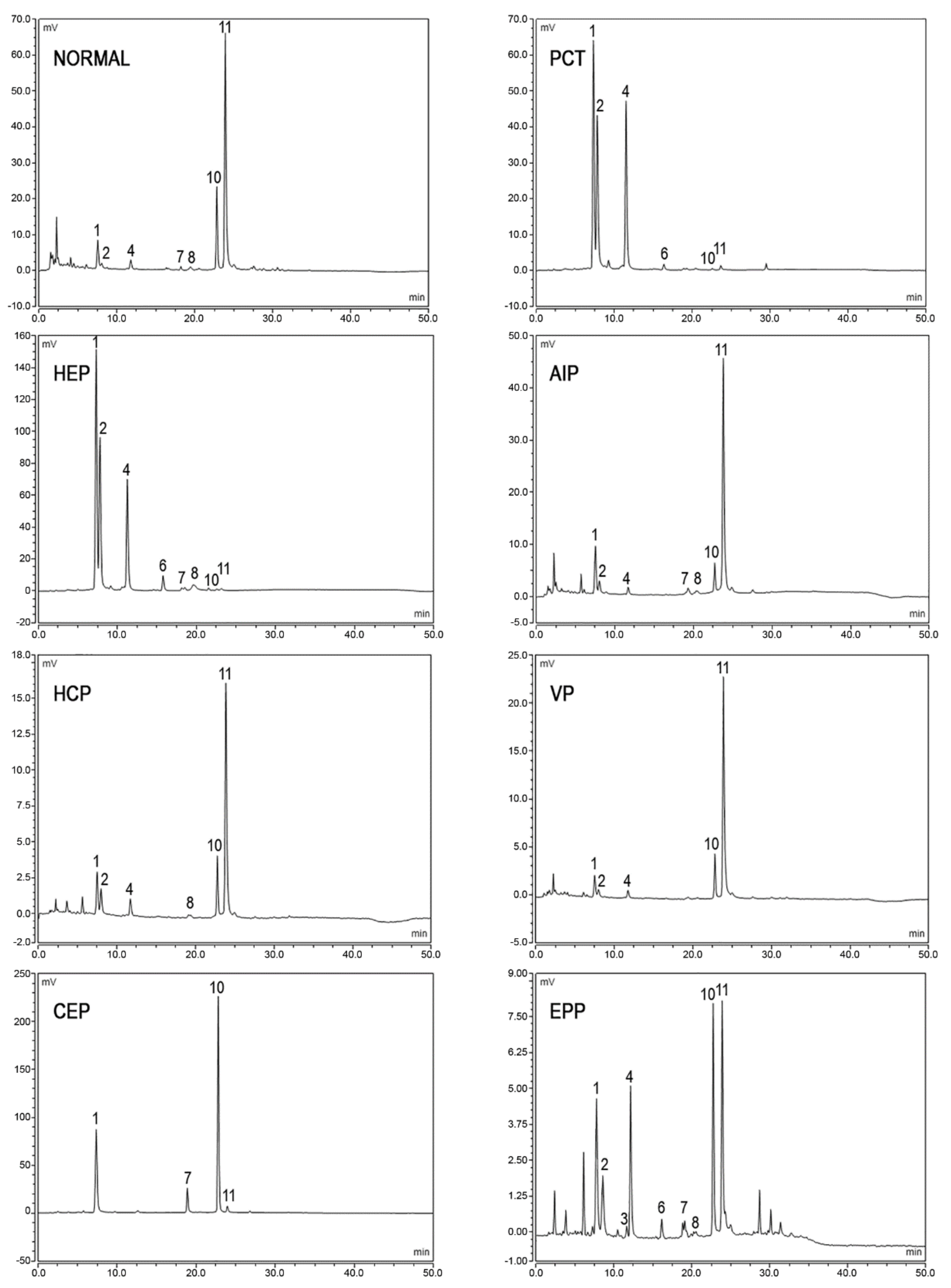
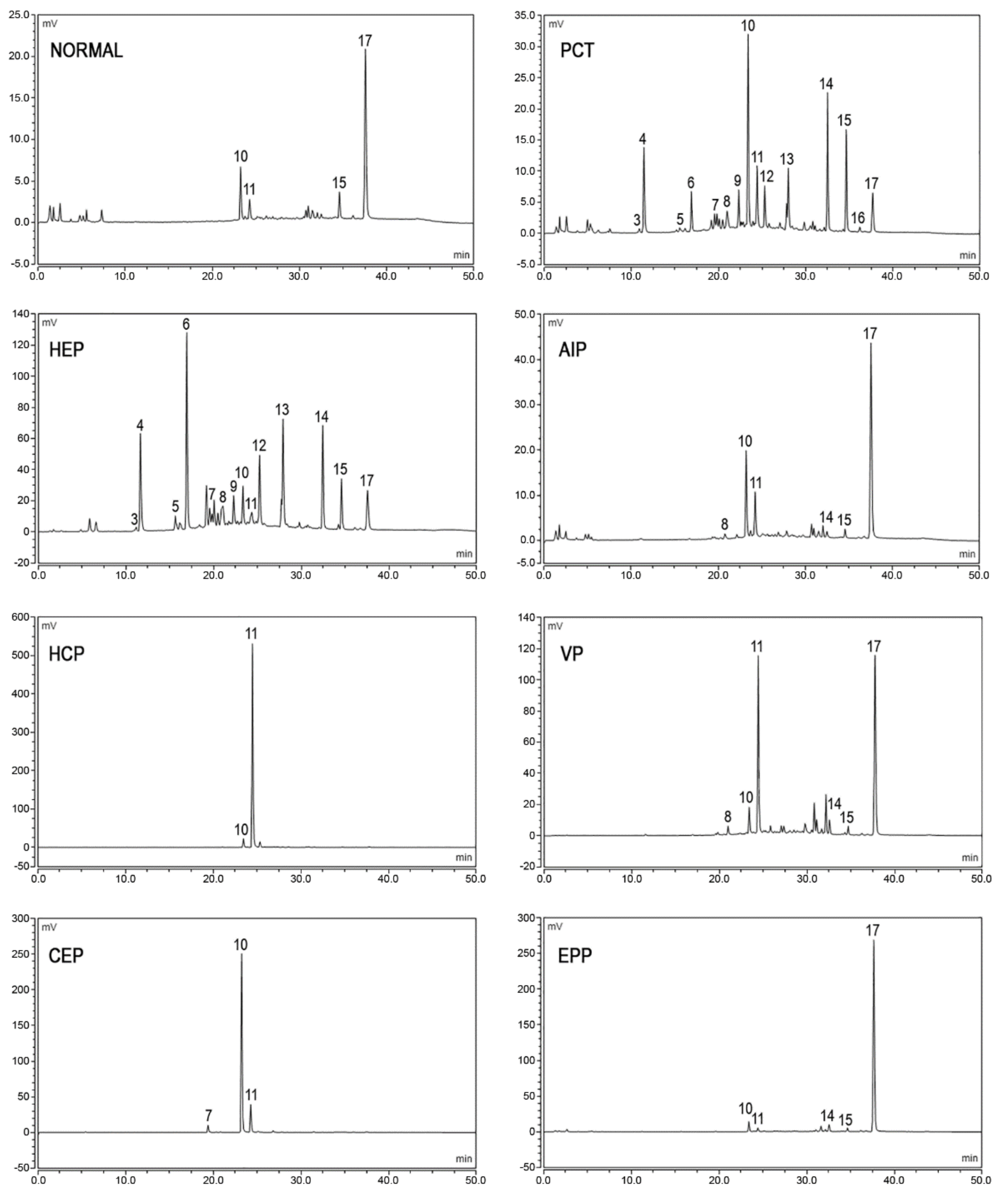
3.2. Measurement of Urine Porphyrins
3.3. Analysis of Fecal Porphyrins
3.4. Erythrocyte Porphyrins Measurement
4. Enzymatic Assays
4.1. ALAD Enzyme Activity
4.2. PBGD Enzyme Activity
5. Genetic Testing
5.1. DNA Sequence Analysis
5.2. Multiplex Ligation-Dependent Probe Amplification (MLPA)
5.3. Next-Generation Sequencing (NGS)
6. European Specialist Porphyria Laboratories
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Balwani, M.; Desnick, R.J. The porphyrias: Advances in Diagnosis and Treatment. Blood 2012, 120, 4496–4504. [Google Scholar] [CrossRef]
- Besur, S.; Hou, W.; Schmeltzer, P.; Bonkovsky, H.L. Clinically Important Features of Porphyrin and Heme Metabolism and the Porphyrias. Metabolites 2014, 4, 977–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolzel, U.; Doss, M.O.; Schuppan, D. Clinical Guide and Update on Porphyrias. Gastroenterology 2019, 157, 365–381. [Google Scholar] [CrossRef] [Green Version]
- Sassa, S. Modern Diagnosis and Management of the Porphyrias. Br. J. Haematol. 2006, 135, 281–292. [Google Scholar] [CrossRef]
- Wang, B.; Rudnick, S.; Cengia, B.; Bonkovsky, H.L. Acute Hepatic Porphyrias: Review and Recent Progress. Hepatol. Commun. 2019, 3, 193–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puy, H.; Gouya, L.; Deybach, J.C. Porphyrias. Lancet 2010, 375, 924–937. [Google Scholar] [CrossRef]
- Linenberger, M.; Fertrin, K.Y. Updates on the Diagnosis and Management of the Most Common Hereditary Porphyrias: AIP and EPP. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Karim, Z.; Lyoumi, S.; Nicolas, G.; Deybach, J.C.; Gouya, L.; Puy, H. Porphyrias: A 2015 Update. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 412–425. [Google Scholar] [CrossRef]
- Bissell, D.M.; Anderson, K.; Bonkovsky, H.L. Porphyria. N. Engl. J. Med. 2017, 377, 862–872. [Google Scholar] [CrossRef]
- Woolf, J.; Marsden, J.T.; Degg, T.; Whatley, S.; Reed, P.; Brazil, N.; Stewart, M.F.; Badminton, M. Best Practice Guidelines on First-Line Laboratory Testing for Porphyria. Ann. Clin. Biochem. 2017, 54, 188–198. [Google Scholar] [CrossRef]
- Ventura, P.; Cappellini, M.D.; Biolcati, G.; Guida, C.C.; Rocchi, E. A Challenging Diagnosis for Potential Fatal Diseases: Rec-Ommendations for Diagnosing Acute Porphyrias. Eur. J. Intern. Med. 2014, 25, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Poblete-Gutiérrez, P.; Wiederholt, T.; Merk, H.F.; Frank, J. The Porphyrias: Clinical Presentation, Diagnosis and Treatment. Eur. J. Dermatol. 2006, 16, 230–240. [Google Scholar] [PubMed]
- Poh-Fitzpatrick, M.B.; Lamola, A.A. Direct Spectrofluorometry of Diluted Erythrocytes and Plasma: A Rapid Diagnostic Method in Primary and Secondary Porphyrinemias. J. Lab. Clin. Med. 1976, 87, 362–370. [Google Scholar] [PubMed]
- Di Pierro, E.; Ventura, P.; Brancaleoni, V.; Moriondo, V.; Marchini, S.; Tavazzi, D.; Nascimbeni, F.; Ferrari, M.C.; Rocchi, E.; Cappellini, M.D. Clinical, Biochemical and Genetic Characteristics of Variegate Porphyria in Italy. Cell. Mol. Biol. 2009, 55, 79–88. [Google Scholar]
- Poh-Fitzpatrick, M.B. A Plasma Porphyrin Fluorescence Marker for Variegate Porphyria. Arch. Dermatol. 1980, 116, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Chularojanamontri, L.; Tuchinda, C.; Srisawat, C.; Neungton, N.; Junnu, S.; Kanyok, S. Utility of Plasma Fluorometric Emission Scanning for Diagnosis of the First 2 Cases Reports of Variegate Porphyria: A Very Rare Type of Porphyrias in Thai. J. Med. Assoc. Thai. 2008, 91, 1915–1919. [Google Scholar]
- Enriquez De, S.R.; Sepulveda, P.; Moran, M.J.; Santos, J.L.; Fontanellas, A.; Hernandez, A. Clinical Utility of Fluorometric Scanning of Plasma Porphyrins for the Diagnosis and Typing of Porphyrias. Clin. Exp. Dermatol. 1993, 18, 128–130. [Google Scholar] [CrossRef]
- Hift, R.J.; Meissner, D.; Meissner, P.N. A Systematic Study of the Clinical and Biochemical Expression of Variegate Porphyria in a Large South African Family. Br. J. Dermatol. 2004, 151, 465–471. [Google Scholar] [CrossRef]
- Da Silva, V.; Simonin, S.; Deybach, J.C.; Puy, H.; Nordmann, Y. Variegate Porphyria: Diagnostic Value of Fluorometric Scanning of Plasma Porphyrins. Clin. Chim. Acta 1995, 238, 163–168. [Google Scholar] [CrossRef]
- Sies, C.W.; Davidson, J.S.; Florkowski, C.M.; Johnson, R.N.; Potter, H.C.; Woollard, G.A.; George, P.M. Plasma Fluorescence Scanning Did Not Detect Latent Variegate Porphyria in Nine Patients with Non-p.R59W Mutations. Pathology 2005, 37, 324–326. [Google Scholar] [CrossRef]
- Zaider, E.; Bickers, D.R. Clinical Laboratory Methods for Diagnosis of the Porphyrias. Clin. Dermatol. 1998, 16, 277–293. [Google Scholar] [CrossRef]
- Osipowicz, K.; Kalinska-Bienias, A.; Kowalewski, C.; Wozniak, K. Development of Bullous Pemphigoid during the Haemodialysis of a Young Man: Case Report and Literature Survey. Int. Wound J. 2017, 14, 288–292. [Google Scholar] [CrossRef]
- Bergler-Czop, B.; Brzezinska-Wcislo, L. Pseudoporphyria Induced by Hemodialysis. Adv. Dermatol. Allergol. 2014, 31, 53–55. [Google Scholar] [CrossRef]
- Handler, N.S.; Handler, M.Z.; Stephany, M.P.; Handler, G.A.; Schwartz, R.A. Porphyria Cutanea Tarda: An Intriguing Genetic Disease and Marker. Int. J. Dermatol. 2017, 56, e106–e117. [Google Scholar] [CrossRef]
- Deacon, A.C.; Elder, G.H. ACP Best Practice No 165: Front Line Tests for the Investigation of Suspected Porphyria. J. Clin. Pathol. 2001, 54, 500–507. [Google Scholar] [CrossRef] [Green Version]
- Rimington, C. Cripps Dj: Biochemical and Fluorescence-Microscopy Screening-Tests for Erythropoietic Protoporphyria. Lancet 1965, 1, 624–626. [Google Scholar] [CrossRef]
- Lau, K.C.; Lam, C.W. Automated Imaging of Circulating Fluorocytes for the Diagnosis of Erythropoietic Protoporphyria: A Pilot Study for Population Screening. J. Med. Screen. 2008, 15, 199–203. [Google Scholar] [CrossRef] [Green Version]
- Piomelli, S.; Lamola, A.A.; Poh-Fitzpatrick, M.F.; Seaman, C.; Harber, L.C. Erythropoietic Protoporphyria and Lead Intoxication: The Molecular Basis for Difference in Cutaneous Photosensitivity. I. Different Rates of Disappearance of Protoporphyrin from the Erythrocytes, Both In Vivo and In Vitro. J. Clin. Investig. 1975, 56, 1519–1527. [Google Scholar] [CrossRef] [Green Version]
- Cordiali, F.P.; Macri, A.; Trento, E.; D’Agosto, G.; Griso, D.; Biolcati, F.; Ameglio, F. Flow Cytometric Analysis of Fluorocytes in Patients with Erythropoietic Porphyria. Eur. J. Histochem. 1997, 41 (Suppl. 2), 9–10. [Google Scholar]
- Brun, A.; Steen, H.B.; Sandberg, S. Erythropoietic Protoporphyria: A Quantitative Determination of Erythrocyte Protoporphyrin in Individual Cells by Flow Cytometry. Scand. J. Clin. Lab. Investig. 1988, 48, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Schleiffenbaum, B.E.; Minder, E.I.; Mohr, P.; Decurtins, M.; Schaffner, A. Cytofluorometry as a Diagnosis of Protoporphyria. Gastroenterology 1992, 102, 1044–1048. [Google Scholar] [CrossRef]
- Anderson, K.E.; Lobo, R.; Salazar, D.; Schloetter, M.; Spitzer, G.; White, A.L.; Young, R.M.; Bonkovsky, H.L.; Frank, E.L.; Mora, J.; et al. Biochemical Diagnosis of Acute Hepatic Porphyria: Updated Expert Recommendations for Primary Care Physicians. Am. J. Med. Sci. 2021. [Google Scholar] [CrossRef]
- Watson, C.J.; Schwartz, S. A Simple Test for Urinary Porphobilinogen. Exp. Biol. Med. 1941, 47, 393. [Google Scholar] [CrossRef]
- Watson, C.J.; Taddeini, L.; Bossenmaier, I. Present Status of the Ehrlich Aldehyde Reaction for Urinary Porphobilinogen. Jama 1964, 190, 501–504. [Google Scholar] [CrossRef]
- With, T. K: Screening Test for Acute Porphyria. Lancet 1970, 2, 1187–1188. [Google Scholar] [CrossRef]
- Bonkovsky, H.L.; Barnard, G.F. Diagnosis of Porphyric Syndromes: A Practical Approach in the Era of Molecular Biology. Semin. Liver Dis. 1998, 18, 57–65. [Google Scholar] [CrossRef]
- Calvo De Mora Almazán, M.; Acuña, M.; Garrido-Astray, C.; Arcos Pulido, B.; Gómez-Abecia, S.; Chicot Llano, M.; González Parra, E.; Gracia Iguacel, C.; Alonso Alonso, P.P.; Egido, J.; et al. Acute Porphyria in an Intensive Care unit. Emergencias 2012, 24, 454–458. [Google Scholar]
- Lamon, J.; With, T.K.; Redeker, A.G. The Hoesch Test: Bedside Screening for Urinary Porphobilinogen in Patients with Suspected Porphyria. Clin. Chem. 1974, 20, 1438–1440. [Google Scholar] [CrossRef]
- McEwen, J.; Paterson, C. Drugs and False-Positive Screening Tests for Porphyria. Br. Med. J. 1972, 1, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddeini, L.; Kay, I.T.; Watson, C.J. Inhibition of the Ehrlich’s Reaction of Porphobilinogen by Indican and Related Compounds. Clin. Chim. Acta 1962, 7, 890–891. [Google Scholar] [CrossRef]
- Castelbón Fernández, F.J.; Solares Fernandez, I.; Arranz Canales, E.; Enríquez De Salamanca, R.; Morales Conejo, M. Protocol for Patients with Suspected Acute Porphyria. Rev. Clin. Esp. 2020, 220, 592–596. [Google Scholar] [CrossRef]
- Aarsand, A.K.; Petersen, P.H.; Sandberg, S. Estimation and Application of Biological Variation of Urinary Delta-Aminolevulinic acid and Porphobilinogen in Healthy Individuals and in Patients with Acute Intermittent Porphyria. Clin. Chem. 2006, 52, 650–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauzerall, D.; Granick, S. The Occurrence and Determination of Delta-Amino-Levulinic Acid and Porphobilinogen in Urine. J. Biol. Chem. 1956, 219, 435–446. [Google Scholar] [CrossRef]
- Kelada, S.N.; Shelton, E.; Kaufmann, R.B.; Khoury, M.J. D-Aminolevulinic Acid Dehydratase Genotype and Lead Toxicity: A HuGE Review. Am. J. Epidemiol. 2001, 154, 1–13. [Google Scholar] [CrossRef]
- Wyss, P.A.; Carter, B.E.; Roth, K.S. Delta-Aminolevulinic Acid Dehydratase: Effects of Succinylacetone in Rat Liver and Kidney in an In Vivo Model of the Renal Fanconi Syndrome. Biochem. Med. Metab. Biol. 1992, 48, 86–89. [Google Scholar] [CrossRef]
- Floderus, Y.; Sardh, E.; Moller, C.; Andersson, C.; Rejkjaer, L.; Andersson, D.E.; Harper, P. Variations in Porphobilinogen and 5-Aminolevulinic Acid Concentrations in Plasma and Urine from Asymptomatic Carriers of the Acute Intermittent Porphyria Gene with Increased Porphyrin Precursor Excretion. Clin. Chem. 2006, 52, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benton, C.M.; Couchman, L.; Marsden, J.T.; Rees, D.C.; Moniz, C.; Lim, C.K. Direct and Simultaneous Quantitation of 5-Aminolaevulinic Acid and Porphobilinogen in Human Serum or Plasma by Hydrophilic Interaction Liquid Chromatography-Atmospheric Pressure Chemical Ionization/Tandem Mass Spectrometry. Biomed. Chromatogr. 2013, 27, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Benton, C.M.; Lim, C.K. Liquid Chromatography and Mass Spectrometry of Haem Biosynthetic Intermediates: A Review. Biomed. Chromatogr. 2012, 26, 1009–1023. [Google Scholar] [CrossRef]
- Zhang, J.; Yasuda, M.; Desnick, R.J.; Balwani, M.; Bishop, D.; Yu, C. A LC-MS/MS Method for the Specific, Sensitive, and Simultaneous Quantification of 5-Aminolevulinic Acid and Porphobilinogen. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2011, 879, 2389–2396. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Habtemarium, B.; Xu, Y.; Simon, A.R.; Kim, J.B.; Robbie, G.J. Normal Reference Ranges for Urinary Delta-Aminolevulinic Acid and Porphobilinogen Levels. JIMD Rep. 2021, 57, 85–93. [Google Scholar] [CrossRef]
- Sardh, E.; Harper, P.; Andersson, D.E.; Floderus, Y. Plasma Porphobilinogen as a Sensitive Biomarker to Monitor the Clinical and Therapeutic Course of Acute Intermittent Porphyria Attacks. Eur. J. Intern. Med. 2009, 20, 201–207. [Google Scholar] [CrossRef]
- Sardh, E.; Rejkjaer, L.; Andersson, D.E.; Harper, P. Safety, Pharmacokinetics and Pharmocodynamics of Recombinant Human Porphobilinogen Deaminase in Healthy Subjects and Asymptomatic Carriers of the Acute Intermittent Porphyria Gene Who Have Increased Porphyrin Precursor Excretion. Clin. Pharmacokinet. 2007, 46, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Li, F.M.; Peters, T.J. High-Performance Liquid Chromatography of Porphyrins. J. Chromatogr. 1988, 429, 123–153. [Google Scholar] [CrossRef]
- Macours, P.; Cotton, F. Improvement in HPLC Separation of Porphyrin Isomers and Application to Biochemical Diagnosis of Porphyrias. Clin. Chem. Lab. Med. 2006, 44, 1433–1440. [Google Scholar] [CrossRef]
- Schreiber, W.E.; Raisys, V.A.; Labbe, R.F. Liquid-Chromatographic Profiles of Urinary Porphyrins. Clin. Chem. 1983, 29, 527–530. [Google Scholar] [CrossRef]
- Lim, C.K.; Rideout, J.M.; Wright, D.J. Separation of Porphyrin Isomers by High-Performance Liquid Chromatography. Biochem. J. 1983, 211, 435–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindmarsh, J.T.; Oliveras, L.; Greenway, D.C. Biochemical Differentiation of the Porphyrias. Clin. Biochem. 1999, 32, 609–619. [Google Scholar] [CrossRef]
- Rimington, C. The Isolation of a Monoacrylic Tri Propionic Porphyrin from Meconium and Its Bearing on the Conversion of Coproporphyrin to Protoporphyrin. S. Afr. Med. J. 1971, 45, 187–189. [Google Scholar]
- Lockwood, W.H.; Poulos, V.; Rossi, E.; Curnow, D.H. Rapid Procedure for Fecal Porphyrin Assay. Clin. Chem. 1985, 31, 1163–1167. [Google Scholar] [CrossRef]
- Pudek, M.R.; Schreiber, W.E.; Jamani, A. Quantitative Fluorometric Screening Test for Fecal Porphyrins. Clin. Chem. 1991, 37, 826–831. [Google Scholar] [CrossRef]
- Lim, C.K.; Peters, T.J. Urine and Faecal Porphyrin Profiles by Reversed-Phase High-Performance Liquid Chromatography in the Porphyrias. Clin. Chim. Acta 1984, 139, 55–63. [Google Scholar] [CrossRef]
- Beukeveld, G.J.; Wolthers, B.G.; Van Saene, J.J.; De Haan, T.H.; De Ruyter-Buitenhuis, L.W.; Van Saene, R.H. Patterns of Porphyrin Excretion in Feces as Determined by Liquid Chromatography; Reference Values and the Effect of Flora Suppression. Clin. Chem. 1987, 33, 2164–2170. [Google Scholar] [CrossRef]
- Zuijderhoudt, F.M.; Kamphuis, J.S.; Kluitenberg, W.E.; Dorresteijn-De, B.J. Precision and Accuracy of a HPLC Method for Measurement of Fecal Porphyrin Concentrations. Clin. Chem. Lab. Med. 2002, 40, 1036–1039. [Google Scholar] [CrossRef]
- Elder, G.H. Identification of a Group of Tetracarboxylate Porphyrins, Containing One Acetate and Three Propionate-Substituents, in Faeces from Patients with Symptomatic Cutaneous Hepatic Porphyria and from Rats with Porphyria Due to Hexachlorobenzene. Biochem. J. 1972, 126, 877–891. [Google Scholar] [CrossRef] [Green Version]
- Rose, I.S.; Young, G.P.; St John, D.J.; Deacon, M.C.; Blake, D.; Henderson, R.W. Effect of Ingestion of Hemoproteins on Fecal Excretion of Hemes and Porphyrins. Clin. Chem. 1989, 35, 2290–2296. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Boeijinga, J.K.; Van Haard, P.M.; Schoemaker, R.C.; Van Vliet-Verbeek, A. Gastrointestinal Blood Loss after Non-Steroidal Anti-Inflammatory Drugs. Measurement by Selective Determination of Faecal Porphyrins. Br. J. Clin. Pharmacol. 1992, 33, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Nordmann, Y.; Grandchamp, B.; De, V.H.; Phung, L.; Cartigny, B.; Fontaine, G. Harderoporphyria: A Variant Hereditary Coproporphyria. J. Clin. Investig. 1983, 72, 1139–1149. [Google Scholar] [CrossRef] [Green Version]
- Kuhnel, A.; Gross, U.; Jacob, K.; Doss, M.O. Studies on Coproporphyrin Isomers in Urine and Feces in the Porphyrias. Clin. Chim. Acta 1999, 282, 45–58. [Google Scholar] [CrossRef]
- Jacob, K.; Doss, M.O. Excretion Pattern of Faecal Coproporphyrin Isomers I-IV in Human Porphyrias. Eur. J. Clin. Chem. Clin. Biochem. 1995, 33, 893–901. [Google Scholar] [CrossRef] [Green Version]
- Heller, S.R.; Labbe, R.F.; Nutter, J. A Simplified Assay for Porphyrins in Whole Blood. Clin. Chem. 1971, 17, 525–528. [Google Scholar] [CrossRef]
- Lamola, A.A.; Joselow, M.; Yamane, T. Zinc Protoporphyrin (ZPP): A simple, Sensitive Fluorometric Screening Test for Lead Poisoning. Clin. Chem. 1975, 21, 93–97. [Google Scholar] [CrossRef]
- Blumberg, W.E.; Eisinger, J.; Lamola, A.A.; Zuckerman, D.M. The Hematofluorometer. Clin. Chem. 1977, 23, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Bailey, G.G.; Needham, L.L. Simultaneous Quantification of Erythrocyte Zinc Protoporphyrin and Protoporphyrin IX by Liquid Chromatography. Clin. Chem. 1986, 32, 2137–2142. [Google Scholar] [CrossRef]
- Chen, Q.; Hirsch, R.E. A Direct and Simultaneous Detection of Zinc Protoporphyrin IX, Free Protoporphyrin IX, and Fluorescent Heme Degradation Product in Red Blood Cell Hemolysates. Free Radical. Research. 2006, 40, 285–294. [Google Scholar] [CrossRef]
- Zhou, P.C.; Huang, W.; Zhang, R.B.; Zou, Z.X.; Luo, H.D.; Falih, A.A.; Li, Y.Q. A Simple and Rapid Fluorimetric Method for Simultaneous Determination of Protoporphyrin IX and Zinc Protoporphyrin IX in Whole Blood. Appl. Spectrosc. 2008, 62, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Sassa, S.; Granick, J.L.; Granick, S.; Kappas, A.; Levere, R.D. Studies in Lead Poisoning. I. Microanalysis of Erythrocyte Protoporphyrin Levels by Spectrophotometry in the Detection of Chronic Lead Intoxication in the Subclinical Range. Biochem. Med. 1973, 8, 135–148. [Google Scholar] [CrossRef]
- Braun, J. Erythrocyte Zinc Protoporphyrin. Kidney Int. Suppl. 1999, 69, S57–S60. [Google Scholar] [CrossRef] [Green Version]
- Anderson, P.M.; Desnick, R.J. Porphobilinogen Deaminase: Methods and Principles of the Enzymatic Assay. Enzyme 1982, 28, 146–157. [Google Scholar] [CrossRef]
- Hindmarsh, J.T. Enzyme Assays and the Porphyrias: Which Tissues and When Indicated. Clin. Dermatol. 1998, 16, 245–250. [Google Scholar] [CrossRef]
- Gouya, L.; Martin-Schmitt, C.; Robreau, A.M.; Austerlitz, F.; Da, S.V.; Brun, P.; Simonin, S.; Lyoumi, S.; Grandchamp, B.; Beaumont, C.; et al. Contribution of a Common Single-Nucleotide Polymorphism to the Genetic Predisposition for Erythropoietic Protoporphyria. Am. J. Hum. Genet. 2006, 78, 2–14. [Google Scholar] [CrossRef] [Green Version]
- Tomokuni, K.; Hirai, Y.; Ichiba, M. Fluorimetric Determination of Hepatic Delta-Aminolevulinic Acid Synthase Activity by High-Performance Liquid Chromatography. J. Chromatogr. 1991, 567, 65–70. [Google Scholar] [CrossRef]
- Bergonia, H.A.; Franklin, M.R.; Kushner, J.P.; Phillips, J.D. A method for Determining Delta-Aminolevulinic Acid Synthase Activity in Homogenized Cells and Tissues. Clin. Biochem. 2015, 48, 788–795. [Google Scholar] [CrossRef] [Green Version]
- Deybach, J.C.; Puy, H.; Robreau, A.M.; Lamoril, J.; Da, S.V.; Grandchamp, B.; Nordmann, Y. Mutations in the Protoporphyrinogen Oxidase Gene in Patients with Variegate Porphyria. Hum. Mol. Genet. 1996, 5, 407–410. [Google Scholar] [CrossRef] [Green Version]
- Lamoril, J.; Martasek, P.; Deybach, J.C.; Da, S.V.; Grandchamp, B.; Nordmann, Y. A Molecular Defect in Coproporphyrinogen Oxidase Gene Causing Harderoporphyria, a Variant Form of Hereditary Coproporphyria. Hum. Mol. Genet. 1995, 4, 275–278. [Google Scholar] [CrossRef]
- Wang, Y.; Gatti, P.; Sadilek, M.; Scott, C.R.; Turecek, F.; Gelb, M.H. Direct Assay of Enzymes in Heme Biosynthesis for the Detection of Porphyrias by Tandem Mass Spectrometry. Uroporphyrinogen Decarboxylase and Coproporphyrinogen III Oxidase. Anal. Chem. 2008, 80, 2599–2605. [Google Scholar] [CrossRef] [Green Version]
- Aarsand, A.K.; Boman, H.; Sandberg, S. Familial and Sporadic Porphyria Cutanea Tarda: Characterization and Diagnostic Strategies. Clin. Chem. 2009, 55, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badenas, C.; To-Figueras, J.; Phillips, J.D.; Warby, C.A.; Munoz, C.; Herrero, C. Identification and Characterization of Novel Uroporphyrinogen Decarboxylase Gene Mutations in a Large Series of Porphyria Cutanea Tarda Patients and Relatives. Clin. Genet. 2009, 75, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Chmielnicka, J.; Szymanska, J.A. Evaluation of Methods for the Estimation of 5-Aminolevulinate Dehydratase for a Broad Range of Lead Concentrations in the Blood of Exposed Workers. J. Clin. Chem. Clin. Biochem. 1979, 17, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.M.; Desnick, R.J. Purification and Properties of Delta-Aminolevulinate Dehydrase from Human Erythrocytes. J. Biol. Chem. 1979, 254, 6924–6930. [Google Scholar] [CrossRef]
- Giampietro, P.F.; Desnick, R.J. Determination of Delta-Aminolevulinate Dehydratase Activity by a Specific Fluorometric Coupled-Enzyme Assay. Anal. Biochem. 1983, 131, 83–92. [Google Scholar] [CrossRef]
- Choiniere, J.R.; Scott, C.R.; Gelb, M.H.; Turecek, F. Direct Assay of Delta-Aminolevulinic Acid Dehydratase in Heme Biosynthesis for the Detection of Porphyrias by Tandem Mass Spectrometry. Anal. Chem. 2010, 82, 6730–6736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gundacker, C.; Gencik, M.; Hengstschlager, M. The Relevance of the Individual Genetic Background for the Toxicokinetics of Two Significant Neurodevelopmental Toxicants: Mercury and Lead. Mutat. Res. 2010, 705, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Sakai T: Biomarkers of lead exposure. Ind. Health. 2000, 38, 127–142. [CrossRef]
- Bonkovsky, H.L.; Dixon, N.; Rudnick, S. Pathogenesis and Clinical Features of the Acute Hepatic Porphyrias (AHPs). Mol. Genet. Metab. 2019, 128, 213–218. [Google Scholar] [CrossRef]
- Puy, H.; Deybach, J.C.; Lamoril, J.; Robreau, A.M.; Da, S.V.; Gouya, L.; Grandchamp, B.; Nordmann, Y. Molecular Epidemiology and Diagnosis of PBG Deaminase gene Defects in Acute Intermittent Porphyria. Am. J. Hum. Genet. 1997, 60, 1373–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlandsen, E.J.; Jorgensen, P.E.; Markussen, S.; Brock, A. Determination of Porphobilinogen Deaminase Activity in Human Erythrocytes: Pertinent Factors in Obtaining Optimal Conditions for Measurements. Scand. J. Clin. Lab. Investig. 2000, 60, 627–634. [Google Scholar]
- Wang, Y.; Scott, C.R.; Gelb, M.H.; Turecek, F. Direct Assay of Enzymes in Heme Biosynthesis for the Detection of Porphyrias by Tandem Mass Spectrometry. Porphobilinogen Deaminase. Anal. Chem. 2008, 80, 2606–2611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandchamp, B.; Puy, H.; Lamoril, J.; Deybach, J.C.; Nordmann, Y. Review: Molecular Pathogenesis of Hepatic Acute Porphyrias. J. Gastroenterol. Hepatol. 1996, 11, 1046–1052. [Google Scholar] [CrossRef]
- Whatley, S.D.; Roberts, A.G.; Llewellyn, D.H.; Bennett, C.P.; Garrett, C.; Elder, G.H. Non-Erythroid Form of Acute Intermittent Porphyria Caused by Promoter and Frameshift Mutations Distant from the Coding Sequence of Exon 1 of the HMBS Gene. Hum. Genet. 2000, 107, 243–248. [Google Scholar] [CrossRef]
- Meissner, P.; Adams, P.; Kirsch, R. Allosteric Inhibition of Human Lymphoblast and Purified Porphobilinogen Deaminase by Protoporphyrinogen and Coproporphyrinogen. A Possible Mechanism for the Acute Attack of Variegate Porphyria. J. Clin. Investig. 1993, 91, 1436–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.N.; Huang, Y.C.; Ro, L.S.; Liao, M.F.; Ning, H.C.; Kuo, H.C. Validation and Evaluation of Two Porphobilinogen Deaminase Activity Assays for Diagnosis of Acute Intermittent Porphyria. Clin. Chim. Acta 2018, 479, 1–6. [Google Scholar] [CrossRef]
- Crossley, B.M.; Bai, J.; Glaser, A.; Maes, R.; Porter, E.; Killian, M.L.; Clement, T.; Toohey-Kurth, K. Guidelines for Sanger Sequencing and Molecular Assay Monitoring. J. Vet. Diagn. Investig. 2020, 32, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Chen, B.; Desnick, R.J. Recent Advances on Porphyria Genetics: Inheritance, Penetrance & Molecular Heterogeneity, Including New Modifying/Causative Genes. Mol. Genet. Metab. 2019, 128, 320–331. [Google Scholar] [PubMed]
- Chen, B.; Solis-Villa, C.; Hakenberg, J.; Qiao, W.; Srinivasan, R.R.; Yasuda, M.; Balwani, M.; Doheny, D.; Peter, I.; Chen, R.; et al. Acute Intermittent Porphyria: Predicted Pathogenicity of HMBS Variants Indicates Extremely Low Penetrance of the Autosomal Dominant Disease. Hum. Mutat. 2016, 37, 1215–1222. [Google Scholar] [CrossRef] [Green Version]
- Lenglet, H.; Schmitt, C.; Grange, T.; Manceau, H.; Karboul, N.; Bouchet-Crivat, F.; Robreau, A.M.; Nicolas, G.; Lamoril, J.; Simonin, S.; et al. From a Dominant to an Oligogenic Model of Inheritance with Environmental Modifiers in Acute Intermittent Porphyria. Hum. Mol. Genet. 2018, 27, 1164–1173. [Google Scholar] [CrossRef]
- Solis, C.; Martinez-Bermejo, A.; Naidich, T.P.; Kaufmann, W.E.; Astrin, K.H.; Bishop, D.F.; Desnick, R.J. Acute Intermittent Porphyria: Studies of the Severe Homozygous Dominant Disease Provides Insights into the Neurologic Attacks in Acute Porphyrias. Arch. Neurol. 2004, 61, 1764–1770. [Google Scholar] [CrossRef] [Green Version]
- Hasanoglu, A.; Balwani, M.; Kasapkara, C.S.; Ezgu, F.S.; Okur, I.; Tumer, L.; Cakmak, A.; Nazarenko, I.; Yu, C.; Clavero, S.; et al. Harderoporphyria Due to Homozygosity for Coproporphyrinogen Oxidase Missense Mutation H327R. J. Inherit. Metab. Dis. 2011, 34, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, R.; Timonen, K.; Von Und Zu, F.M.; Laitinen, E.; Ahola, H.; Tenhunen, R.; Taketani, S.; Mustajoki, P. Homozygous Variegate Porphyria: 20 y Follow-Up and Characterization of Molecular Defect. J. Investig. Dermatol. 2001, 116, 610–613. [Google Scholar] [CrossRef] [Green Version]
- Balwani, M.; Bloomer, J.; Desnick, R. Erythropoietic Protoporphyria, Autosomal Recessive. 1993. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK100826/ (accessed on 26 July 2021).
- Phillips, J.D.; Steensma, D.P.; Pulsipher, M.A.; Spangrude, G.J.; Kushner, J.P. Congenital Erythropoietic Porphyria Due to a Mutation in GATA1: The First Trans-Acting Mutation Causative for a Human Porphyria. Blood 2007, 109, 2618–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pierro, E.; Russo, R.; Karakas, Z.; Brancaleoni, V.; Gambale, A.; Kurt, I.; Winter, S.S.; Granata, F.; Czuchlewski, D.R.; Langella, C.; et al. Congenital Erythropoietic Porphyria Linked to GATA1-R216W Mutation: Challenges for Diagnosis. Eur. J. Haematol. 2015, 94, 491–497. [Google Scholar] [CrossRef]
- Whatley, S.D.; Ducamp, S.; Gouya, L.; Grandchamp, B.; Beaumont, C.; Badminton, M.N.; Elder, G.H.; Holme, S.A.; Anstey, A.V.; Parker, M.; et al. C-Terminal Deletions in the ALAS2 Gene Lead to Gain of Function and Cause X-linked Dominant Protoporphyria without Anemia or Iron Overload. Am. J. Hum. Genet. 2008, 83, 408–414. [Google Scholar] [CrossRef] [Green Version]
- Brancaleoni, V.; Balwani, M.; Granata, F.; Graziadei, G.; Missineo, P.; Fiorentino, V.; Fustinoni, S.; Cappellini, M.D.; Naik, H.; Desnick, R.J.; et al. X-Chromosomal Inactivation Directly Influences the Phenotypic Manifestation of X-linked Protoporphyria. Clin. Genet. 2016, 89, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Weiss, Y.; Chen, B.; Yasuda, M.; Nazarenko, I.; Anderson, K.E.; Desnick, R.J. Porphyria Cutanea Tarda and Hepatoerythropoietic Porphyria: Identification of 19 Novel Uroporphyrinogen III Decarboxylase Mutations. Mol. Genet. Metab. 2019, 128, 363–366. [Google Scholar] [CrossRef]
- Loskove, Y.; Yasuda, M.; Chen, B.; Nazarenko, I.; Cody, N.; Desnick, R.J. Acute Hepatic Porphyrias: Identification of 46 Hydroxymethylbilane Synthase, 11 Coproporphyrinogen Oxidase, and 20 Protoporphyrinogen Oxidase Novel Mutations. Mol. Genet. Metab. 2019, 128, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Whatley, S.D.; Mason, N.G.; Holme, S.A.; Anstey, A.V.; Elder, G.H.; Badminton, M.N. Molecular Epidemiology of Erythropoietic Protoporphyria in the U.K. Br. J. Dermatol. 2010, 162, 642–646. [Google Scholar] [CrossRef]
- Weiss, Y.; Balwani, M.; Chen, B.; Yasuda, M.; Nazarenko, I.; Desnick, R.J. Congenital Erythropoietic Porphyria and Erythropoietic Protoporphyria: Identification of 7 Uroporphyrinogen III Synthase and 20 Ferrochelatase Novel Mutations. Mol. Genet. Metab. 2019, 128, 358–362. [Google Scholar] [CrossRef]
- Moran-Jimenez, M.J.; Borrero-Corte, M.J.; Jara-Rubio, F.; Garcia-Pastor, I.; Diaz-Diaz, S.; Castelbon-Fernandez, F.J.; Enriquez-De-Salamanca, R.; Mendez, M. Molecular Analysis of 55 Spanish Patients with Acute Intermittent Porphyria. Genes 2020, 11, 924. [Google Scholar] [CrossRef]
- Ventura, P.; Brancaleoni, V.; Di Pierro, E.; Graziadei, G.; Macri, A.; Carmine, G.C.; Nicolli, A.; Rossi, M.T.; Granata, F.; Fiorentino, V.; et al. Clinical and Molecular Epidemiology of Erythropoietic Protoporphyria in Italy. Eur. J. Dermatol. 2020, 30, 532–540. [Google Scholar] [CrossRef]
- Floderus, Y.; Shoolingin-Jordan, P.M.; Harper, P. Acute Intermittent Porphyria in Sweden. Molecular, Functional and Clinical Consequences of Some New Mutations Found in the Porphobilinogen Deaminase Gene. Clin. Genet. 2002, 62, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Paradisi, I.; Arias, S. Marked Geographic Aggregation of Acute Intermittent Porphyria Families Carrying Mutation Q180X in Venezuelan Populations, with Description of Further Mutations. J. Inherit. Metab. Dis. 2010, 33 (Suppl. 3), S455–S463. [Google Scholar] [CrossRef] [PubMed]
- Van Tuyll Van Serooskerken, A.M.; Drogemoller, B.I.; Te, V.K.; Bladergroen, R.S.; Steijlen, P.M.; Poblete-Gutierrez, P.; Van, G.M.; Van Heerden, C.J.; Warnich, L.; Frank, J. Extended Haplotype Studies in South African and Dutch Variegate Porphyria Families Carrying the Recurrent p.R59W Mutation Confirm a Common Ancestry. Br. J. Dermatol. 2012, 166, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Yin, X.; Hergersberg, M.; Goldgar, D.E.; Rufenacht, U.B.; Schuurmans, M.M.; Puy, H.; Deybach, J.C. Minder EI: Ancestral Founder of Mutation W283X in the Porphobilinogen Deaminase Gene Among Acute Intermittent Porphyria Patients. Hum. Hered. 2002, 54, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Granata, B.X.; Parera, V.E.; Batlle, A.; Rossetti, M. V: Haplotype Study in Argentinean Variegate Porphyria Patients. Hum. Hered. 2015, 80, 139–143. [Google Scholar] [CrossRef]
- Chen, B.; Whatley, S.; Badminton, M.; Aarsand, A.K.; Anderson, K.E.; Bissell, D.M.; Bonkovsky, H.L.; Cappellini, M.D.; Floderus, Y.; Friesema, E.C.H.; et al. International Porphyria Molecular Diagnostic Collaborative: An Evidence-Based Database of Verified Pathogenic and Benign Variants for the Porphyrias. Genet. Med. 2019, 21, 2605–2613. [Google Scholar] [CrossRef]
- Wood, L.H.; Whatley, S.D.; McKenna, K.; Badminton, M.N. Exonic Deletions as a Cause of Erythropoietic Protoporphyria. Ann. Clin. Biochem. 2006, 43, 229–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whatley, S.D.; Mason, N.G.; Holme, S.A.; Anstey, A.V.; Elder, G.H.; Badminton, M.N. Gene Dosage Analysis Identifies Large Deletions of the FECH Gene in 10% of Families with Erythropoietic Protoporphyria. J. Investig. Dermatol. 2007, 127, 2790–2794. [Google Scholar] [CrossRef] [Green Version]
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative Quantification of 40 Nucleic Acid Sequences By Multiplex Ligation-Dependent Probe Amplification. Nucleic Acids Res. 2002, 30, e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, R.F.; Roberts, R.G.; Mann, K.; Yau, S.C.; Berg, J.; Ogilvie, C.M. Multiplex Ligation-Dependent Probe Amplification Using a Completely Synthetic Probe Set. Biotechniques 2004, 37, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, E.; Brancaleoni, V.; Besana, V.; Cappellini, M.D. Multiplex Ligation-Dependent Probe Amplification: A Novel Approach for Genetic Diagnosis of Porphyria. J. Hum. Genet. 2009, 54, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Whatley, S.D.; Mason, N.G.; Woolf, J.R.; Newcombe, R.G.; Elder, G.H.; Badminton, M.N. Diagnostic Strategies for Autosomal Dominant Acute Porphyrias: Retrospective Analysis of 467 Unrelated Patients Referred for Mutational Analysis of the HMBS, CPOX, or PPOX Gene. Clin. Chem. 2009, 55, 1406–1414. [Google Scholar] [CrossRef] [Green Version]
- Di Pierro, E.; Besana, V.; Moriondo, V.; Brancaleoni, V.; Tavazzi, D.; Casalgrandi, G.; Ventura, P.; Rocchi, E. Cappellini MD: A large Deletion on Chromosome 11 in Acute Intermittent Porphyria. Blood Cells Mol. Dis. 2006, 37, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Cerbino, G.N.; Gerez, E.N.; Varela, L.S.; Melito, V.A.; Parera, V.E.; Batlle, A.; Rossetti, M.V. Acute Intermittent Porphyria in Argentina: An update. Biomed. Res. Int. 2015, 2015, 946387. [Google Scholar] [CrossRef]
- Granata, F.; Mendez, M.; Brancaleoni, V.; Castelbon, F.J.; Graziadei, G.; Ventura, P.; Di Pierro, E. Molecular Characterization, by Digital PCR Analysis of Four HMBS Gene Mutations Affecting the Ubiquitous Isoform of Porphobilinogen Deaminase (PBGD) in Patients with Acute Intermittent Porphyria (AIP). Mol. Genet. Metab. 2018, 125, 295–301. [Google Scholar] [CrossRef]
- Li, C.; Di Pierro, E.; Brancaleoni, V.; Cappellini, M.D.; Steensma, D.P. A Novel Large Deletion and Three Polymorphisms in the FECH Gene Associated with Erythropoietic Protoporphyria. Clin. Chem. Lab. Med. 2009, 47, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, E.; Brancaleoni, V.; Moriondo, V.; Besana, V.; Cappellini, M.D. Co-Existence of Two Functional Mutations on the Same Allele of the Human Ferrochelatase Gene in Erythropoietic Protoporphyria. Clin. Genet. 2007, 71, 84–88. [Google Scholar] [CrossRef]
- Di Pierro, E.; Brancaleoni, V.; Besana, V.; Ausenda, S.; Drury, S.; Cappellini, M.D. A 10376 bp Deletion of FECH Gene Responsible for Erythropoietic Protoporphyria. Blood Cells Mol. Dis. 2008, 40, 233–236. [Google Scholar] [CrossRef]
- Magness, S.T.; Tugores, A.; Christensen, S.R.; Wagner-Mcpherson, C.; Evans, G.A.; Naylor, E.W.; Brenner, D.A. Deletion of the Ferrochelatase Gene in a Patient with Protoporphyria. Hum. Mol. Genet. 1994, 3, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- De Verneuil, H.; Bourgeois, F.; De Rooij, F.; Siersema, P.D.; Wilson, J.H.; Grandchamp, B.; Nordmann, Y. Characterization of a New Mutation (R292G) and a Deletion at the Human Uroporphyrinogen Decarboxylase Locus in Two Patients with Hepatoerythropoietic Porphyria. Hum. Genet. 1992, 89, 548–552. [Google Scholar]
- Mendez, M.; Sorkin, L.; Rossetti, M.V.; Astrin, K.H.; Del, C.B.A.; Parera, V.E.; Aizencang, G.; Desnick, R.J. Familial Porphyria Cutanea Tarda: Characterization of Seven Novel Uroporphyrinogen Decarboxylase Mutations and Frequency of Common Hemochromatosis Alleles. Am. J. Hum. Genet. 1998, 63, 1363–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbaro, M.; Kotajarvi, M.; Harper, P.; Floderus, Y. Identification of an AluY-Mediated Deletion of Exon 5 in the CPOX Gene by MLPA Analysis in Patients with Hereditary Coproporphyria. Clin. Genet. 2012, 81, 249–256. [Google Scholar] [CrossRef]
- Barbaro, M.; Kotajarvi, M.; Harper, P.; Floderus, Y. Partial Protoporphyrinogen Oxidase (PPOX) Gene Deletions, due to Different Alu-Mediated Mechanisms, Identified by MLPA Analysis in Patients with Variegate Porphyria. Orphanet J. Rare Dis. 2013, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Katugampola, R.P.; Badminton, M.N.; Finlay, A.Y.; Whatley, S.; Woolf, J.; Mason, N.; Deybach, J.C.; Puy, H.; Ged, C.; De, V.H.; et al. Congenital Erythropoietic Porphyria: A Single-Observer Clinical Study of 29 Cases. Br. J. Dermatol. 2012, 167, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Whatley, S.D.; Badminton, M.N. Role of Genetic Testing in the Management of Patients with Inherited Porphyria and Their Families. Ann. Clin. Biochem. 2013, 50, 204–216. [Google Scholar] [CrossRef]
- To-Figueras, J.; Ducamp, S.; Clayton, J.; Badenas, C.; Delaby, C.; Ged, C.; Lyoumi, S.; Gouya, L.; De, V.H.; Beaumont, C.; et al. ALAS2 Acts as a Modifier Gene in Patients with Congenital Erythropoietic Porphyria. Blood 2011, 118, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Yien, Y.Y.; Ducamp, S.; Van Der Vorm, L.N.; Kardon, J.R.; Manceau, H.; Kannengiesser, C.; Bergonia, H.A.; Kafina, M.D.; Karim, Z.; Gouya, L.; et al. Mutation in Human CLPX Elevates Levels of Delta-Aminolevulinate Synthase and Protoporphyrin IX to Promote Erythropoietic Protoporphyria. Proc. Natl. Acad. Sci. USA 2017, 114, E8045–E8052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiara, M.; Primon, I.; Tarantini, L.; Agnelli, L.; Brancaleoni, V.; Granata, F.; Bollati, V.; Di Pierro, E. Targeted Resequencing of FECH Locus Reveals that a Novel Deep Intronic Pathogenic Variant and eQTLs May Cause Erythropoietic Protoporphyria (EPP) through a Methylation-Dependent Mechanism. Genet. Med. 2020, 22, 35–43. [Google Scholar] [CrossRef]
- Barman-Aksozen, J.; Suter, L.; Wegmann, F.; Meienberg, J.; Minder, A.E.; Beer, M.; Komminoth, P.; Minder, E.I.; Schneider-Yin, X. A Next-Generation-Sequencing Panel for Mutational Analysis of Dominant Acute Hepatic Porphyrias. Scand. J. Clin. Lab. Invest. 2019, 79, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Tollanes, M.C.; Aarsand, A.K.; Villanger, J.H.; Stole, E.; Deybach, J.C.; Marsden, J.; To-Figueras, J.; Sandberg, S. Establishing a Network of Specialist Porphyria Centres-Effects on Diagnostic Activities and Services. Orphanet J. Rare Dis. 2012, 7, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarsand, A.K.; Villanger, J.H.; Stole, E.; Deybach, J.C.; Marsden, J.; To-Figueras, J.; Badminton, M.; Elder, G.H.; Sandberg, S. European Specialist Porphyria Laboratories: Diagnostic Strategies, Analytical Quality, Clinical Interpretation, and Reporting as Assessed by an External Quality Assurance Program. Clin. Chem. 2011, 57, 1514–1523. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Porphyrias | ADP/AIP/HCP | VP | PCT/HEP/CEP | EPP/XLP |
|---|---|---|---|---|
| Plasma peak (nm) | 618–622 | 626–628 | 618–620 | 632–636 |
| Porphyrins | Unit | Normal Subjects | Porphyrias | |
|---|---|---|---|---|
| EPP | XLP | |||
| Total | µg/g Hb | <3 | >3 | |
| ZPP | % | 85–100% | 10–15% | 20–40% |
| PP-IX | <15% | 85–90% | 60–80% | |
| Porphyria | OMIM | Inheritance | Gene | Chr. | kb | RefSeq | Exons | Isoforms |
|---|---|---|---|---|---|---|---|---|
| XLP | 300752 | XL | ALAS2 | Xp11.21 | 22 | NM_000032.4 | 11 | Ery |
| ADP | 612740 | AR | ALAD | 9q32 | 15 | NM_000031.5 | 13 | Ubi/Ery |
| AIP | 176000 | AD | HMBS | 11q23.3 | 9 | NM_000190.3 | 15 | Ubi/Ery |
| CEP | 263700 | AR | UROS | 10q26.2 | 38 | NM_000375.2 | 10 | Ubi/Ery |
| CEP/XLTT | 314050 | XL | GATA1 | Xp11.23 | 8 | NM_002049.4 | 6 | Ubi |
| PCT/HEP | 176100 | AD/AR | UROD | 1p34.1 | 3 | NM_000374.4 | 10 | Ubi |
| HCP | 121300 | AD | CPOX | 3q11.2 | 14 | NM_000097.5 | 7 | Ubi |
| VP | 176200 | AD | PPOX | 1q23.3 | 5 | NM_000309.3 | 13 | Ubi |
| EPP | 177000 | AR | FECH | 18q21.31 | 42 | NM_000140.3 | 11 | Ubi |
| EPP2 | 618015 | AD | CLPX | 15q22.31 | 37 | NM_006660.5 | 14 | Ubi |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Pierro, E.; De Canio, M.; Mercadante, R.; Savino, M.; Granata, F.; Tavazzi, D.; Nicolli, A.M.; Trevisan, A.; Marchini, S.; Fustinoni, S. Laboratory Diagnosis of Porphyria. Diagnostics 2021, 11, 1343. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11081343
Di Pierro E, De Canio M, Mercadante R, Savino M, Granata F, Tavazzi D, Nicolli AM, Trevisan A, Marchini S, Fustinoni S. Laboratory Diagnosis of Porphyria. Diagnostics. 2021; 11(8):1343. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11081343
Chicago/Turabian StyleDi Pierro, Elena, Michele De Canio, Rosa Mercadante, Maria Savino, Francesca Granata, Dario Tavazzi, Anna Maria Nicolli, Andrea Trevisan, Stefano Marchini, and Silvia Fustinoni. 2021. "Laboratory Diagnosis of Porphyria" Diagnostics 11, no. 8: 1343. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11081343