
Endothelial Dysfunction in Acute Hepatic Porphyrias

 , , , ,
, , , ,
Abstract
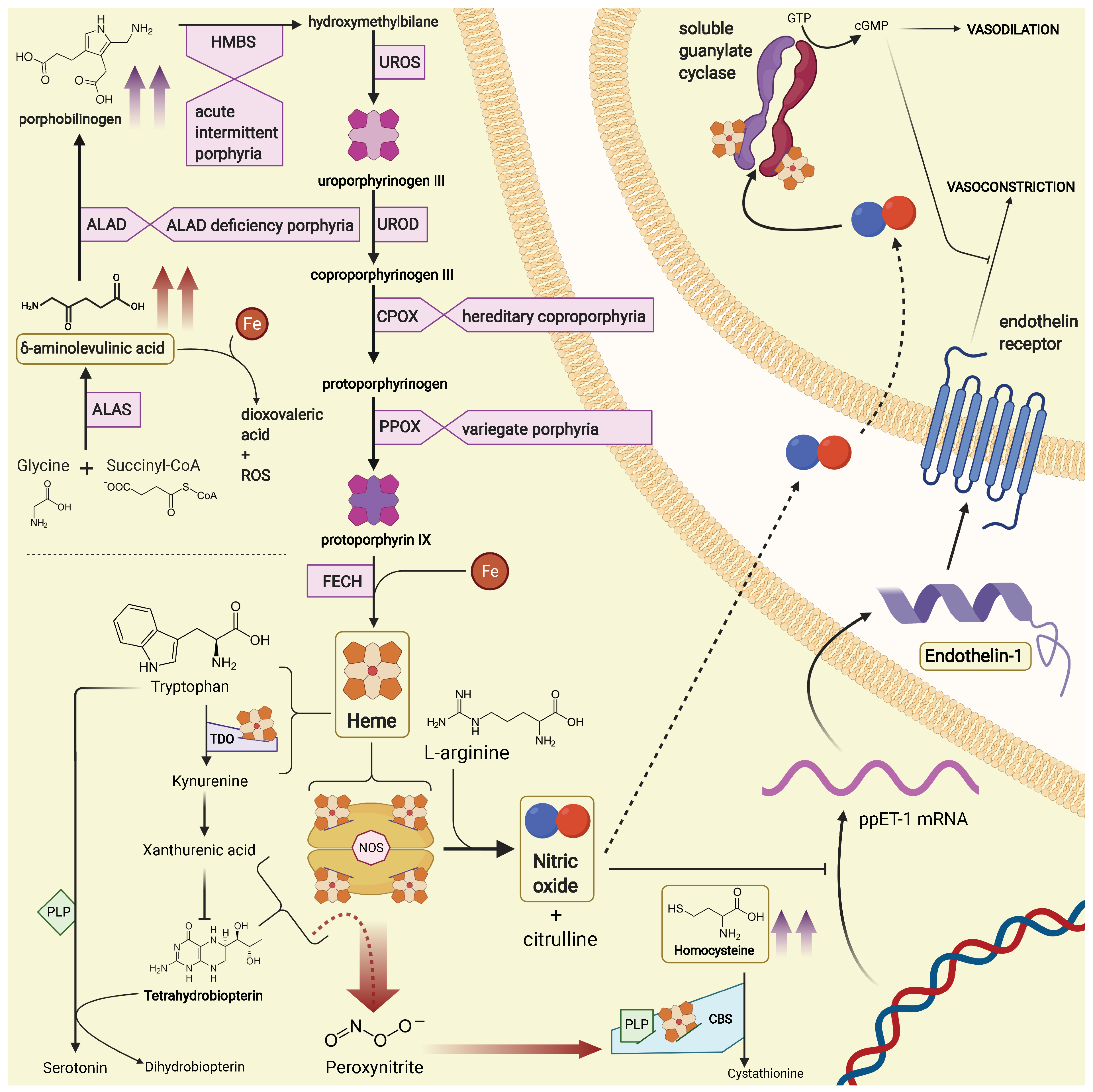
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Biochemical Assessment
2.3. Statistical Analysis
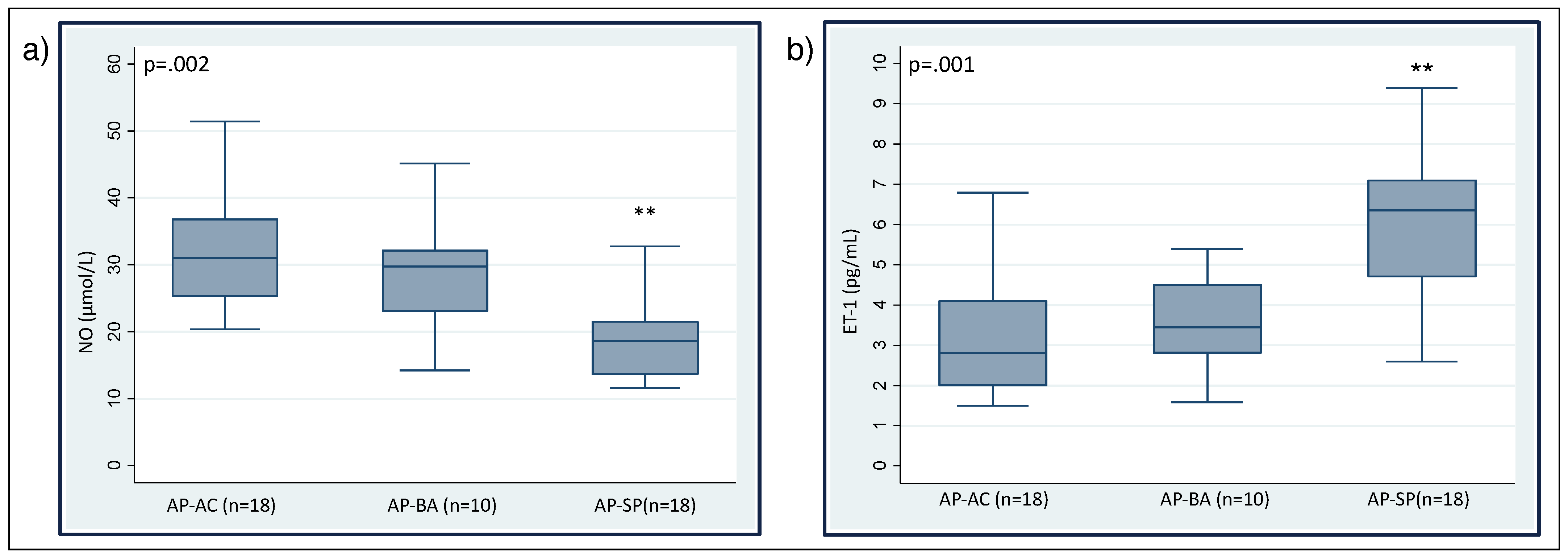
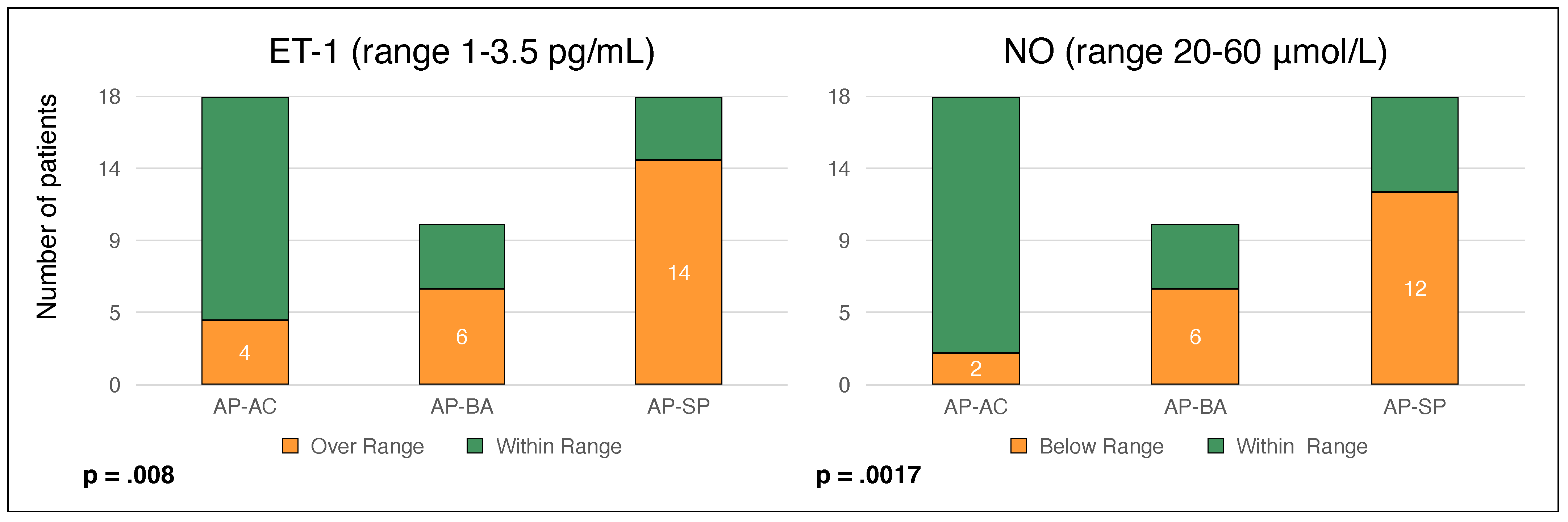
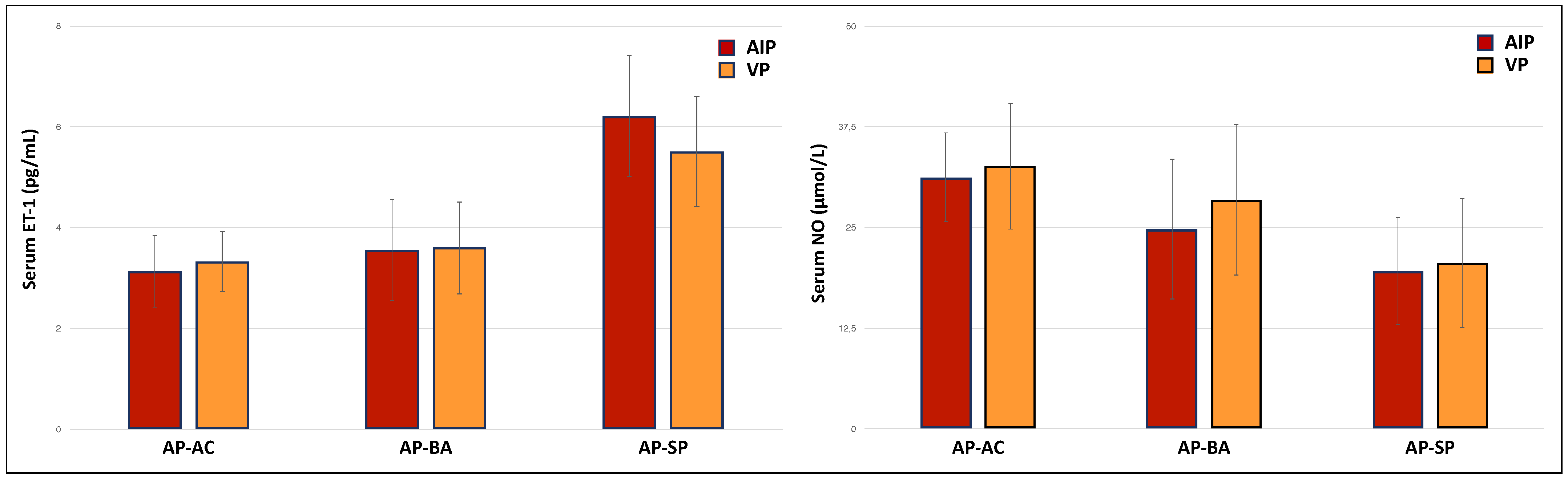
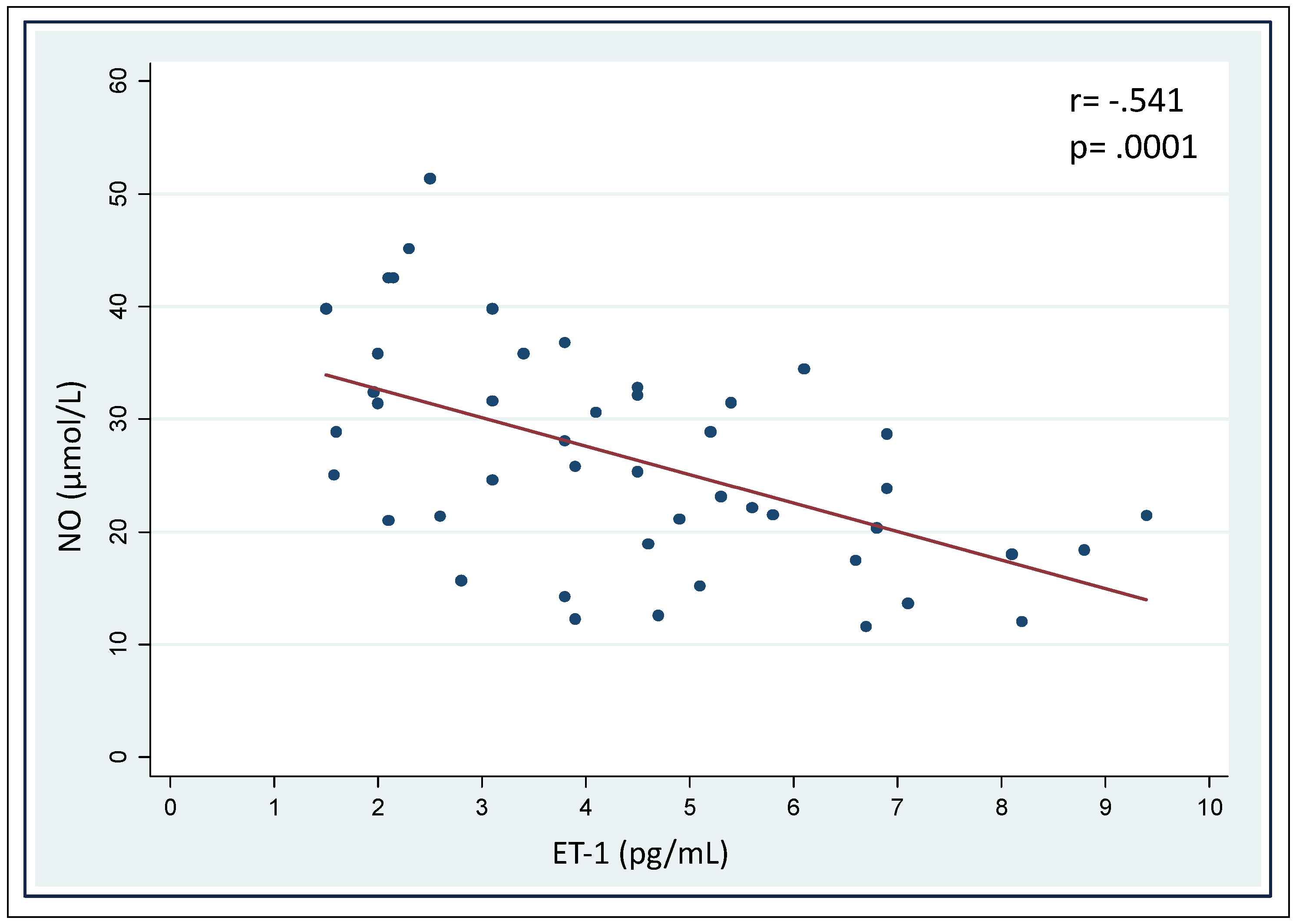
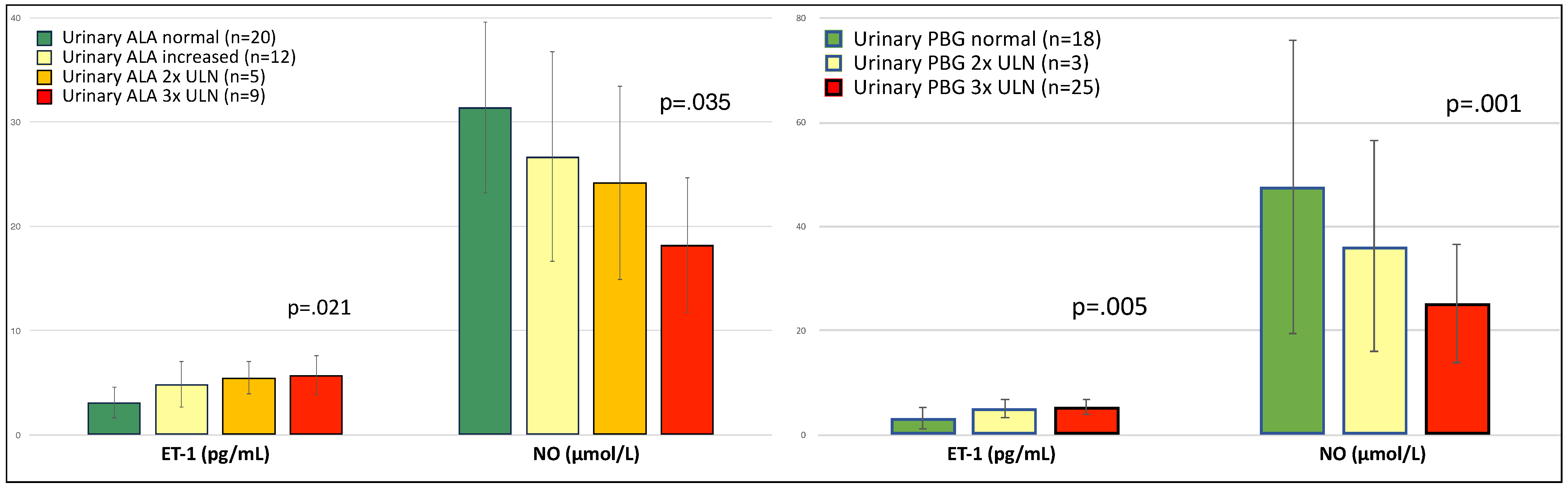
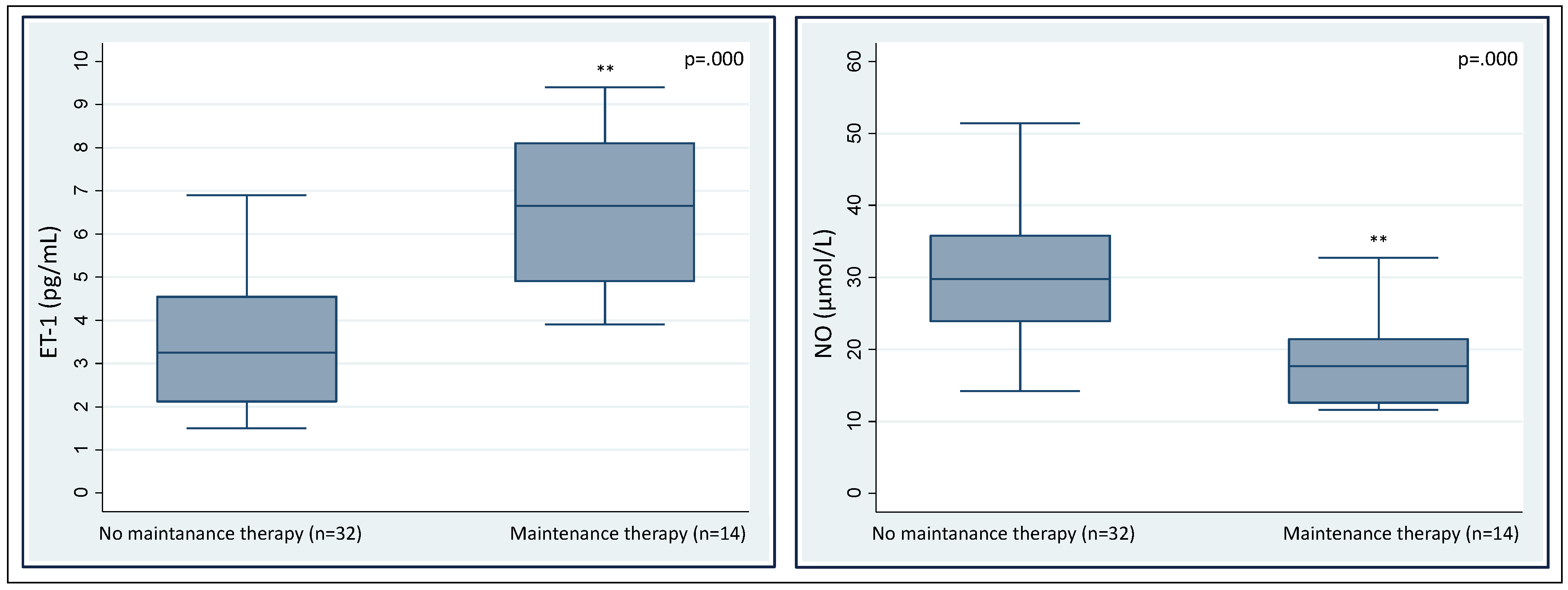
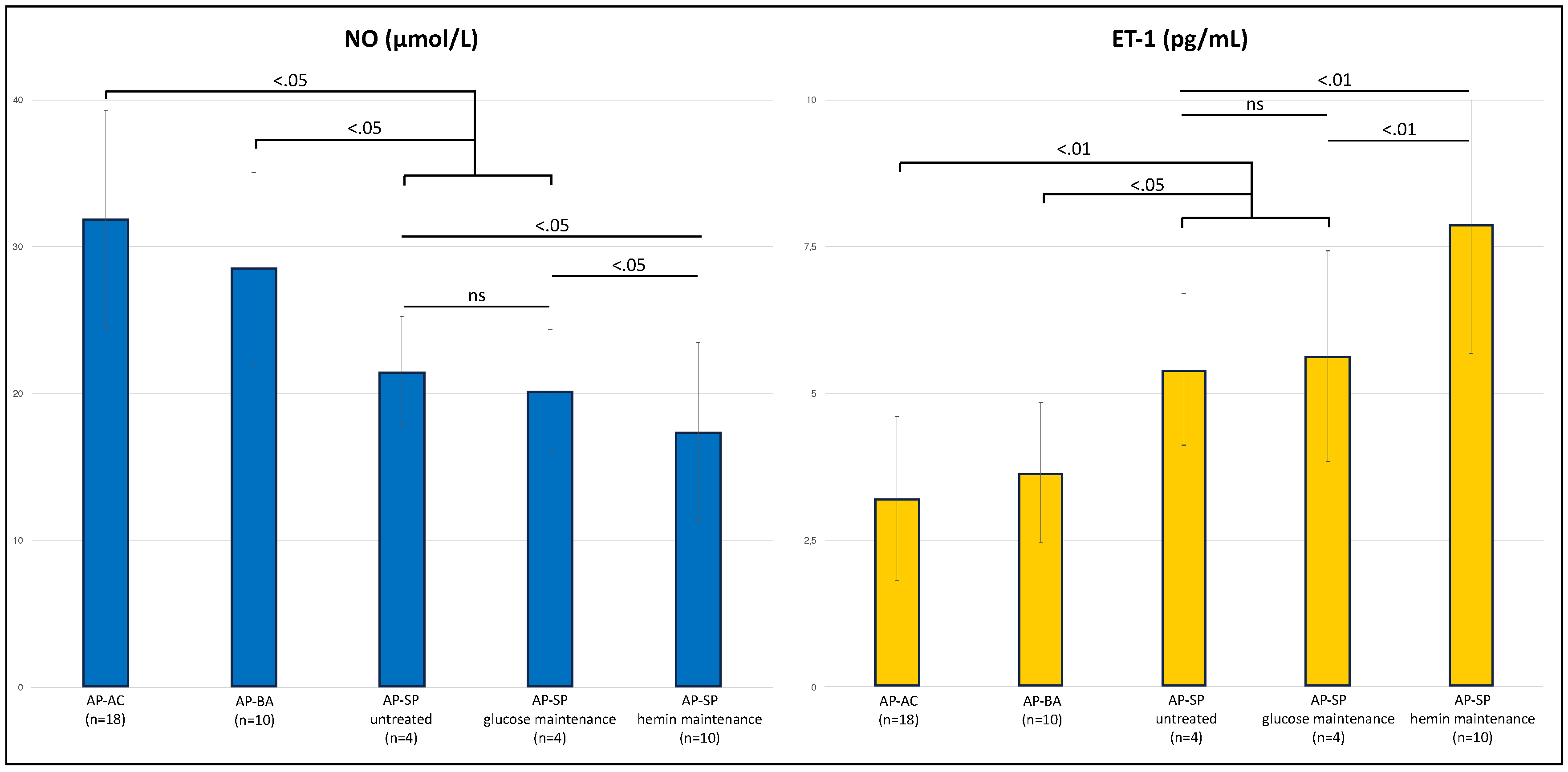
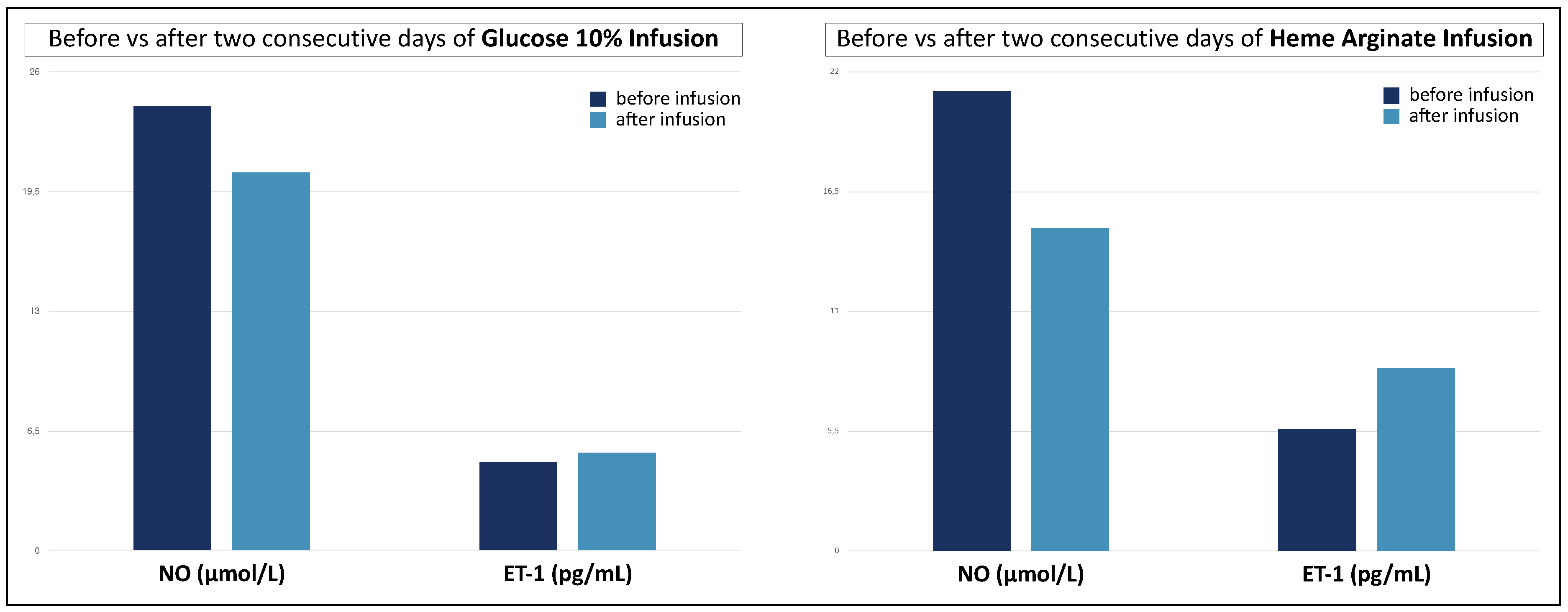
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AIP | Acute intermittent porphyria |
| AHP | Acute Hepatic Porphyrias |
| ALA | -aminolevulinic acid |
| ALAS1 | ALA synthase 1 |
| APA | acute porphyric attack |
| AP-AC | AHP patient—asymptomatic carrier |
| AP-BA | AHP patient—asymptomatic with biochemical alterations |
| AP-SP | AHP patient—symptomatic |
| ED | endothelial dysfunction |
| ET1 | serum endothelin-1 |
| HMBS | hydroxymethylbilane synthase |
| IV | intravenous |
| IU | International Units |
| NO | nitric oxide |
| NOS | NO synthase |
| PAKD | porphyria-associated kidney disease |
| PBG | porphobilinogen |
| PPOX | protoporphyrinogen oxidase |
| PRES | posterior reversible encephalopathy syndrome |
| sGC | soluble guanylate cyclase |
| ULN | upper level of normal |
| VP | Variegate Porphyria |
References
- Bassenge, E. Endothelial function in different organs. Prog. Cardiovasc. Dis. 1996, 39, 209–228, The Endothelium and Cardiocirculatory Function, Part I. [Google Scholar] [CrossRef]
- Lüscher, T.F.; Barton, M. Biology of the endothelium. Clin. Cardiol. 1997, 20, II-3. [Google Scholar] [CrossRef]
- Petty, R.G.; Pearson, J.D. Endothelium—The axis of vascular health and disease. J. R. Coll. Physicians Lond. 1989, 23, 92. [Google Scholar] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, A.J.; Gano, L.B.; Eskurza, I.; Silver, A.E.; Gates, P.E.; Jablonski, K.; Seals, D.R. Vascular endothelial dysfunction with aging: Endothelin-1 and endothelial nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H425–H432. [Google Scholar] [CrossRef] [Green Version]
- Hadi, H.A.; Al Suwaidi, J. Endothelial dysfunction in diabetes mellitus. Vasc. Health Risk Manag. 2007, 3, 853. [Google Scholar]
- Davignon, J.; Ganz, P. Role of Endothelial Dysfunction in Atherosclerosis. Circulation 2004, 109, III-27–III-32. [Google Scholar] [CrossRef] [Green Version]
- Caballero, A.E. Endothelial Dysfunction in Obesity and Insulin Resistance: A Road to Diabetes and Heart Disease. Obes. Res. 2003, 11, 1278–1289. [Google Scholar] [CrossRef]
- Kauppinen, R. Porphyrias. Lancet 2005, 365, 241–252. [Google Scholar] [CrossRef]
- Puy, H.; Gouya, L.; Deybach, J.C. Porphyrias. Lancet 2010, 375, 924–937. [Google Scholar] [CrossRef]
- Andersson, C.; Lithner, F. Hypertension and renal disease in patients with acute intermittent porphyria. J. Intern. Med. 1994, 236, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.F. Review of hepatocellular cancer, hypertension and renal impairment as late complications of acute porphyria and recommendations for patient follow-up. J. Clin. Pathol. 2012, 65, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Pallet, N.; Mami, I.; Schmitt, C.; Karim, Z.; François, A.; Rabant, M.; Nochy, D.; Gouya, L.; Deybach, J.C.; Xu-Dubois, Y.; et al. High prevalence of and potential mechanisms for chronic kidney disease in patients with acute intermittent porphyria. Kidney Int. 2015, 88, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Gouya, L.; Ventura, P.; Balwani, M.; Bissell, D.M.; Rees, D.C.; Stölzel, U.; Phillips, J.D.; Kauppinen, R.; Langendonk, J.G.; Desnick, R.J.; et al. EXPLORE: A prospective, multinational, natural history study of patients with acute hepatic porphyria with recurrent attacks. Hepatology 2020, 71, 1546–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, P.; Cappellini, M.D.; Rocchi, E. The acute porphyrias: A diagnostic and therapeutic challenge in internal and emergency medicine. Intern. Emerg. Med. 2009, 4, 297–308. [Google Scholar] [CrossRef]
- Elder, G.H.; Hift, R.J.; Meissner, P.N. The acute porphyrias. Lancet 1997, 349, 1613–1617. [Google Scholar] [CrossRef]
- Ricci, A.; Guida, C.C.; Manzini, P.; Cuoghi, C.; Ventura, P. Kidney Involvement in Acute Hepatic Porphyrias: Pathophysiology and Diagnostic Implications. Diagnostics 2021, 11, 2324. [Google Scholar] [CrossRef]
- Ricci, A.; Di Pierro, E.; Marcacci, M.; Ventura, P. Mechanisms of Neuronal Damage in Acute Hepatic Porphyrias. Diagnostics 2021, 11, 2205. [Google Scholar] [CrossRef]
- Marcacci, M.; Ricci, A.; Cuoghi, C.; Marchini, S.; Pietrangelo, A.; Ventura, P. Challenges in diagnosis and management of acute hepatic porphyrias: From an uncommon pediatric onset to innovative treatments and perspectives. Orphanet J. Rare Dis. 2022, 17, 1–10. [Google Scholar] [CrossRef]
- Handschin, C.; Lin, J.; Rhee, J.; Peyer, A.K.; Chin, S.; Wu, P.H.; Meyer, U.A.; Spiegelman, B.M. Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1α. Cell 2005, 122, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Freidja, M.L.; Vessieres, E.; Clere, N.; Desquiret, V.; Guihot, A.L.; Toutain, B.; Loufrani, L.; Jardel, A.; Procaccio, V.; Faure, S.; et al. Heme oxygenase-1 induction restores high-blood-flow-dependent remodeling and endothelial function in mesenteric arteries of old rats. J. Hypertens. 2011, 29, 102–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balla, J.; Vercellotti, G.M.; Nath, K.; Yachie, A.; Nagy, E.; Eaton, J.W.; Balla, G. Haem, haem oxygenase and ferritin in vascular endothelial cell injury. Nephrol. Dial. Transplant. 2003, 18, v8–v12. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, K.T.; Chang, H.; Ardehali, H. Role of Heme in Cardiovascular Physiology and Disease. J. Am. Heart Assoc. 2015, 4, e001138. [Google Scholar] [CrossRef] [Green Version]
- Higdon, A.N.; Benavides, G.A.; Chacko, B.K.; Ouyang, X.; Johnson, M.S.; Landar, A.; Zhang, J.; Darley-Usmar, V.M. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: The protective role of autophagy. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1394–H1409. [Google Scholar] [CrossRef] [Green Version]
- Rocchi, E.; Ventura, P.; Ronzoni, A.; Rosa, M.C.; Gozzi, C.; Marri, L.; Casalgrandi, G.; Cappellini, M.D. Pro-oxidant and antioxidant factors in acute intermittent porphyria: Family studies. J. Inherit. Metab. Dis. 2004, 27, 251–266. [Google Scholar] [CrossRef]
- Ahamed, M.; Verma, S.; Kumar, A.; Siddiqui, M.K. Delta-aminolevulinic acid dehydratase inhibition and oxidative stress in relation to blood lead among urban adolescents. Hum. Exp. Toxicol. 2006, 25, 547–553. [Google Scholar] [CrossRef]
- Bourque, S.L.; Davidge, S.T.; Adams, M.A. The interaction between endothelin-1 and nitric oxide in the vasculature: New perspectives. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1288–R1295. [Google Scholar] [CrossRef] [Green Version]
- Cowley, A.W.; Mori, T.; Mattson, D.; Zou, A.P. Role of renal NO production in the regulation of medullary blood flow. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R1355–R1369. [Google Scholar] [CrossRef] [Green Version]
- Andersson, C.; Thunell, S.; Floderus, Y.; Forsell, C.; Lundin, G.; Anvret, M.; Lannfelt, L.; Wetterberg, L.; Lithner, F. Diagnosis of acute intermittent porphyria in northern Sweden: An evaluation of mutation analysis and biochemical methods. J. Intern. Med. 1995, 237, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.K.C.; Lam, C.W.K.; Chan, Y.W. High-performance thin-layer chromatography of free porphyrins for diagnosis of porphyria. Clin. Chem. 1994, 40 Pt 1, 2026–2029. [Google Scholar] [CrossRef] [PubMed]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar] [CrossRef]
- Nolte, W.; Ehrenreich, H.; Wiltfang, J.; Pahl, K.; Unterberg, K.; Kamrowski-Kruck, H.; Schindler, C.G.; Figulla, H.R.; Buchwald, A.B.; Hartmann, H.; et al. Systemic and splanchnic endothelin-1 plasma levels in liver cirrhosis before and after transjugular intrahepatic portosystemic shunt (TIPS). Liver 2000, 20, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.; Lüscher, T.F. Release of endothelin from the porcine aorta. Inhibition by endothelium-derived nitric oxide. J. Clin. Investig. 1990, 85, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Khimji, A.K.; Rockey, D.C. Endothelin—Biology and disease. Cell. Signal. 2010, 22, 1615–1625. [Google Scholar] [CrossRef]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.; Castro, A.F., III; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef]
- Church, S.; Moore, M.; Youngs, G. Hypertension and renal impairment as complications of acute porphyria. Nephrol. Dial. Transplant. 1992, 7, 986–990. [Google Scholar]
- Pallet, N.; Karras, A.; Thervet, E.; Gouya, L.; Karim, Z.; Puy, H. Porphyria and kidney diseases. Clin. Kidney J. 2018, 11, 191–197. [Google Scholar] [CrossRef]
- Ventura, P.; Corradini, E.; Di Pierro, E.; Marchini, S.; Marcacci, M.; Cuoghi, C.; Buzzetti, E.; Pietrangelo, A. Hyperhomocysteinemia in patients with acute porphyrias: A potentially dangerous metabolic crossroad? Eu. J. Intern. Med. 2020, 79, 101–107. [Google Scholar] [CrossRef]
- Towns, C.; Balakrishnan, S.; Florkowski, C.; Davies, A.; Barrington-Ward, E. High penetrance, recurrent attacks and thrombus formation in a family with hereditary coproporphyria. JIMD Rep. 2022, 63, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo-Calle, D.A.; Solano, J.M.; Rabinstein, A.A.; Bonkovsky, H.L. Porphyria-induced posterior reversible encephalopathy syndrome and central nervous system dysfunction. Mol. Genet. Metab. 2019, 128, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Thachil, J. L-Asparaginase, nitric oxide and posterior reversible encephalopathy syndrome. Ann. Hematol. 2013, 92, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Bredt, D.S.; Hwang, P.M.; Snyder, S.H. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 1990, 347, 768–770. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O.; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Hill, A.; Wang, X.; Sapsford, R.J.; Russell, R.P.; Farrell, A.L.; Jessop, H.A.; McGawley, G.M.; Oxborough, D.L.; Pleasants, P.; Richards, S.J.; et al. Nitric oxide consumption and pulmonary hypertension in patients with paroxysmal nocturnal hemoglobinuria. Blood 2005, 106, 1046. [Google Scholar] [CrossRef]
- Ginsberg, L. Vasculitis and the peripheral nervous system. Rheumatology 2020, 59, iii55–iii59. [Google Scholar] [CrossRef]
- Gomez-Gomez, A.; Marcos, J.; Aguilera, P.; To-Figueras, J.; Pozo, O.J. Comprehensive analysis of the tryptophan metabolome in urine of patients with acute intermittent porphyria. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017, 1060, 347–354. [Google Scholar] [CrossRef]
- Gomez-Gomez, A.; Aguilera, P.; Langohr, K.; Casals, G.; Pavon, C.; Marcos, J.; To-Figueras, J.; Pozo, O.J. Evaluation of Metabolic Changes in Acute Intermittent Porphyria Patients by Targeted Metabolomics. Int. J. Mol. Sci. 2022, 23, 3219. [Google Scholar] [CrossRef] [PubMed]
- Homedan, C.; Laafi, J.; Schmitt, C.; Gueguen, N.; Lefebvre, T.; Karim, Z.; Desquiret-Dumas, V.; Wetterwald, C.; Deybach, J.C.; Gouya, L.; et al. Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model. Int. J. Biochem. Cell. Biol. 2014, 51, 93–101. [Google Scholar] [CrossRef] [Green Version]
- To-Figueras, J.; Lopez, R.M.; Deulofeu, R.; Herrero, C. Preliminary report: Hyperhomocysteinemia in patients with acute intermittent porphyria. Metabolism 2010, 59, 1809–1810. [Google Scholar] [CrossRef] [PubMed]
- Klatt, P.; Pfeiffer, S.; List, B.M.; Lehner, D.; Glatter, O.; Bächinger, H.P.; Werner, E.R.; Schmidt, K.; Mayer, B. Characterization of Heme-deficient Neuronal Nitric-oxide Synthase Reveals a Role for Heme in Subunit Dimerization and Binding of the Amino Acid Substrate and Tetrahydrobiopterin. J. Biol. Chem. 1996, 271, 7336–7342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, E.D.; Rezende, B.A.; Cortes, S.F.; Lemos, V.S. Neuronal Nitric Oxide Synthase in Vascular Physiology and Diseases. Front. Physiol. 2016, 7, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignarro, L.J. Regulation of cytosolic guanylyl cyclase by porphyrins and metalloporphyrins. Adv. Pharmacol. 1994, 26, 35–65. [Google Scholar]
- Dai, Y.; Faul, E.M.; Ghosh, A.; Stuehr, D.J. NO rapidly mobilizes cellular heme to trigger assembly of its own receptor. Proc. Natl. Acad. Sci. USA 2022, 119, e2115774119. [Google Scholar] [CrossRef]
- Lavandera, J.; Rodríguez, J.; Ruspini, S.; Meiss, R.; Zuccoli, J.R.; Martínez, M.D.C.; Gerez, E.; Batlle, A.; Buzaleh, A.M. Pleiotropic effects of 5-aminolevulinic acid in mouse brain. Biochem. Cell Biol. 2016, 94, 297–305. [Google Scholar] [CrossRef]
- Jover, R.; Hoffmann, F.; Scheffler-Koch, V.; Lindberg, R.L. Limited heme synthesis in porphobilinogen deaminase-deficient mice impairs transcriptional activation of specific cytochrome P450 genes by phenobarbital. Eur. J. Biochem. 2000, 267, 7128–7137. [Google Scholar] [CrossRef]
- Buzaleh, A.; Meiss, R.; Lavandera, J.; Vallecorsa, P.; Ruspini, S.; Batlle, A. Óxido Nítrico Sintasa y Hemo Oxigenasa en encéfalo de ratones tratados con anestésicos volátiles y otros agentes porfirinogénicos: Estudio inmunohistoquímico de la expresión proteica. Medicina 2012, 72, 121. [Google Scholar]
- Soong, J.; Adams, M.A.; Nakatsu, K. Acute depletion of heme by succinylacetone alters vascular responses but does not induce hypertension. Can. J. Physiol. Pharmacol. 2008, 86, 613–619. [Google Scholar] [CrossRef]
- Bourque, S.L.; Benjamin, C.D.; Adams, M.A.; Nakatsu, K. Lack of hemodynamic effects after extended heme synthesis inhibition by succinylacetone in rats. J. Pharmacol. Exp. Ther. 2010, 333, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N. Eng. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.; Ventura, P. Givosiran for the treatment of acute hepatic porphyria. Expert Rev. Clin. Pharmacol. 2022, 1–11. [Google Scholar] [CrossRef] [PubMed]
- To-Figueras, J.; Wijngaard, R.; García-Villoria, J.; Aarsand, A.K.; Aguilera, P.; Deulofeu, R.; Brunet, M.; Gómez-Gómez, À.; Pozo, O.J.; Sandberg, S. Dysregulation of homocysteine homeostasis in acute intermittent porphyria patients receiving heme arginate or givosiran. J. Inherit. Metab. Dis. 2021, 44, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Petrides, P.E.; Klein, M.; Schuhmann, E.; Torkler, H.; Molitor, B.; Loehr, C.; Obermeier, Z.; Beykirch, M.K. Severe homocysteinemia in two givosiran-treated porphyria patients: Is free heme deficiency the culprit? Ann. Hematol. 2021, 100, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Fontanellas, A.; Ávila, M.A.; Arranz, E.; Enríquez de Salamanca, R.; Morales-Conejo, M. Acute intermittent porphyria, givosiran, and homocysteine. J. Inherit. Metab. Dis. 2021, 44, 790–791. [Google Scholar] [CrossRef]
- Vassiliou, D.; Sardh, E. Homocysteine elevation in givosiran treatment: Suggested ALAS1 siRNA effect on cystathionine beta-synthase. J. Int. Med. 2021, 290, 928–930. [Google Scholar] [CrossRef]
- Ricci, A.; Marcacci, M.; Cuoghi, C.; Pietrangelo, A.; Ventura, P. Hyperhomocysteinemia in patients with acute porphyrias: A possible effect of ALAS1 modulation by siRNAm therapy and its control by vitamin supplementation. Eur. J. Intern. Med. 2021, 92, 121–123. [Google Scholar] [CrossRef]
- Gomá-Garcés, E.; Pérez-Gómez, M.V.; Ortíz, A. Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 383, 1989. [Google Scholar] [CrossRef]
- Lazareth, H.; Poli, A.; Bignon, Y.; Mirmiran, A.; Rabant, M.; Schmitt, C.; Puy, H.; Karras, A.; Gouya, L.; Pallet, N.; et al. Renal function decline with small interfering RNA silencing ALAS1. Kidney Int. Rep. 2021, 6, 1904. [Google Scholar] [CrossRef]
- Wang, H.; Sun, Q.; Zhou, Y.; Zhang, H.; Luo, C.; Xu, J.; Dong, Y.; Wu, Y.; Liu, H.; Wang, W. Nitration-mediated deficiency of cystathionine β-synthase activity accelerates the progression of hyperhomocysteinemia. Free. Radic. Biol. Med. 2017, 113, 519–529. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AP-AC group (n = 18) | AP-BA group (n = 10) | AP-SP group (n = 18) | p | |
|---|---|---|---|---|
| Age (years) | 45 ± 13 | 42 ± 11 | 48 ± 25 | .455 |
| Sex (M/F) | 9/9 | 5/5 | 8/10 | .934 |
| AHP diagnosis (AIP/VP) | 8/10 | 7/3 | 16/2 | .017 |
| Active smoking status (yes/no) | 2/16 | 1/9 | 1/17 | .090 |
| Urinary ALA∘ (mol/mmol creatinine) | 2.95 ± 1.66 | 8.39 ± 3.22 | 13.9 ± 5.09 | .000 *; .000**; .001*** |
| Urinary PBG∘ (mol/mmol creatinine) | 1.14 ± 0.53 | 19.2 ± 13.1 | 33.7 ± 12.7 | .001*; .000**; .002*** |
| Urinary total porphyrins∘ (g/g creatinine) | 96.2 ± 24.1 | 329 ± 156 | 797 ± 414 | .085 *; .000 **; .001 *** |
| Creatinine clearance (mL/min) | 79.1 ± 9.51 | 71.2 ± 5.66 | 69.8 ± 8.44 | .052 *; .005 **; .923 *** |
| SGPT (IU/L) | 25.6 ± 8.61 | 30.5 ± 5.69 | 32.5 ± 9.71 | .059 |
| SGOT (IU/L) | 28.7 ± 7.43 | 29.2 ± 6.74 | 31.5 ± 10.4 | .559 |
| Serum albumin (g/L) | 3.82 ± 0.25 | 3.85 ± 0.31 | 3.75 ± 0.38 | .687 |
| ET-1 (pg/mL) | 3.21 ± 1.51 | 3.56 ± 1.24 | 6.16 ± 1.81 | .564 *; .000 **; .022 *** |
| NO (mol/L) | 31.9 ± 8.48 | 28.6 ± 9.68 | 19.7 ± 6.81 | .842 *; .000 **; .000 *** |
| n | Treatment | ||
|---|---|---|---|
| AP-AC | 1 (5.5%) | ||
| Hypertension | AP-BA | 1 (10%) | |
| AP-SP | 6 (33.3%) | 5 hemin/1 glucose | |
| AP-AC | 0 (0%) | ||
| Kidney Impairment | AP-BA | 1 (10%) | |
| AP-SP | 4 (22%) | 3 hemin/1 glucose | |
| AP-AC | 0 (0%) | ||
| Thrombosis | AP-BA | 0 (0%) | |
| AP-SP | 3 (16.7%) | 3 hemin |
| Treated | Untreated | ||
|---|---|---|---|
| Diagnosis (AIP/VP) | 13/1 | 18/14 | |
| Clinical Status | |||
| AP-AC | 0 | 18 | |
| AP-BA | 0 | 10 | |
| AP-SP | 14 | 4 | |
| Treatment (hemin/glucose) | 10/4 | ||
| Hemin treatment frequency (times per week) | |||
| 1 | 6 | ||
| >1 | 4 |
| Correlation with NO Levels | Correlation with ET-1 Levels | |||
|---|---|---|---|---|
| Parameter | r | p | r | p |
| ALA | −0.237 | .117 | 0.284 | .056 |
| PBG | −0.277 | .061 | 0.255 | .086 |
| Total porphyrins | −0.313 | .071 | 0.239 | .108 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricci, A.; Sandri, G.; Marcacci, M.; Di Pierro, E.; Granata, F.; Cuoghi, C.; Marchini, S.; Pietrangelo, A.; Ventura, P. Endothelial Dysfunction in Acute Hepatic Porphyrias. Diagnostics 2022, 12, 1303. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics12061303
Ricci A, Sandri G, Marcacci M, Di Pierro E, Granata F, Cuoghi C, Marchini S, Pietrangelo A, Ventura P. Endothelial Dysfunction in Acute Hepatic Porphyrias. Diagnostics. 2022; 12(6):1303. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics12061303
Chicago/Turabian StyleRicci, Andrea, Gilda Sandri, Matteo Marcacci, Elena Di Pierro, Francesca Granata, Chiara Cuoghi, Stefano Marchini, Antonello Pietrangelo, and Paolo Ventura. 2022. "Endothelial Dysfunction in Acute Hepatic Porphyrias" Diagnostics 12, no. 6: 1303. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics12061303