
Comprehensive Profiling of Genomic and Transcriptomic Differences between Risk Groups of Lung Adenocarcinoma and Lung Squamous Cell Carcinoma



Abstract
:1. Introduction
2. Materials and Methods
2.1. Data
2.2. Gene Expression Signature Analysis
2.3. Differential Expression Analysis
2.4. Active Subnetwork Analysis
2.5. Copy Number Variation Analysis
2.6. Simple Nucleotide Variations Analysis
2.7. Visualization
3. Results
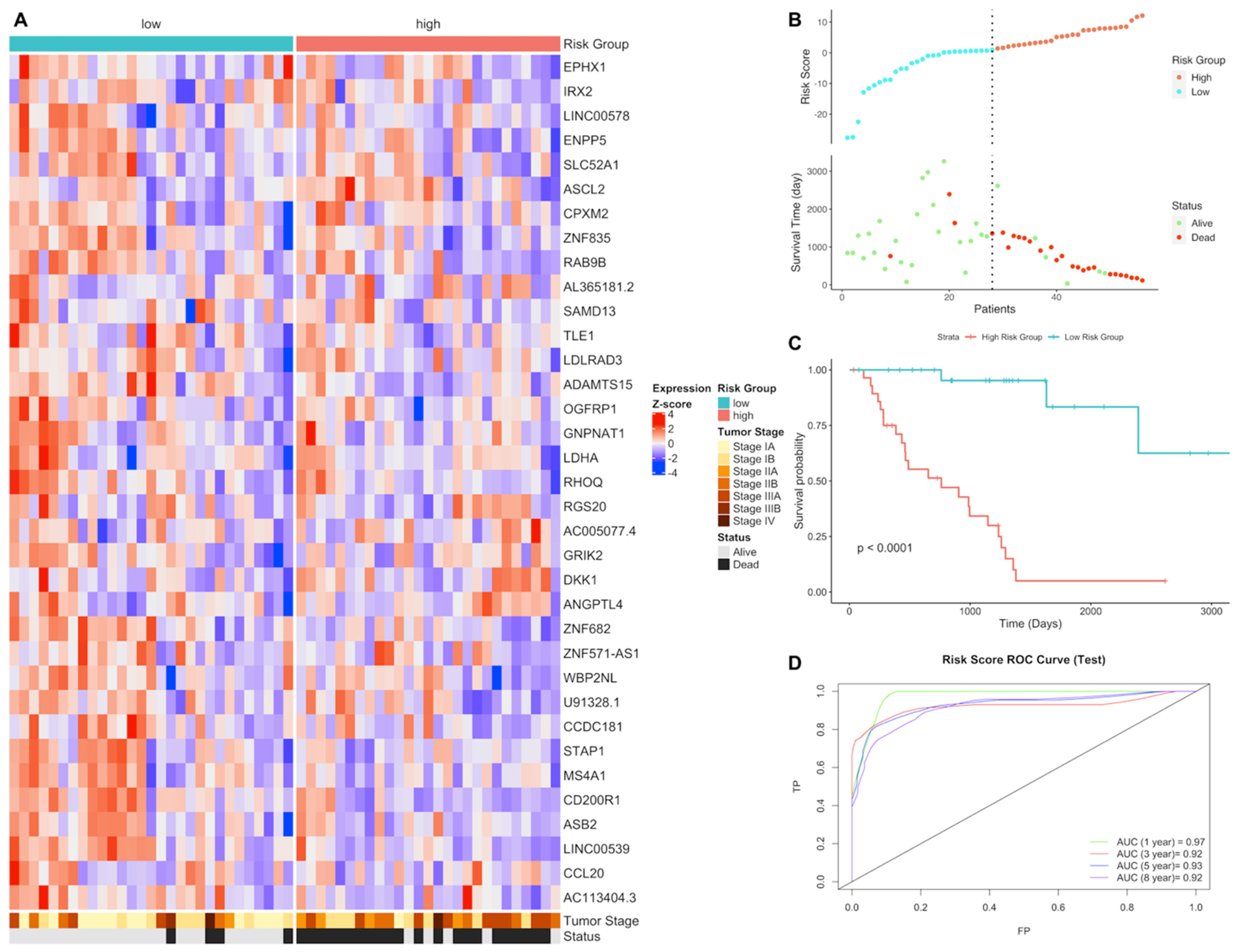
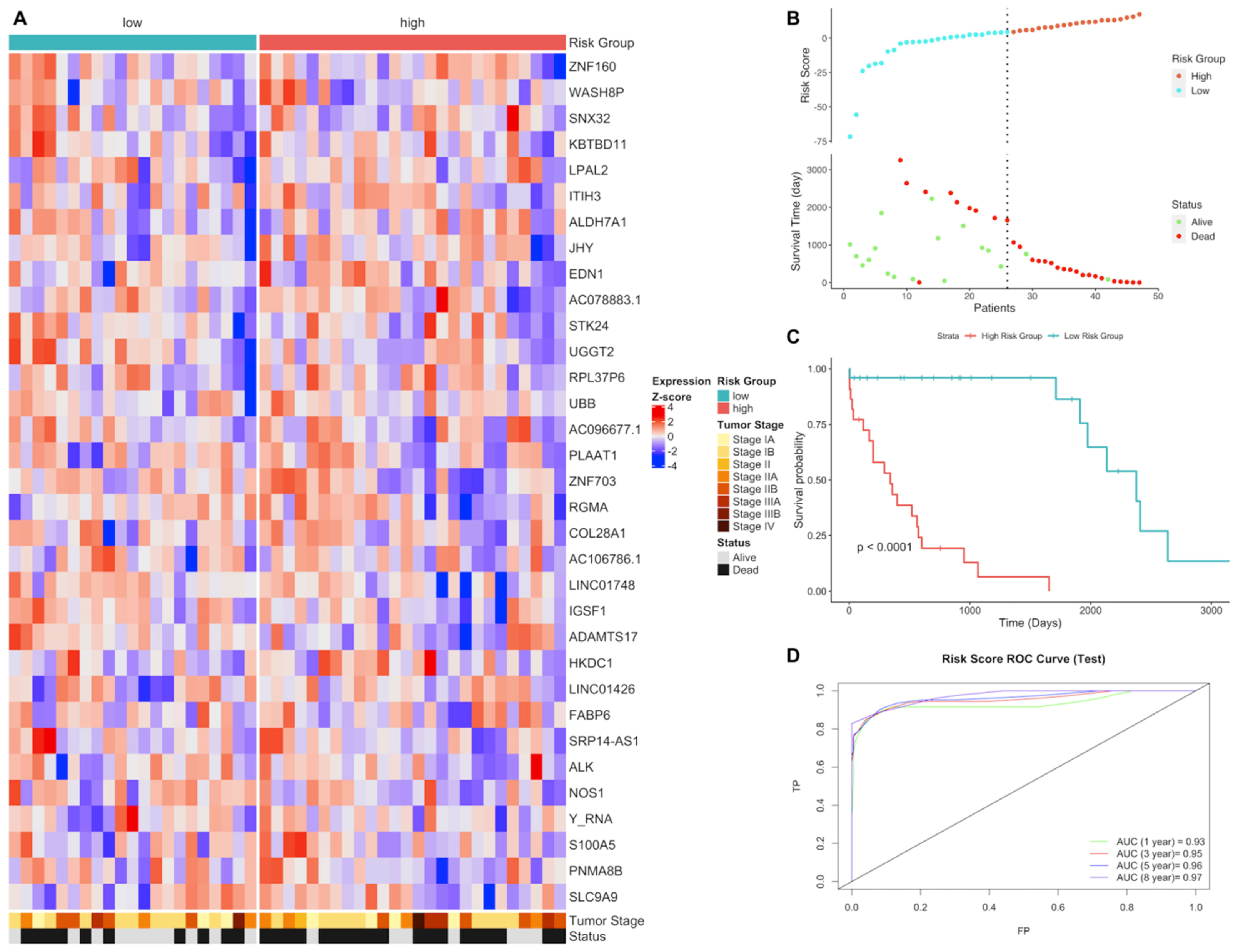
3.1. Gene Expression Signature Analysis of LUAD and LUSC Patients
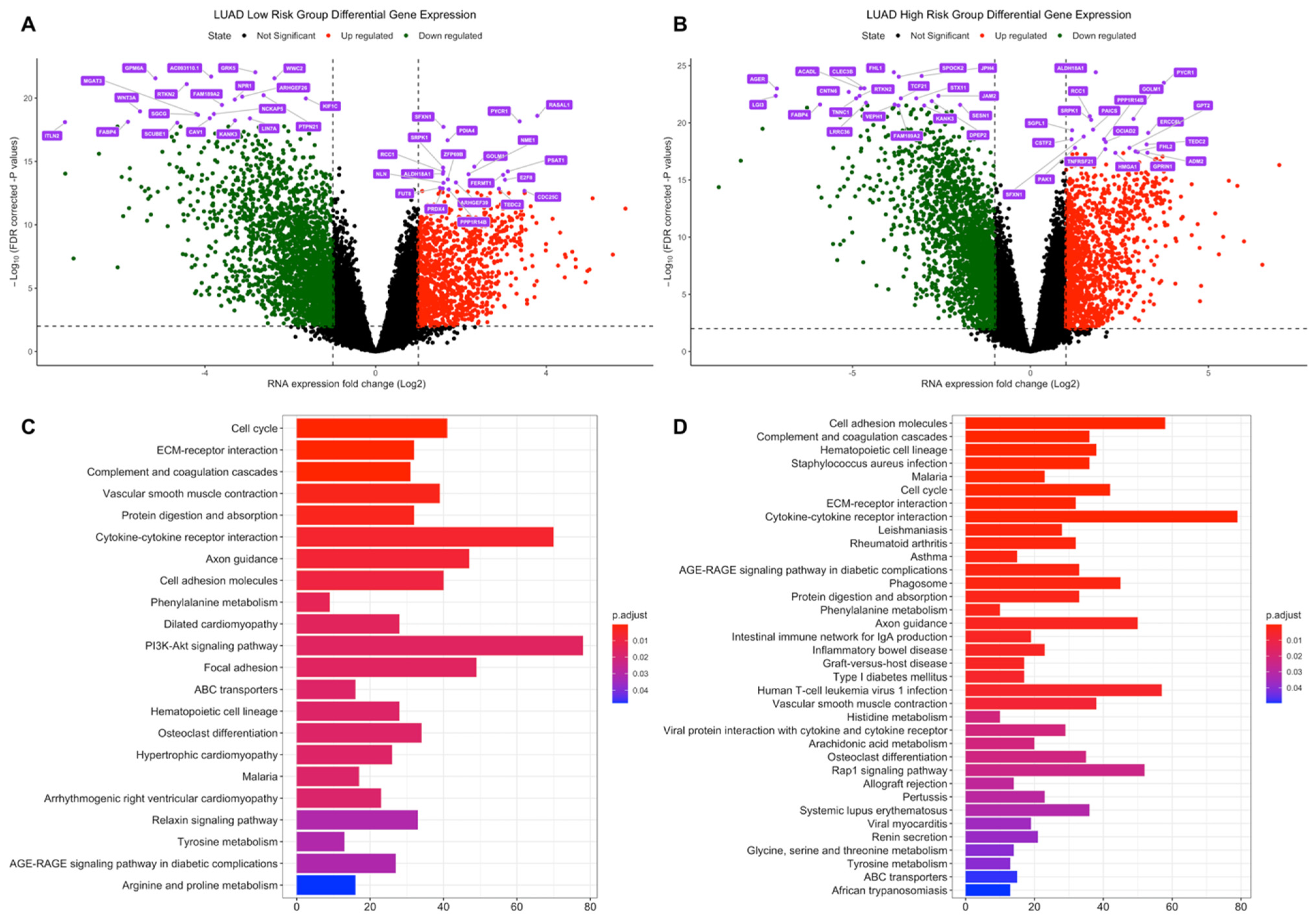
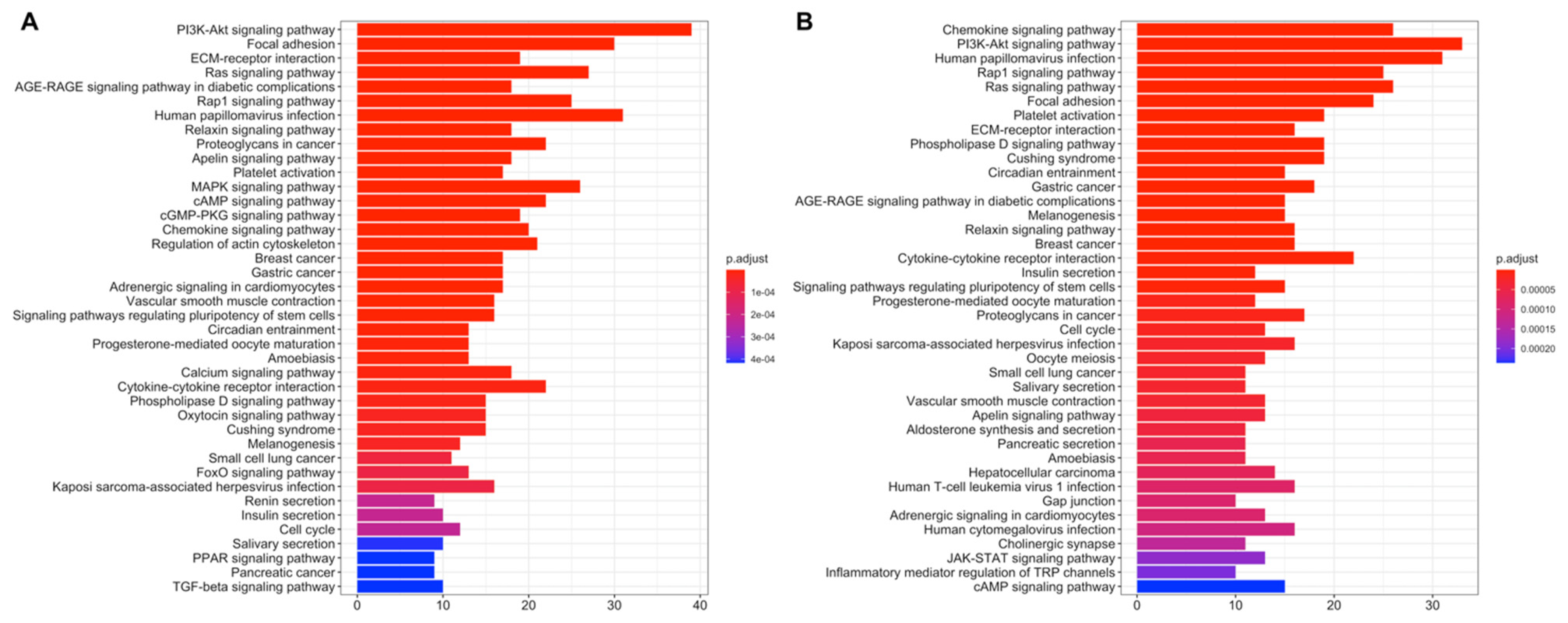
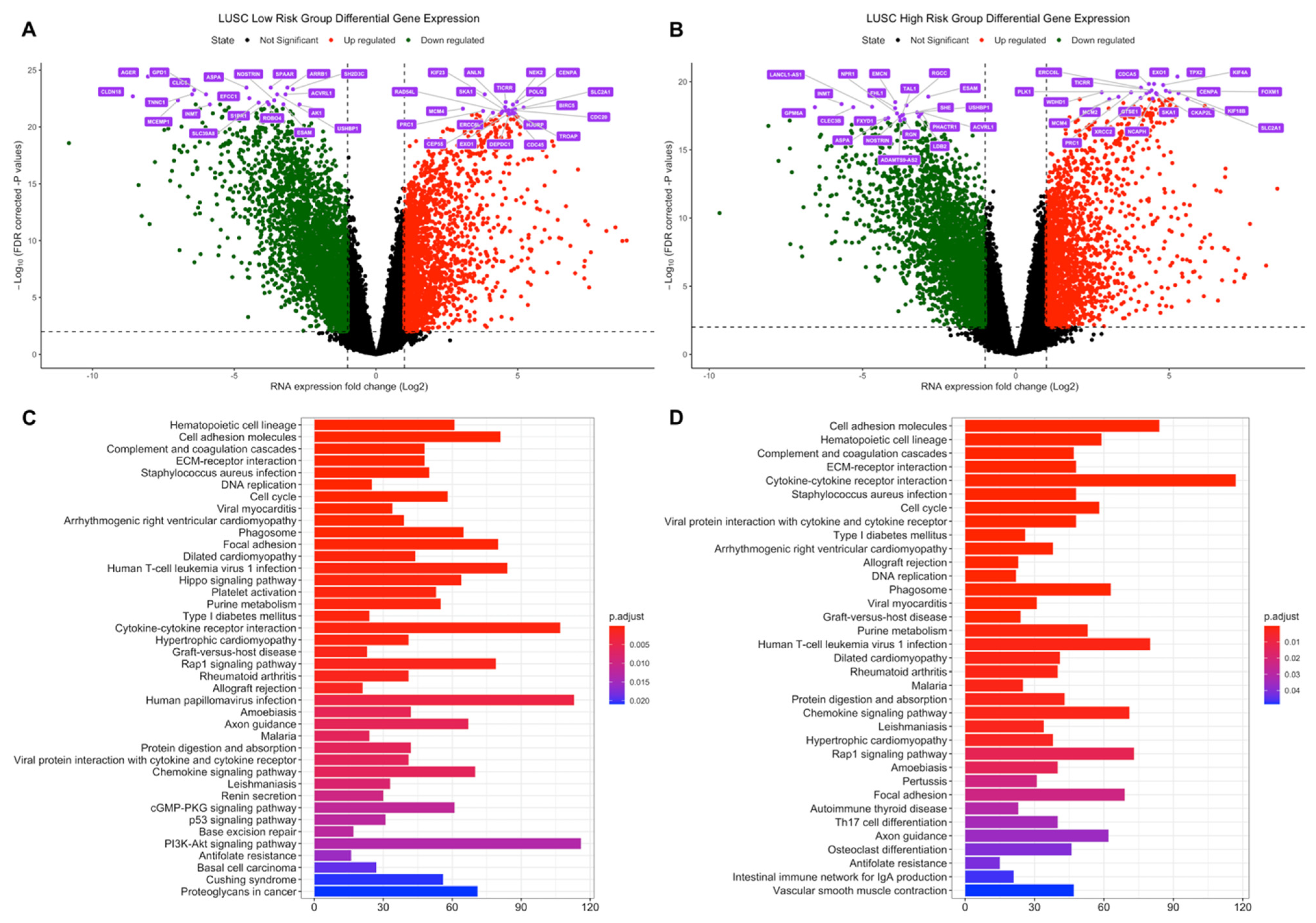
3.2. Differential Expression and Active Subnetwork Analysis of Risk Groups
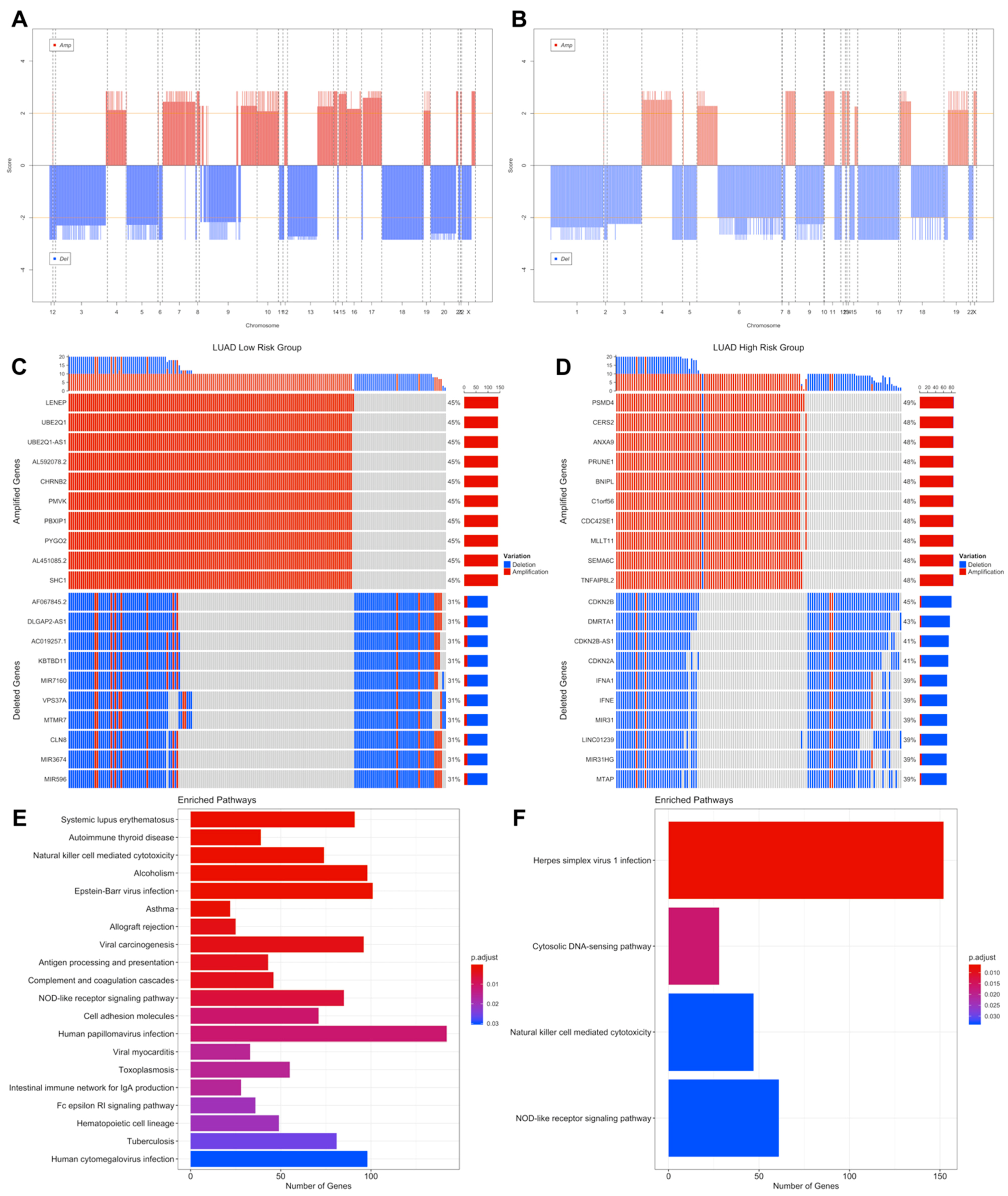
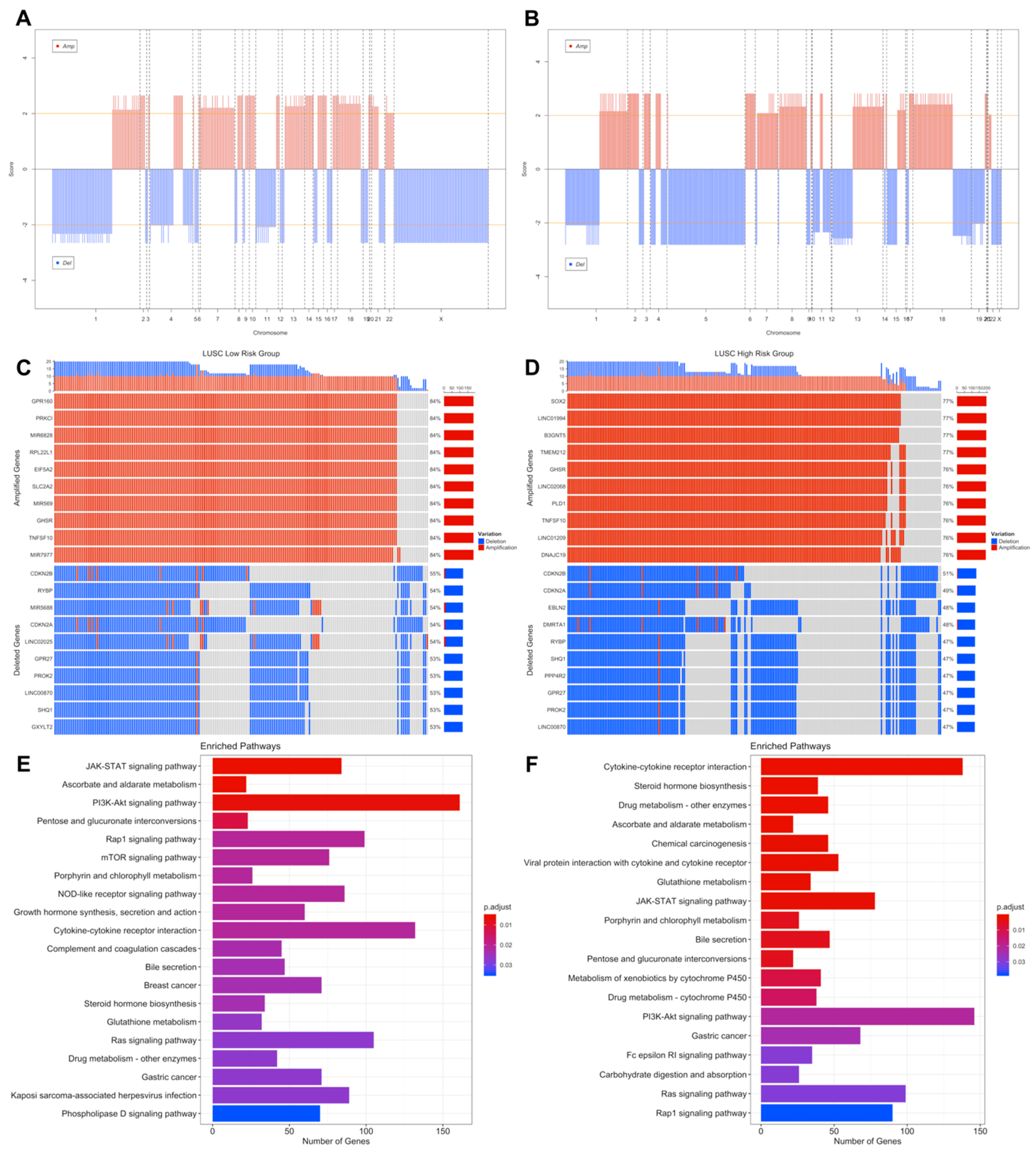
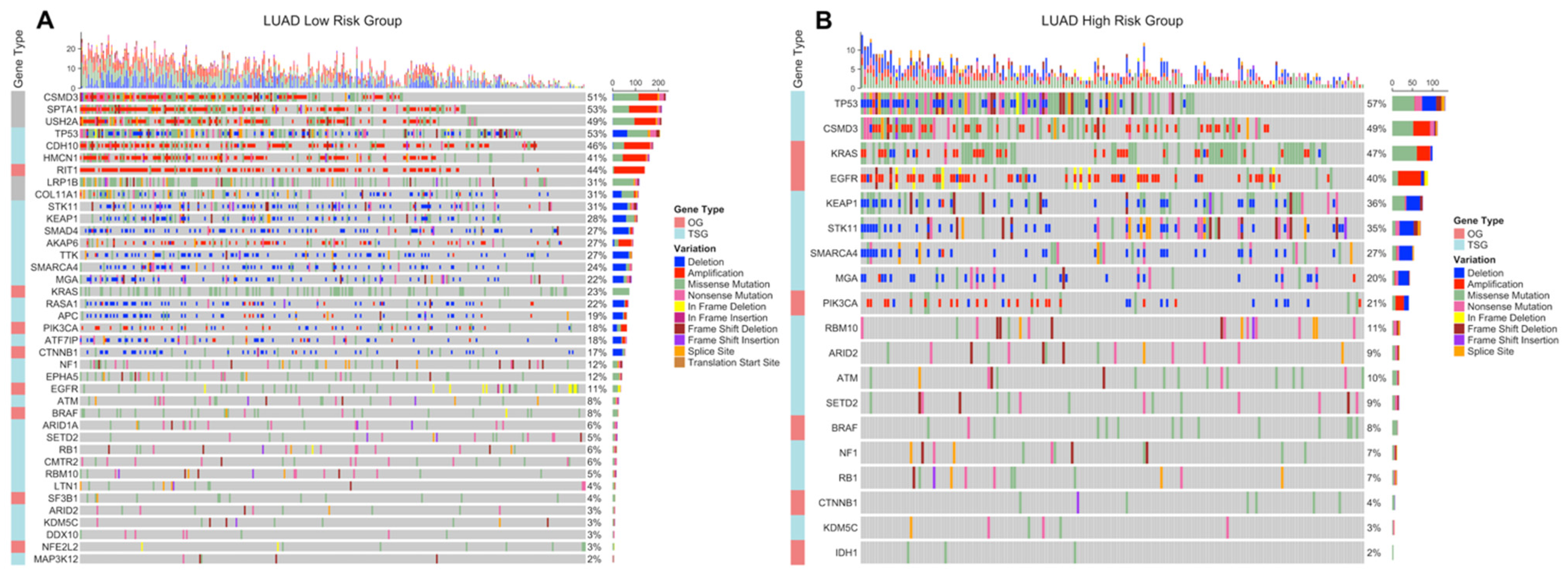
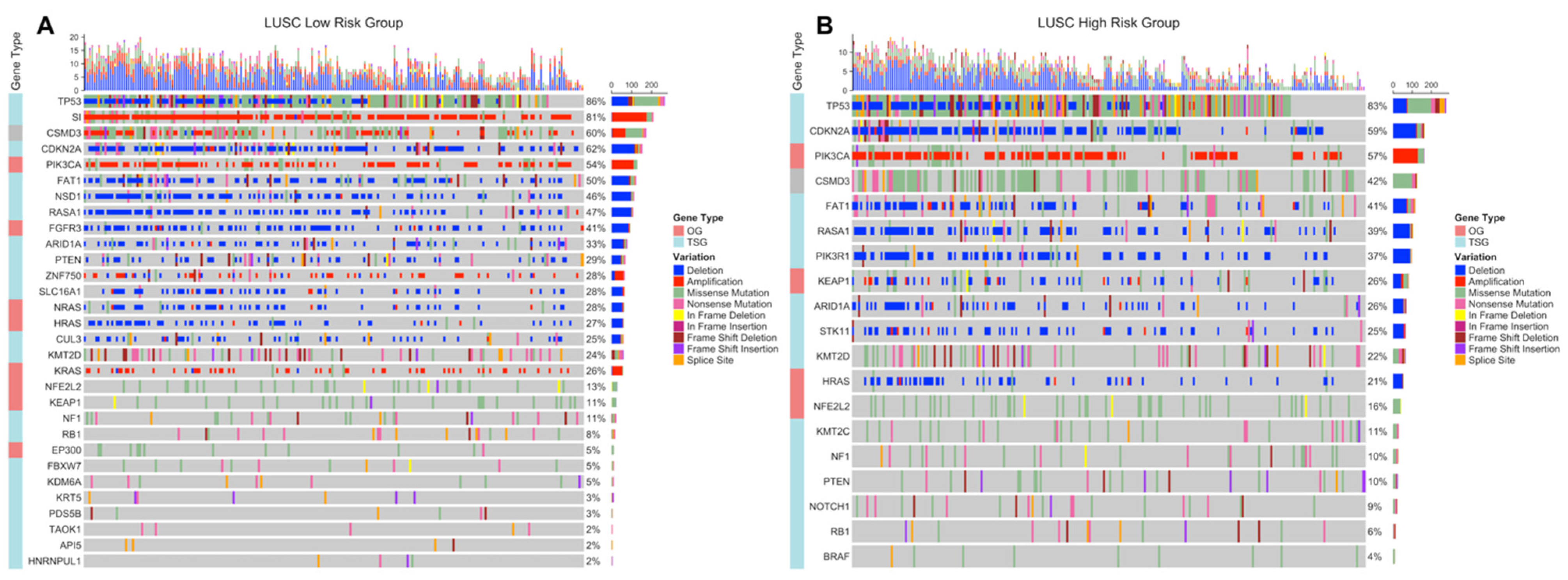
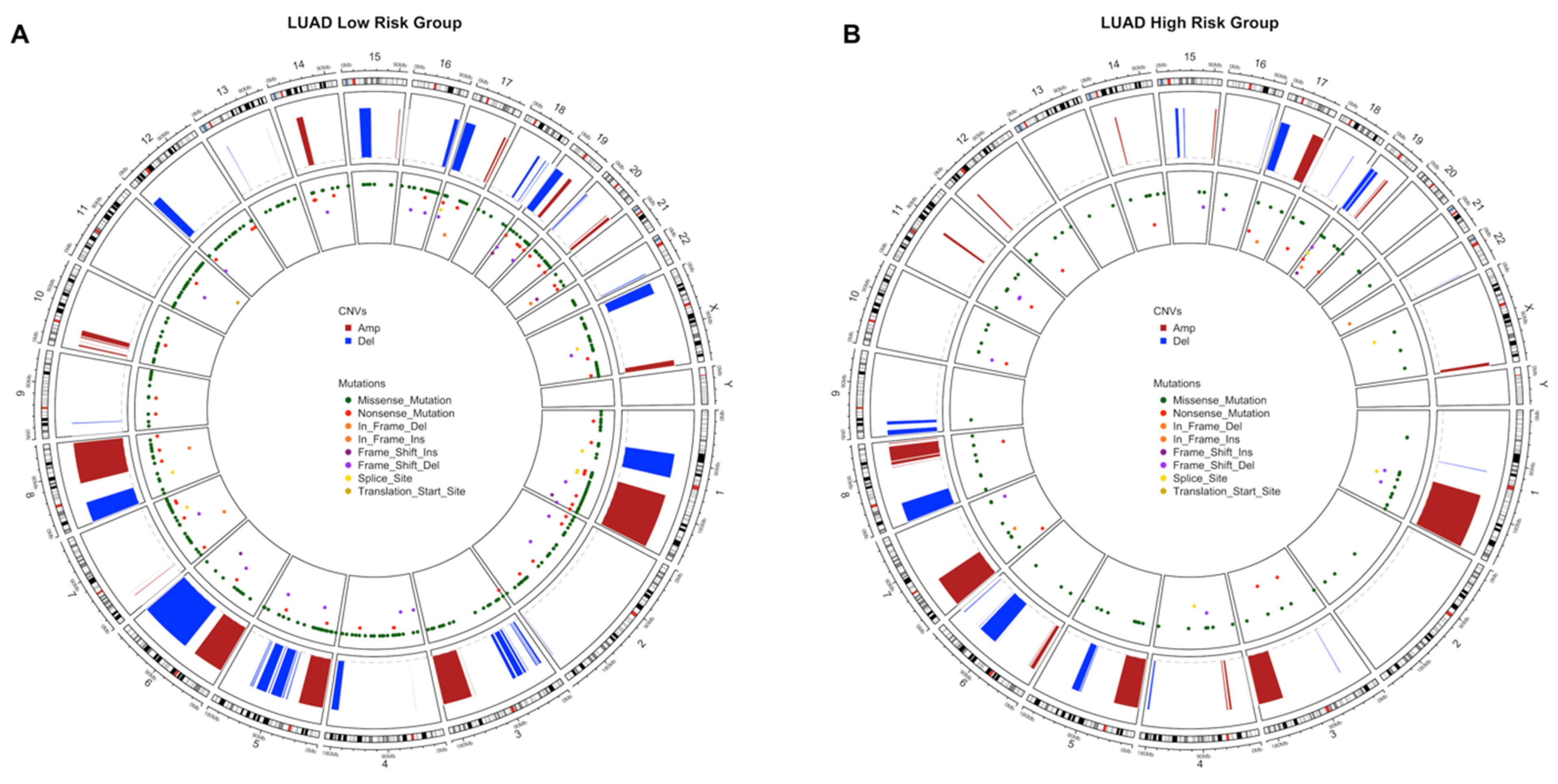
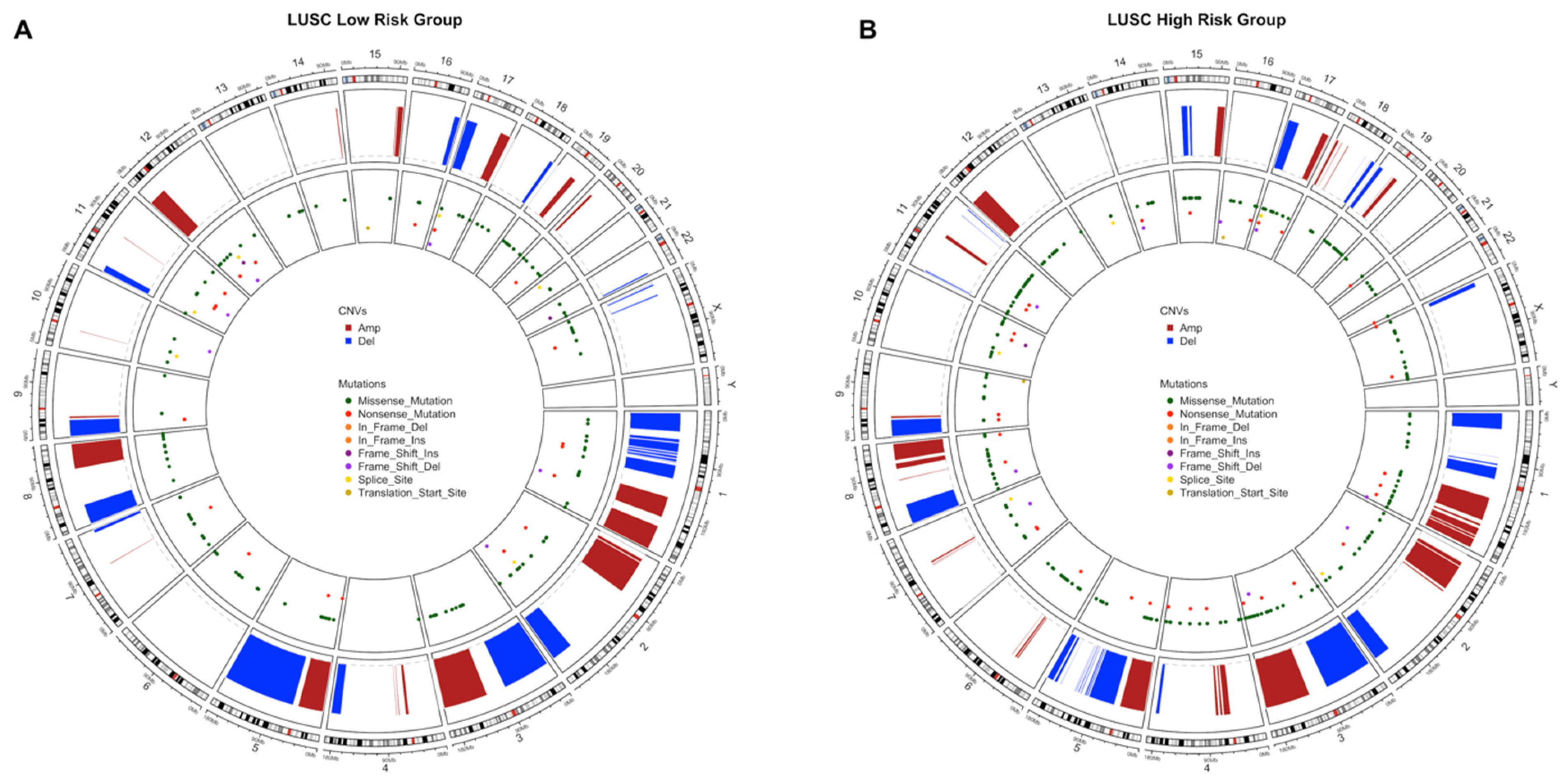
3.3. Copy Number Variations Analysis
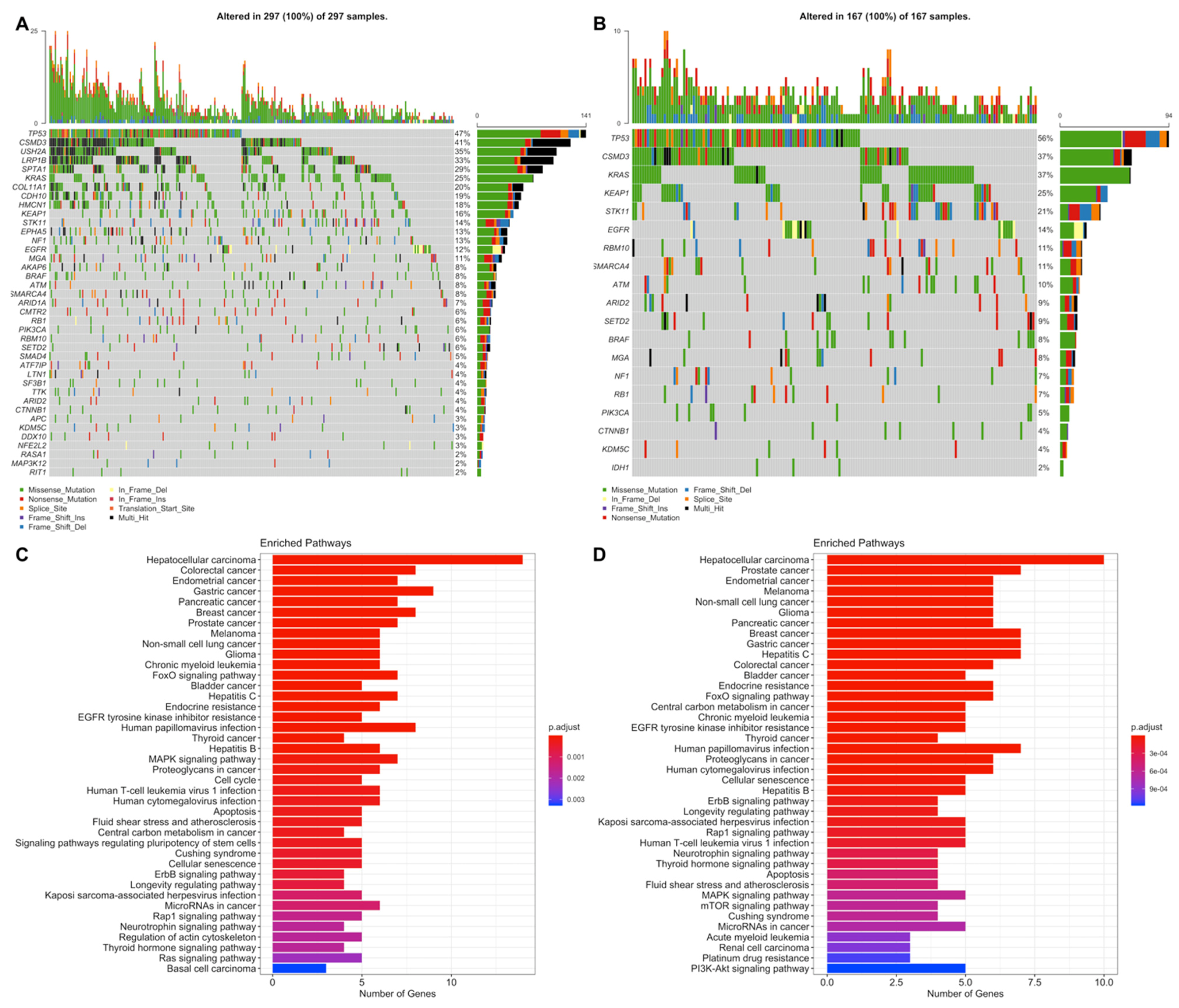
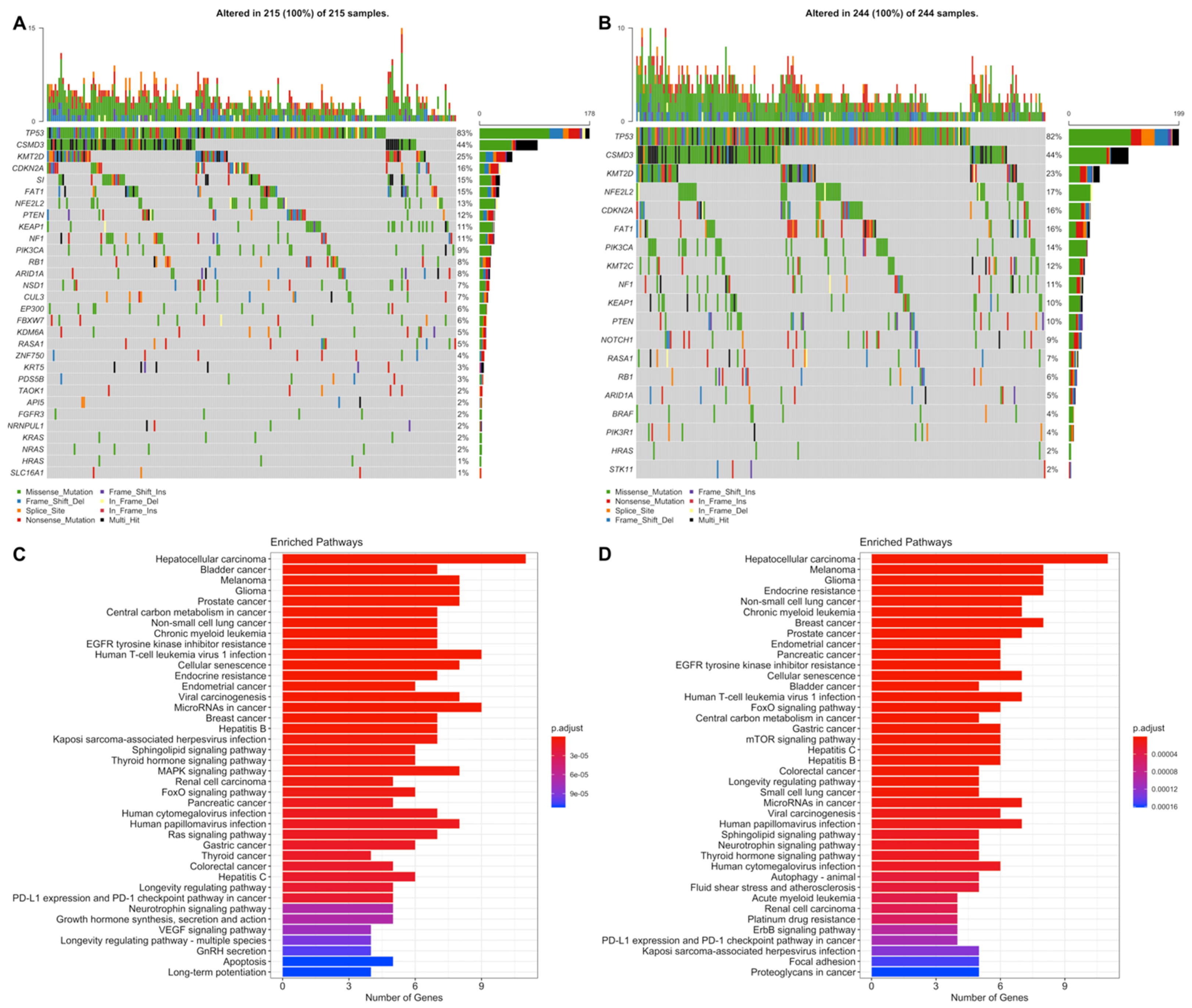
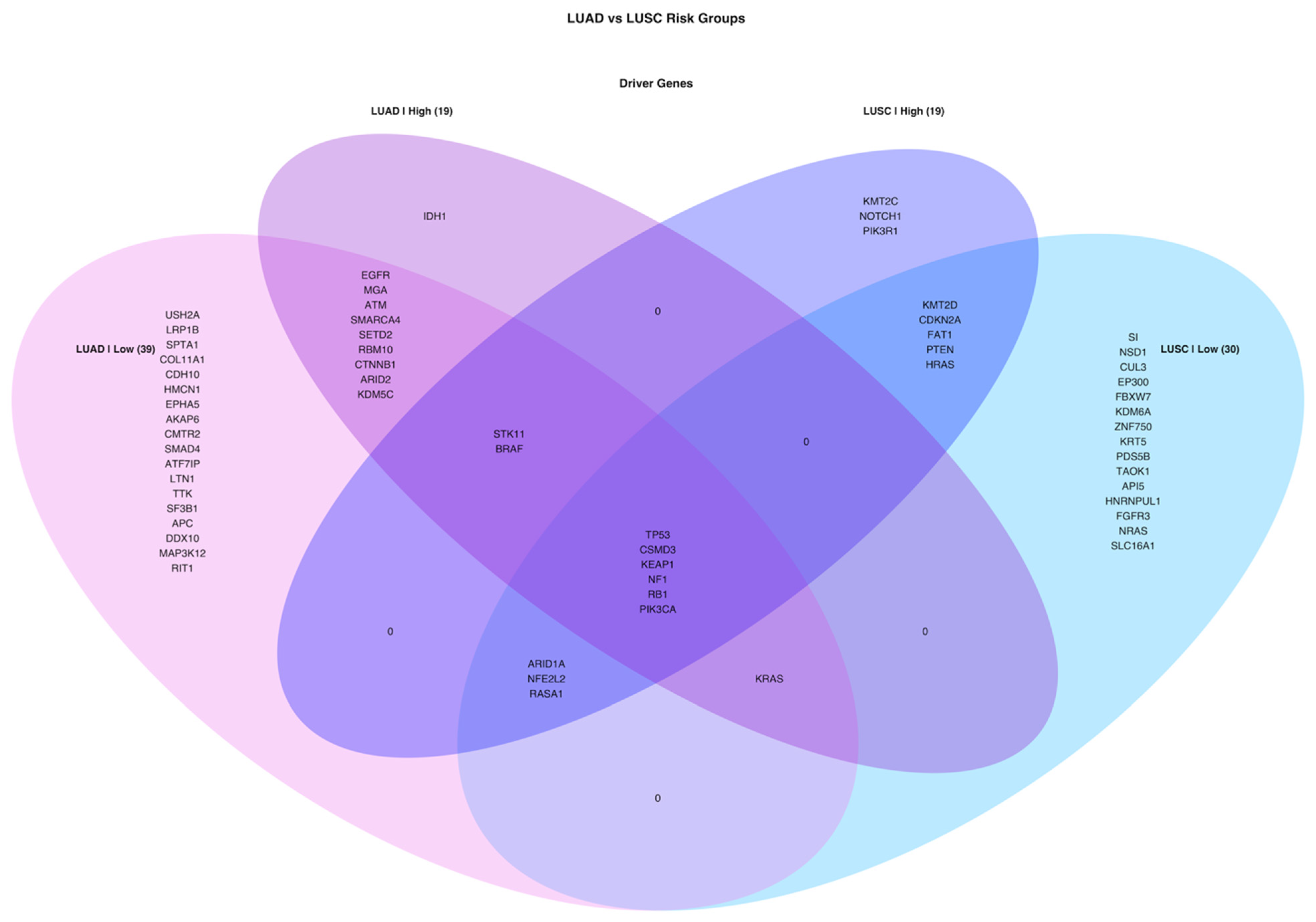
3.4. Simple Nucleotide Variations Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GLOBOCAN 2020: Cancer Today. Available online: https://gco.iarc.fr/today/home (accessed on 29 December 2020).
- Alexander, M.; Kim, S.Y.; Cheng, H. Update 2020: Management of Non-Small Cell Lung Cancer. Lung 2020, 198, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Chansky, K.; Detterbeck, F.C.; Nicholson, A.G.; Rusch, V.W.; Vallières, E.; Groome, P.; Kennedy, C.; Krasnik, M.; Peake, M.; Shemanski, L.; et al. The IASLC Lung Cancer Staging Project: External Validation of the Revision of the TNM Stage Groupings in the Eighth Edition of the TNM Classification of Lung Cancer. J. Thorac. Oncol. 2017, 12, 1109–1121. [Google Scholar] [CrossRef] [Green Version]
- Camidge, D.R.; Doebele, R.C.; Kerr, K.M. Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat. Rev. Clin. Oncol. 2019, 16, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-Y.; Huang, J.-Y.; Chen, H.-C.; Lin, C.-H.; Lin, S.-H.; Hung, W.-H.; Cheng, Y.-F. The comparison between adenocarcinoma and squamous cell carcinoma in lung cancer patients. J. Cancer Res. Clin. Oncol. 2019, 146, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D. Lung Cancer Pathology. Clin. Chest Med. 2020, 41, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fujimoto, J.; Wedge, D.C.; Song, X.; Seth, S.; Chow, C.-W.; Cao, Y.; Gumbs, C.; Gold, K.A.; Kalhor, N.; et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nat. Cell Biol. 2014, 511, 543–550. [CrossRef]
- The Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nat. Cell Biol. 2012, 489, 519–525. [CrossRef]
- Krzystanek, M.; Moldvay, J.; Szüts, D.; Szallasi, Z.; Eklund, A.C. A robust prognostic gene expression signature for early stage lung adenocarcinoma. Biomark. Res. 2016, 4, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Evans, J.R.; Malik, R.; Feng, F.Y.; Dhanasekaran, S.M.; Cao, X.; Chen, G.; Beer, D.G.; Jiang, H.; Chinnaiyan, A.M. Development of a RNA-Seq Based Prognostic Signature in Lung Adenocarcinoma. J. Natl. Cancer Inst. 2017, 109, 200. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qi, F.; Li, F. Establishment of a Gene Signature to Predict Prognosis for Patients with Lung Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 8479. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.-Q.; Strumpf, D.; Li, C.-Y.; Li, Q.; Liu, N.; Der, S.; Shepherd, F.A.; Tsao, M.-S.; Jurisica, I. Prognostic Gene Expression Signature for Squamous Cell Carcinoma of Lung. Clin. Cancer Res. 2010, 16, 5038–5047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, J.; Chen, Y.; Yang, L.; Chen, S. A prognostic 4-gene expression signature for squamous cell lung carcinoma. J. Cell. Physiol. 2017, 232, 3702–3713. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Chen, H.; Shan, Z.; Yang, L. Identification of differentially expressed genes between lung adenocarcinoma and lung squamous cell carcinoma by gene expression profiling. Mol. Med. Rep. 2016, 14, 1483–1490. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Wang, L.; Feng, F.; Tian, S. Weighted gene expression profiles identify diagnostic and prognostic genes for lung adenocarcinoma and squamous cell carcinoma. J. Int. Med Res. 2020, 48, 0300060519893837. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T. A Package for Survival Analysis in R. R Package Version 3.2-7. 2020. Available online: https://cran.r-project.org/package=survival (accessed on 21 May 2020).
- Simon, N.; Friedman, J.H.; Hastie, T.; Tibshirani, R. Regularization Paths for Cox’s Proportional Hazards Model via Coordinate Descent. J. Stat. Softw. 2011, 39, 1–13. [Google Scholar] [CrossRef]
- Gerds, T.A.; Ozenne, B. RiskRegression: Risk Regression Models and Prediction Scores for Survival Analysis with Competing Risks. R Package Version 2020.12.08. 2020. Available online: https://cran.r-project.org/package=riskRegression (accessed on 21 May 2020).
- Zhang, J.; Jin, Z. Cutoff: Seek the Significant Cutoff Value. R Package Version 1.3. 2019. Available online: https://cran.r-project.org/package=cutoff (accessed on 21 May 2020).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Vrahatis, A.G.; Balomenos, P.; Tsakalidis, A.K.; Bezerianos, A. DEsubs: An R package for flexible identification of differentially expressed subpathways using RNA-seq experiments. Bioinformatics 2016, 32, 3844–3846. [Google Scholar] [CrossRef] [Green Version]
- Morganella, S.; Pagnotta, S.M.; Ceccarelli, M. GAIA: An R Package for Genomic Analysis of Significant Chromosomal Aberrations. R Package Version 2.32.0. 2020. Available online: https://bioconductor.org/packages/gaia (accessed on 21 May 2020).
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Silva, T.C.; Colaprico, A.; Olsen, C.; D’Angelo, F.; Bontempi, G.; Ceccarelli, M.; Noushmehr, H. TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Research 2016, 5, 1542. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eynden, J.V.D.; Fierro, A.C.; Verbeke, L.P.C.; Marchal, K. SomInaClust: Detection of cancer genes based on somatic mutation patterns of inactivation and clustering. BMC Bioinform. 2015, 16, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jin, Z. Ggrisk: Risk Score Plot for Cox Regression. R Package Version 1.2. 2020. Available online: https://cran.r-project.org/package=ggrisk (accessed on 21 May 2020).
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves Using “ggplot2”. R Package Version 0.4.8. 2020. Available online: https://cran.r-project.org/package=survminer (accessed on 21 May 2020).
- Heagerty, P.J.; Saha-Chaudhuri, P. survivalROC: Time-Dependent ROC Curve Estimation from Censored Survival Data. R Package Version 1.0.3. 2013. Available online: https://cran.r-project.org/package=survivalROC (accessed on 21 May 2020).
- Kennedy, N. Forestmodel: Forest Plots from Regression Models. R Package Version 0.6.2. 2020. Available online: https://cran.r-project.org/package=forestmodel (accessed on 21 May 2020).
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Yu, G. Enrichplot: Visualization of Functional Enrichment Result. R Package Version 1.8.1. 2020. Available online: https://github.com/GuangchuangYu/enrichplot (accessed on 21 May 2020).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, D.; Zeileis, A.; Hornik, K. Vcd: Visualizing Categorical Data. R Package Version 1.4-8. 2020. Available online: https://cran.r-project.org/package=vcd (accessed on 21 May 2020).
- Meyer, D.; Zeileis, A.; Hornik, K. The Strucplot Framework: Visualizing Multi-way Contingency Tables withvcd. J. Stat. Softw. 2006, 17, 1–48. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H. VennDiagram: Generate High-Resolution Venn and Euler Plots. R Package Version 1.6.20. 2018. Available online: https://cran.r-project.org/package=VennDiagram (accessed on 21 May 2020).
- Kumar, S.; Rao, N.; Ge, R. Emerging Roles of ADAMTSs in Angiogenesis and Cancer. Cancers 2012, 4, 1252. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N 6 -Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Hu, Z.; Qu, X.; Zhu, J.; Li, L.; Ring, B.Z.; Su, L. Putative EPHX1 Enzyme Activity Is Related with Risk of Lung and Upper Aerodigestive Tract Cancers: A Comprehensive Meta-Analysis. PLoS ONE 2011, 6, e14749. [Google Scholar] [CrossRef]
- Zhu, X.; Guo, X.; Wu, S.; Wei, L. ANGPTL4 Correlates with NSCLC Progression and Regulates Epithelial-Mesenchymal Transition via ERK Pathway. Lung 2016, 194, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.-G.; Chen, L.; Wang, Q.-L.; Zhao, X.-L.; Tan, J.; Cui, Y.-H.; Liu, X.-D.; Zhang, X.; Bian, X.-W. Elevated expression of ASCL2 is an independent prognostic indicator in lung squamous cell carcinoma. J. Clin. Pathol. 2015, 69, 313–318. [Google Scholar] [CrossRef]
- Kadomoto, S.; Izumi, K.; Mizokami, A. The CCL20-CCR6 Axis in Cancer Progression. Int. J. Mol. Sci. 2020, 21, 5186. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Zhao, X.; Jiang, M.; Gu, M.; Wang, Z.; Yue, W. DKK1 promotes migration and invasion of non–small cell lung cancer via β-catenin signaling pathway. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, R.; Hirohashi, Y.; Kitamura, H.; Nishida, S.; Murai, A.; Takaya, A.; Yamamoto, E.; Matsuki, M.; Tanaka, T.; Kubo, T.; et al. GRIK2 has a role in the maintenance of urothelial carcinoma stem-like cells, and its expression is associated with poorer prognosis. Oncotarget 2017, 8, 28826–28839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Hou, L.; Cui, H.; Zhang, L.; Tan, X.; Leng, X.; Li, Y. LDHA upregulation independently predicts poor survival in lung adenocarcinoma, but not in lung squamous cell carcinoma. Futur. Oncol. 2018, 14, 2483–2492. [Google Scholar] [CrossRef]
- Yang, L.; Lee, M.M.; Leung, M.M.; Wong, Y.H. Regulator of G protein signaling 20 enhances cancer cell aggregation, migration, invasion and adhesion. Cell. Signal. 2016, 28, 1663–1672. [Google Scholar] [CrossRef]
- Han, S.-W.; Kim, H.-P.; Shin, J.-Y.; Jeong, E.-G.; Lee, W.-C.; Kim, K.Y.; Park, S.Y.; Lee, D.-W.; Won, J.-K.; Jeong, S.-Y.; et al. RNA editing in RHOQ promotes invasion potential in colorectal cancer. J. Exp. Med. 2014, 211, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Yuan, D.; Yang, X.; Yuan, Z.; Zhao, Y.; Guo, J. TLE1 function and therapeutic potential in cancer. Oncotarget 2016, 8, 15971–15976. [Google Scholar] [CrossRef]
- Tabatabaeian, H.; Rao, A.; Ramos, A.; Chu, T.; Sudol, M.; Lim, Y.P. The emerging roles of WBP2 oncogene in human cancers. Oncogene 2020, 39, 4621–4635. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Suzuki, Y.; Inoue, Y.; Tsuchiya, K.; Karayama, M.; Iwashita, Y.; Kahyo, T.; Kawase, A.; Tanahashi, M.; Ogawa, H.; et al. CD200 and CD200R1 are differentially expressed and have differential prognostic roles in non-small cell lung cancer. OncoImmunology 2020, 9, 1746554. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Zhuang, J.; Li, H.; Liu, C.; Zhou, C.; Liu, L.; Sun, C. Exploration of methylation-driven genes for monitoring and prognosis of patients with lung adenocarcinoma. Cancer Cell Int. 2018, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Li, Y.; Ma, C.; Zhang, J.; Zhang, Y.; Fu, Z.; Luo, H. Independent Prognostic Potential of GNPNAT1 in Lung Adenocarcinoma. BioMed Res. Int. 2020, 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qiu, X. Comprehensive Analysis of the Expression and Prognosis for IRXs in Non-small Cell Lung Cancer. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Puderecki, M.; Szumiło, J.; Marzec-Kotarska, B. Novel prognostic molecular markers in lung cancer (Review). Oncol. Lett. 2020, 20, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Ding, D.; Yu, W.; Zhu, C.; Ding, Y. The Lung Adenocarcinoma Microenvironment Mining and Its Prognostic Merit. Technol. Cancer Res. Treat. 2020, 19. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, H.; Xu, Y.; Li, J.; Deng, C.; Deng, Y.; Bai, J.; Li, X.; Xiao, Y.; Zhang, Y. Systematic identification of lincRNA-based prognostic biomarkers by integrating lincRNA expression and copy number variation in lung adenocarcinoma. Int. J. Cancer 2019, 144, 1723–1734. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.M.; Francis, S.M.S.; Tan, M.E.; Martins, M.U.; Winterford, C.; Davidson, M.R.; Duhig, E.E.; Clarke, B.E.; Hayward, N.K.; Yang, I.A.; et al. MS4A1 Dysregulation in Asbestos-Related Lung Squamous Cell Carcinoma Is Due to CD20 Stromal Lymphocyte Expression. PLoS ONE 2012, 7, e34943. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Mu, D. MicroRNAs and lung cancers: From pathogenesis to clinical implications. Front. Med. 2012, 6, 134–155. [Google Scholar] [CrossRef] [Green Version]
- Sage, A.P.; Ng, K.W.; Marshall, E.A.; Stewart, G.L.; Minatel, B.C.; Enfield, K.S.S.; Martin, S.D.; Brown, C.J.; Abraham, N.; Lam, W.L. Assessment of long non-coding RNA expression reveals novel mediators of the lung tumour immune response. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Tang, L.-X.; Chen, G.-H.; Li, H.; He, P.; Zhang, Y.; Xu, X.-W. Long non-coding RNA OGFRP1 regulates LYPD3 expression by sponging miR-124-3p and promotes non-small cell lung cancer progression. Biochem. Biophys. Res. Commun. 2018, 505, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Giacalone, N.J.; Den, R.B.; Eisenberg, R.; Chen, H.; Olson, S.J.; Massion, P.P.; Carbone, D.P.; Lu, B. ALDH7A1 expression is associated with recurrence in patients with surgically resected non-small-cell lung carcinoma. Futur. Oncol. 2013, 9, 737–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Shen, Q.; Shi, Q.; Yu, B.; Wang, X.; Cheng, K.; Lu, G.; Zhou, X. Detection of ALK protein expression in lung squamous cell carcinomas by immunohistochemistry. J. Exp. Clin. Cancer Res. 2014, 33, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldrini, L.; Gisfredi, S.; Ursino, S.; Faviana, P.; Lucchi, M.; Melfi, F.; Mussi, A.; Basolo, F.; Fontanini, G. Expression of endothelin-1 is related to poor prognosis in non-small cell lung carcinoma. Eur. J. Cancer 2005, 41, 2828–2835. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, X.; Deng, L.; Li, X.; Wang, G.; Li, Y.; Chen, M. High expression of FABP4 and FABP6 in patients with colorectal cancer. World J. Surg. Oncol. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Wang, X.; Shi, B.; Zhao, Y.; Lu, Q.; Fei, X.; Lu, C.; Li, C.; Chen, H. HKDC1 promotes the tumorigenesis and glycolysis in lung adenocarcinoma via regulating AMPK/mTOR signaling pathway. Cancer Cell Int. 2020, 20, 1–12. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, Y.; Bhandari, A.; Xia, E.; Wang, O. IGSF1: A novel oncogene regulates the thyroid cancer progression. Cell Biochem. Funct. 2019, 37, 516–524. [Google Scholar] [CrossRef]
- Gong, J.; Tian, J.; Lou, J.; Wang, X.; Ke, J.; Li, J.; Yang, Y.; Gong, Y.; Zhu, Y.; Zou, D.; et al. A polymorphic MYC response element in KBTBD11 influences colorectal cancer risk, especially in interaction with an MYC-regulated SNP rs6983267. Ann. Oncol. 2017, 29, 632–639. [Google Scholar] [CrossRef]
- Zou, Z.; Li, X.; Sun, Y.; Li, L.; Zhang, Q.; Zhu, L.; Zhong, Z.; Wang, M.; Wang, Q.; Liu, Z.; et al. NOS1 expression promotes proliferation and invasion and enhances chemoresistance in ovarian cancer. Oncol. Lett. 2020, 19, 2989–2995. [Google Scholar] [CrossRef] [Green Version]
- Ueda, M.; Iguchi, T.; Masuda, T.; Komatsu, H.; Nambara, S.; Sakimura, S.; Hirata, H.; Uchi, R.; Eguchi, H.; Ito, S.; et al. Up-regulation of SLC9A9 Promotes Cancer Progression and Is Involved in Poor Prognosis in Colorectal Cancer. Anticancer Res. 2017, 37, 2255–2263. [Google Scholar] [CrossRef] [Green Version]
- Huang, N.; Lin, W.; Shi, X.; Tao, T. STK24 expression is modulated by DNA copy number/methylation in lung adenocarcinoma and predicts poor survival. Futur. Oncol. 2018, 14, 2253–2263. [Google Scholar] [CrossRef]
- Tang, Y.; Geng, Y.; Luo, J.; Shen, W.; Zhu, W.; Meng, C.; Li, M.; Zhou, X.; Zhang, S.; Cao, J. Downregulation of ubiquitin inhibits the proliferation and radioresistance of non-small cell lung cancer cells in vitro and in vivo. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Baykara, O.; Dalay, N.; Kaynak, K.; Buyru, N. ZNF703 Overexpression may act as an oncogene in non-small cell lung cancer. Cancer Med. 2016, 5, 2873–2878. [Google Scholar] [CrossRef]
- Li, J.; Ye, L.; Mansel, R.E.; Jiang, W.G. Potential prognostic value of repulsive guidance molecules in breast cancer. Anticancer Res. 2011, 31, 1703–1711. [Google Scholar] [PubMed]
- Chong, P.K.; Lee, H.; Zhou, J.; Liu, S.-C.; Loh, M.C.S.; Wang, T.T.; Chan, S.P.; Smoot, D.T.; Ashktorab, H.; So, J.B.Y.; et al. ITIH3 Is a Potential Biomarker for Early Detection of Gastric Cancer. J. Proteome Res. 2010, 9, 3671–3679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cui, J.; Tang, Y.-L.; Huang, L.; Zhou, C.-Y.; Xu, J.-X. Prognostic Roles of mRNA Expression of S100 in Non-Small-Cell Lung Cancer. BioMed Res. Int. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Z.; Gao, S.; M’Rabet, N.; De Geyter, C.; Zhang, H. Sp1 Is Necessary for Gene Activation of Adamts17 by Estrogen. J. Cell. Biochem. 2014, 115, 1829–1839. [Google Scholar] [CrossRef]
- Li, R.; Yang, Y.-E.; Jin, J.; Zhang, M.-Y.; Liu, X.-X.; Yin, Y.-H.; Qu, Y.-Q. Identification of lncRNA biomarkers in lung squamous cell carcinoma using comprehensive analysis of lncRNA mediated ceRNA network. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3246–3258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.-W.; Ye, H.; Wei, P.-P.; He, B.; Han, C.; Chen, Z.-H.; Chen, Y.-Q.; Wang, W.-T. Global identification and characterization of lncRNAs that control inflammation in malignant cholangiocytes. BMC Genom. 2018, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Liu, H.; Yan, X.; Wang, J. In Silico Analysis Identifies Differently Expressed lncRNAs as Novel Biomarkers for the Prognosis of Thyroid Cancer. Comput. Math. Methods Med. 2020, 2020, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Ye, Y.J.; Ren, X.W.; Huang, J.; Shen, Z.L. Detection of preoperative chemoradiotherapy sensitivity molecular characteristics of rectal cancer by transcriptome second generation sequencing. J. Peking Univ. Health Sci. 2019, 51, 542–547. [Google Scholar] [CrossRef]
- Tian, B.; Han, X.; Li, G.; Jiang, H.; Qi, J.; Li, J.; Tian, Y.; Wang, C. A Long Intergenic Non-coding RNA, LINC01426, Promotes Cancer Progression via AZGP1 and Predicts Poor Prognosis in Patients with LUAD. Mol. Ther. Methods Clin. Dev. 2020, 18, 765–780. [Google Scholar] [CrossRef]
- Yang, H.; Jin, L.; Sun, X. A thirteen-gene set efficiently predicts the prognosis of glioblastoma. Mol. Med. Rep. 2019, 19, 1613–1621. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, L.M.; Yeung, S.J.; Lee, M.-H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar]
- Keenan, M.M.; Chi, J.-T. Alternative Fuels for Cancer Cells. Cancer J. 2015, 21, 49–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, K.; Kohno, T.; Kawanishi, M.; Ohwada, S.; Yokota, J. Association ofCDKN2A (p16)/CDKN2B (p15) alterations and homozygous chromosome arm 9p deletions in human lung carcinoma. Genes, Chromosom. Cancer 1998, 22, 232–240. [Google Scholar] [CrossRef]
- Schuster, K.; Venkateswaran, N.; Rabellino, A.; Girard, L.; Peña-Llopis, S.; Scaglioni, P.P. Nullifying the CDKN2AB Locus Promotes Mutant K-ras Lung Tumorigenesis. Mol. Cancer Res. 2014, 12, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Gu, Y.; Liu, J.; Wu, R.; Fu, L.; Zhao, J.; Guan, Y. Coexistence of p16/CDKN2A homozygous deletions and activating EGFR mutations in lung adenocarcinoma patients signifies a poor response to EGFR-TKIs. Lung Cancer 2016, 102, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Bass, A.J.; Watanabe, H.; Mermel, C.H.; Yu, S.; Perner, S.; Verhaak, R.G.; Kim, S.Y.; Wardwell, L.; Tamayo, P.; Gat-Viks, I.; et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 2009, 41, 1238–1242. [Google Scholar] [CrossRef]
- Maier, S.; Wilbertz, T.; Braun, M.; Scheble, V.; Reischl, M.; Mikut, R.; Menon, R.; Nikolov, P.; Petersen, K.; Beschorner, C.; et al. SOX2 amplification is a common event in squamous cell carcinomas of different organ sites. Hum. Pathol. 2011, 42, 1078–1088. [Google Scholar] [CrossRef]
- Fejzo, M.S.; Anderson, L.; Chen, H.-W.; Guandique, E.; Kalous, O.; Conklin, D.; Slamon, D.J. Proteasome ubiquitin receptor PSMD4 is an amplification target in breast cancer and may predict sensitivity to PARPi. Genes, Chromosom. Cancer 2017, 56, 589–597. [Google Scholar] [CrossRef]
- Cai, M.-J.; Cui, Y.; Fang, M.; Wang, Q.; Zhang, A.-J.; Kuai, J.-H.; Pang, F.; Cui, X.-D. Inhibition of PSMD4 blocks the tumorigenesis of hepatocellular carcinoma. Gene 2019, 702, 66–74. [Google Scholar] [CrossRef]
- Cheng, Y.-M.; Lin, P.-L.; Wu, D.-W.; Wang, L.; Huang, C.-C.; Lee, H. PSMD4 is a novel therapeutic target in chemoresistant colorectal cancer activated by cytoplasmic localization of Nrf2. Oncotarget 2018, 9, 26342–26352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Türkoğlu, S.A.; Dayi, G.; Köçkar, F. Upregulation of PSMD4 Gene By Hypoxia in Prostate Cancer Cells. Turk. J. Boil. 2020, 44, 275–283. [Google Scholar] [CrossRef]
- O’Leary, C.; Gasper, H.; Sahin, K.B.; Tang, M.; Kulasinghe, A.; Adams, M.N.; Richard, D.J.; O’Byrne, K.J. Epidermal Growth Factor Receptor (EGFR)-Mutated Non-Small-Cell Lung Cancer (NSCLC). Pharmaceuticals 2020, 13, 273. [Google Scholar] [CrossRef]
- Mathsyaraja, H.; Catchpole, J.; Eastwood, E.; Babaeva, E.; Geuenich, M.; Cheng, P.F.; Freie, B.; Ayers, J.; Yu, M.; Wu, N.; et al. Loss of MGA mediated Polycomb repression promotes tumor progression and invasiveness. bioRxiv 2020. [Google Scholar] [CrossRef]
- Xue, Y.; Meehan, B.; Fu, Z.; Wang, X.Q.D.; Fiset, P.O.; Rieker, R.; Levins, C.; Kong, T.; Zhu, X.; Morin, G.; et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, P.; Lv, X.; Zhang, L.; Zhang, J. The role of the ataxia telangiectasia mutated gene in lung cancer: Recent advances in research. Ther. Adv. Respir. Dis. 2017, 11, 375–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Jia, M.; Sun, W.; Feng, L.; Gu, C.; Wu, T. Functional role of RBM10 in lung adenocarcinoma proliferation. Int. J. Oncol. 2018, 54, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Yim, S.; Park, H. The cancer driver genes IDH1/2, JARID1C/ KDM5C, and UTX/ KDM6A: Crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, K.W.; Zhang, W.; Soh, J.; Stastny, V.; Chen, M.; Sun, H.; Thu, K.; Rios, J.J.; Yang, C.; Marconett, C.N.; et al. CDKN2A/p16 Inactivation Mechanisms and Their Relationship to Smoke Exposure and Molecular Features in Non–Small-Cell Lung Cancer. J. Thorac. Oncol. 2013, 8, 1378–1388. [Google Scholar] [CrossRef] [Green Version]
- Gkountakos, A.; Sartori, G.; Falcone, I.; Piro, G.; Ciuffreda, L.; Carbone, C.; Tortora, G.; Scarpa, A.; Bria, E.; Milella, M.; et al. PTEN in Lung Cancer: Dealing with the Problem, Building on New Knowledge and Turning the Game Around. Cancers 2019, 11, 1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pązik, M.; Michalska, K.; Żebrowska-Nawrocka, M.; Zawadzka, I.; Łochowski, M.; Balcerczak, E. Clinical significance of HRAS and KRAS genes expression in patients with non–small-cell lung cancer—Preliminary Findings. BMC Cancer 2021, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Han, Y.; Li, J.; Chai, R.; Bai, C. Prognostic value of KRAS/TP53/PIK3CA in non-small cell lung cancer. Oncol. Lett. 2019, 17, 3233–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Morrison, C.; Wang, L.; Xiong, D.; Vedell, P.; Cui, P.; Hua, X.; Ding, F.; Lu, Y.; James, M.; et al. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis 2012, 33, 1270–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Fleur, L.; Falk-Sörqvist, E.; Smeds, P.; Berglund, A.; Sundström, M.; Mattsson, J.S.; Brandén, E.; Koyi, H.; Isaksson, J.; Brunnström, H.; et al. Mutation patterns in a population-based non-small cell lung cancer cohort and prognostic impact of concomitant mutations in KRAS and TP53 or STK11. Lung Cancer 2019, 130, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LUAD | LUSC | |||
|---|---|---|---|---|
| Category | Train Group (n: 436) | Test Group (n: 56) | Train Group (n: 431) | Test Group (n: 47) |
| Age at diagnosis (median; range) | 66; 33–88 | 66.5; 42–86 | 68; 39–90 | 69; 45–85 |
| Gender | ||||
| Female | 232 | 33 | 112 | 14 |
| Male | 204 | 23 | 319 | 33 |
| Tumor stage | ||||
| I | 241 | 28 | 211 | 25 |
| II | 106 | 13 | 138 | 16 |
| III | 68 | 13 | 76 | 5 |
| IV | 23 | 2 | 6 | 1 |
| Vital status | ||||
| Alive | 284 | 30 | 275 | 18 |
| Dead | 152 | 26 | 156 | 29 |
| Smoked years (median; range) | 33; 2–61 | 31.5; 4–64 | 40; 8–62 | 40; 10–60 |
| Smoked packs per year (median; range) | 40; 0.15–154 | 48; 5–94.5 | 50; 1–240 | 50; 2–157.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zengin, T.; Önal-Süzek, T. Comprehensive Profiling of Genomic and Transcriptomic Differences between Risk Groups of Lung Adenocarcinoma and Lung Squamous Cell Carcinoma. J. Pers. Med. 2021, 11, 154. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11020154
Zengin T, Önal-Süzek T. Comprehensive Profiling of Genomic and Transcriptomic Differences between Risk Groups of Lung Adenocarcinoma and Lung Squamous Cell Carcinoma. Journal of Personalized Medicine. 2021; 11(2):154. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11020154
Chicago/Turabian StyleZengin, Talip, and Tuğba Önal-Süzek. 2021. "Comprehensive Profiling of Genomic and Transcriptomic Differences between Risk Groups of Lung Adenocarcinoma and Lung Squamous Cell Carcinoma" Journal of Personalized Medicine 11, no. 2: 154. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11020154