
Is HSD17B13 Genetic Variant a Protector for Liver Dysfunction? Future Perspective as a Potential Therapeutic Target

 , , ,
, , ,
Abstract
:1. Introduction
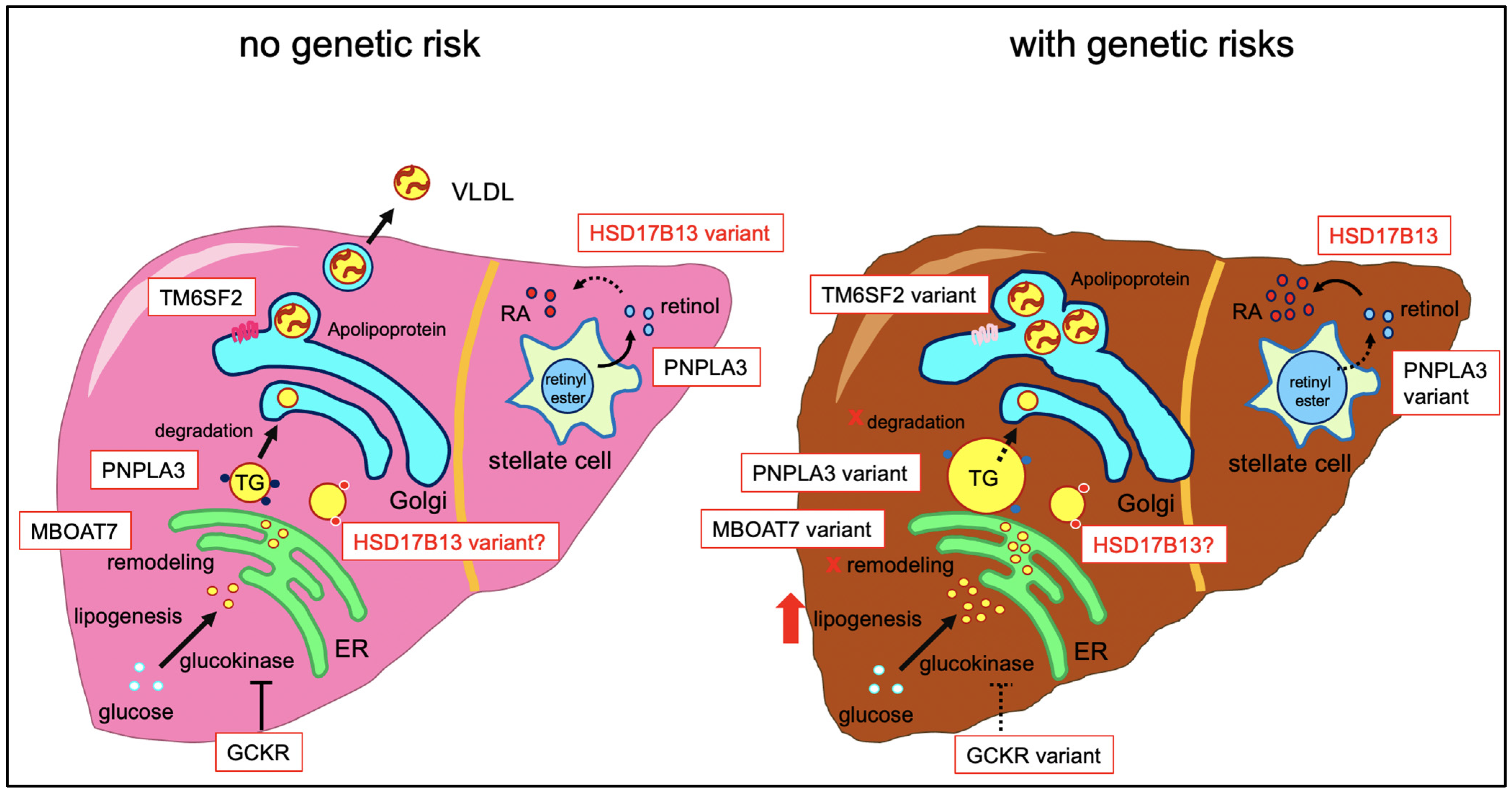
2. Molecular Function of HSD17B13
3. Role of HSD17B13 Genetic Variants in Liver Disease
3.1. Fatty Liver Disease
3.2. Viral Hepatitis
3.3. Hepatocellular Carcinoma (HCC)
3.4. Simple Steatosis
4. Role of HSD17B13 Genetic Variants in the Other Diseases
5. HSD17B13 as a Potential Therapeutic Target
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meringer, H.; Shibolet, O.; Deutsch, L. Hepatocellular carcinoma in the post-hepatitis C virus era: Should we change the paradigm? World J. Gastroenterol. 2019, 25, 3929–3940. [Google Scholar] [CrossRef]
- Chen, L.; Abou-Alfa, G.K.; Zheng, B.; Liu, J.-F.; Bai, J.; Du, L.-T.; Qian, Y.-S.; Fan, R.; Liu, X.-L.; Wu, L.; et al. Genome-scale profiling of circulating cell-free DNA signatures for early detection of hepatocellular carcinoma in cirrhotic patients. Cell Res. 2021, 31, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Association for the Study of the Liver. EASL Recommendations on Treatment of Hepatitis C 2018. J. Hepatol. 2018, 69, 461–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belperio, P.S.; Shahoumian, T.A.; Loomis, T.P.; Mole, L.A.; Backus, L.I. Real-world effectiveness of daclatasvir plus sofosbuvir and velpatasvir/sofosbuvir in hepatitis C genotype 2 and 3. J. Hepatol. 2019, 70, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-H.; Hsu, Y.-C.; Chen, T.-H.; Ji, F.; Chen, I.-S.; Tsai, Y.-N.; Hai, H.; Thuy, L.T.T.; Hosaka, T.; Sezaki, H.; et al. Hepatocellular carcinoma incidence with tenofovir versus entecavir in chronic hepatitis B: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 1039–1052. [Google Scholar] [CrossRef]
- Mirdad, R.S.; Hyer, J.M.; Diaz, A.; Tsilimigras, D.I.; Azap, R.A.; Paro, A.; Pawlik, T.M. Postoperative imaging surveillance for hepatocellular carcinoma: How much is enough? J. Surg. Oncol. 2021, 123, 1568–1577. [Google Scholar] [CrossRef]
- Valenti, L.; Pelusi, S. Redefining fatty liver disease classification in 2020. Liver Int. 2020, 40, 1016–1017. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Valenti, L.; Bugianesi, E.; Pajvani, U.; Targher, G. Nonalcoholic fatty liver disease: Cause or consequence of type 2 diabetes? Liver Int. 2016, 36, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Tacke, F.; Arrese, M.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.-S.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespo, G. Moving Forward in the Stratification of Cardiac Risk in Liver Transplantation Candidates. Liver Transplant. 2021. [Google Scholar] [CrossRef]
- Yen, Y.-H.; Cheng, Y.-F.; Wang, J.-H.; Lin, C.-C.; Wang, C.-C. Characteristics and etiologies of hepatocellular carcinoma in patients without cirrhosis: When East meets West. PLoS ONE 2021, 16, e0244939. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pingitore, P.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Indiveri, C.; Pujia, A.; Montalcini, T.; Hedfalk, K.; Romeo, S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 574–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prill, S.; Caddeo, A.; Baselli, G.; Jamialahmadi, O.; Dongiovanni, P.; Rametta, R.; Kanebratt, K.P.; Pujia, A.; Pingitore, P.; Mancina, R.M.; et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci. Rep. 2019, 9, 11585. [Google Scholar] [CrossRef] [Green Version]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, L.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 7, e1001324. [Google Scholar] [CrossRef]
- Raimondo, A.; Rees, M.G.; Gloyn, A.L. Glucokinase regulatory protein: Complexity at the crossroads of triglyceride and glucose metabolism. Curr. Opin. Lipidol. 2015, 26, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Buch, S.; Stickel, F.; Trépo, E.; Way, M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.; Rosendahl, J.; Berg, T.; Ridinger, M.; et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230. [Google Scholar] [CrossRef] [Green Version]
- Thangapandi, V.R.; Knittelfelder, O.; Brosch, M.; Patsenker, E.; Vvedenskaya, O.; Buch, S.; Hinz, S.; Hendricks, A.; Nati, M.; Herrmann, A.; et al. Loss of hepatic Mboat7 leads to liver fibrosis. Gut 2021, 70, 940–950. [Google Scholar] [CrossRef]
- Chambers, J.C.; Alcohol Genome-wide Association (AlcGen) Consortium; Zhang, W.; Sehmi, J.; Li, X.; Wass, M.; Van Der Harst, P.; Holm, H.; Sanna, S.; Kavousi, M.; et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat. Genet. 2011, 43, 1131–1138. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-TruncatingHSD17B13Variant and Protection from Chronic Liver Disease. New Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Darlay, R.; Cockell, S.; Meroni, M.; Govaere, O.; Tiniakos, D.; Burt, A.D.; Bedossa, P.; Palmer, J.; Liu, Y.-L.; et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort. J. Hepatol. 2020, 73, 505–515. [Google Scholar] [CrossRef]
- Ma, Y.; Belyaeva, O.V.; Brown, P.M.; Fujita, K.; Valles, K.; Karki, S.; De Boer, Y.S.; Koh, C.; Chen, Y.; Du, X.; et al. 17-Beta Hydroxysteroid Dehydrogenase 13 Is a Hepatic Retinol Dehydrogenase Associated With Histological Features of Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 1504–1519. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, J.K.; Kastaniotis, A.J.; Autio, K.J.; Jiang, G.; Chen, Z.; Glumoff, T. 17B-hydroxysteroid dehydrogenases as acyl thioester metabolizing enzymes. Mol. Cell. Endocrinol. 2019, 489, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Moeller, G.; Adamski, J. Multifunctionality of human 17β-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2006, 248, 47–55. [Google Scholar] [CrossRef]
- Matsuura, K.; Shiraishi, H.; Hara, A.; Sato, K.; Deyashiki, Y.; Ninomiya, M.; Sakai, S. Identification of a Principal mRNA Species for Human 3α-Hydroxysteroid Dehydrogenase Isoform (AKR1C3) That Exhibits High Prostaglandin D2 11-Ketoreductase Activity. J. Biochem. 1998, 124, 940–946. [Google Scholar] [CrossRef]
- Liu, S.; Huang, C.; Li, D.; Ren, W.; Zhang, H.; Qi, M.; Li, X.; Yu, L. Molecular cloning and expression analysis of a new gene for short-chain dehydrogenase/reductase 9. Acta Biochim. Pol. 2007, 54, 213–218. [Google Scholar] [CrossRef]
- Horiguchi, Y.; Araki, M.; Motojima, K. 17beta-Hydroxysteroid dehydrogenase type 13 is a liver-specific lipid droplet-associated protein. Biochem. Biophys. Res. Commun. 2008, 370, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Wang, Y.; Jia, X.; Wu, W.; Li, L.; Tian, X.; Li, S.; Wang, C.; Xu, H.; Cao, J.; et al. Comparative proteomic study reveals 17β-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2014, 111, 11437–11442. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Peng, J.; Li, S.; Dai, Y.-B.; Wang, C.-J.; Xu, H.; Gao, M.; Ruan, X.-Z.; Gustafsson, J.-Å.; Guan, Y.-F.; et al. Liver X receptor α induces 17β-hydroxysteroid dehydrogenase-13 expression through SREBP-1c. Am. J. Physiol. Metab. 2017, 312, E357–E367. [Google Scholar] [CrossRef]
- Namjou, B.; Network, T.E.; Lingren, T.; Huang, Y.; Parameswaran, S.; Cobb, B.L.; Stanaway, I.B.; Connolly, J.J.; Mentch, F.D.; Benoit, B.; et al. GWAS and enrichment analyses of non-alcoholic fatty liver disease identify new trait-associated genes and pathways across eMERGE Network. BMC Med. 2019, 17, 135. [Google Scholar] [CrossRef]
- Kozlitina, J. Genetic Risk Factors and Disease Modifiers of Nonalcoholic Steatohepatitis. Gastroenterol. Clin. N. Am. 2020, 49, 25–44. [Google Scholar] [CrossRef]
- Carlsson, B.; Lindén, D.; Brolén, G.; Liljeblad, M.; Bjursell, M.; Romeo, S.; Loomba, R. Review article: The emerging role of genetics in precision medicine for patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2020, 51, 1305–1320. [Google Scholar] [CrossRef]
- Pirola, C.J.; Garaycoechea, M.; Flichman, D.; Arrese, M.; Martino, J.S.; Gazzi, C.; Castaño, G.O.; Sookoian, S. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J. Lipid Res. 2019, 60, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Kallwitz, E.; Tayo, B.O.; Kuniholm, M.H.; Daviglus, M.; Zeng, D.; Isasi, C.R.; Cotler, S.J. Association of HSD17B13 rs72613567:TA with non-alcoholic fatty liver disease in Hispanics/Latinos. Liver Int. 2020, 40, 889–893. [Google Scholar] [CrossRef]
- Seko, Y.; Yamaguchi, K.; Tochiki, N.; Yano, K.; Takahashi, A.; Okishio, S.; Kataoka, S.; Okuda, K.; Umemura, A.; Moriguchi, M.; et al. Attenuated effect of PNPLA3 on hepatic fibrosis by HSD17B13 in Japanese patients with non-alcoholic fatty liver disease. Liver Int. 2020, 40, 1686–1692. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Y.; Guo, T.; Yang, F.; Mao, Y.; Li, L.; Liu, C.; Gao, H.; Jin, Y.; Che, Y.; et al. Genetic variant rs72613567 ofHSD17B13gene reduces alcohol-related liver disease risk in Chinese Han population. Liver Int. 2020, 40, 2194–2202. [Google Scholar] [CrossRef]
- Raja, A.M.; Ciociola, E.; Ahmad, I.N.; Dar, F.S.; Naqvi, S.M.S.; Moaeen-Ud-Din, M.; Raja, G.K.; Romeo, S.; Mancina, R.M. Genetic Susceptibility to Chronic Liver Disease in Individuals from Pakistan. Int. J. Mol. Sci. 2020, 21, 3558. [Google Scholar] [CrossRef]
- Anderson, E.L.; Howe, L.D.; Jones, H.E.; Higgins, J.; Lawlor, D.A.; Fraser, A. The Prevalence of Non-Alcoholic Fatty Liver Disease in Children and Adolescents: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0140908. [Google Scholar] [CrossRef] [Green Version]
- Di Sessa, A.; Umano, G.R.; Cirillo, G.; Marzuillo, P.; Arienzo, M.R.; Pedullà, M.; del Giudice, E.M. The rs72613567:TA Variant in the Hydroxysteroid 17-beta Dehydrogenase 13 Gene Reduces Liver Damage in Obese Children. J. Pediatr. Gastroenterol. Nutr. 2020, 70, 371–374. [Google Scholar] [CrossRef]
- About, F.; Abel, L.; Cobat, A. HCV-Associated Liver Fibrosis and HSD17B13. N. Engl. J. Med. 2018, 379, 1875–1876. [Google Scholar] [CrossRef]
- Yang, J.; Trépo, E.; Nahon, P.; Cao, Q.; Moreno, C.; Letouzé, E.; Imbeaud, S.; Bayard, Q.; Gustot, T.; Deviere, J.; et al. A 17-Beta-Hydroxysteroid Dehydrogenase 13 Variant Protects from Hepatocellular Carcinoma Development in Alcoholic Liver Disease. Hepatology 2019, 70, 231–240. [Google Scholar] [CrossRef]
- Enomoto, H.; Aizawa, N.; Hasegawa, K.; Ikeda, N.; Sakai, Y.; Yoh, K.; Takata, R.; Yuri, Y.; Kishino, K.; Shimono, Y.; et al. Possible Relevance of PNPLA3 and TLL1 Gene Polymorphisms to the Efficacy of PEG-IFN Therapy for HBV-Infected Patients. Int. J. Mol. Sci. 2020, 21, 3089. [Google Scholar] [CrossRef]
- Fukuhara, T.; Wada, M.; Nakamura, S.; Ono, C.; Shiokawa, M.; Yamamoto, S.; Motomura, T.; Okamoto, T.; Okuzaki, D.; Yamamoto, M.; et al. Amphipathic α-Helices in Apolipoproteins Are Crucial to the Formation of Infectious Hepatitis C Virus Particles. PLoS Pathog. 2014, 10, e1004534. [Google Scholar] [CrossRef]
- Gellert-Kristensen, H.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Stender, S. High Risk of Fatty Liver Disease Amplifies the Alanine Transaminase-Lowering Effect of a HSD17B13 Variant. Hepatology 2020, 71, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Stickel, F.; Lutz, P.; Buch, S.; Nischalke, H.D.; Silva, I.; Rausch, V.; Fischer, J.; Weiss, K.H.; Gotthardt, D.; Rosendahl, J.; et al. Genetic Variation in HSD17B13 Reduces the Risk of Developing Cirrhosis and Hepatocellular Carcinoma in Alcohol Misusers. Hepatology 2020, 72, 88–102. [Google Scholar] [CrossRef]
- De Benedittis, C.; Bellan, M.; Crevola, M.; Boin, E.; Barbaglia, M.N.; Mallela, V.R.; Ravanini, P.; Ceriani, E.; Fangazio, S.; Sainaghi, P.P.; et al. Interplay of PNPLA3 and HSD17B13 Variants in Modulating the Risk of Hepatocellular Carcinoma among Hepatitis C Patients. Gastroenterol. Res. Pract. 2020, 2020, 4216451. [Google Scholar]
- Wang, X.; Liao, X.; Yang, C.; Huang, K.; Yu, T.; Yu, L.; Han, C.; Zhu, G.; Zeng, X.; Liu, Z.; et al. Identification of prognostic biomarkers for patients with hepatocellular carcinoma after hepatectomy. Oncol. Rep. 2019, 41, 1586–1602. [Google Scholar] [CrossRef]
- Chen, J.; Zhuo, J.-Y.; Yang, F.; Liu, Z.-K.; Zhou, L.; Xie, H.-Y.; Xu, X.; Zheng, S.-S. 17-beta-hydroxysteroid dehydrogenase 13 inhibits the progression and recurrence of hepatocellular carcinoma. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 220–226. [Google Scholar] [CrossRef]
- Rotroff, D.; Pijut, S.S.; Marvel, S.W.; Jack, J.R.; Havener, T.; Pujol, A.; Schluter, A.; Graf, G.A.; Ginsberg, H.N.; Shah, H.S.; et al. Genetic Variants in HSD17B3, SMAD3, and IPO11 Impact Circulating Lipids in Response to Fenofibrate in Individuals with Type 2 Diabetes. Clin. Pharmacol. Ther. 2018, 103, 712–721. [Google Scholar] [CrossRef]
- Xu, L.; Jiang, C.Q.; Lam, T.H.; Zhang, W.S.; Zhu, F.; Jin, Y.L.; Thomas, G.N.; Cheng, K.K.; Schooling, C.M. Mendelian randomization estimates of alanine aminotransferase with cardiovascular disease: Guangzhou Biobank Cohort study. Hum. Mol. Genet. 2016, 26, 430–437. [Google Scholar] [CrossRef]
- Di Sessa, A.; Umano, G.R.; Cirillo, G.; Passaro, A.P.; Verde, V.; Cozzolino, D.; Guarino, S.; Marzuillo, P.; Del Giudice, E.M. Pediatric non-alcoholic fatty liver disease and kidney function: Effect of HSD17B13 variant. World J. Gastroenterol. 2020, 26, 5474–5483. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; LaVine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Mathurin, P.; Hollebecque, A.; Arnalsteen, L.; Buob, D.; Leteurtre, E.; Caiazzo, R.; Pigeyre, M.; Verkindt, H.; Dharancy, S.; Louvet, A.; et al. Prospective Study of the Long-Term Effects of Bariatric Surgery on Liver Injury in Patients without Advanced Disease. Gastroenterology 2009, 137, 532–540. [Google Scholar] [CrossRef]
- Lassailly, G.; Caiazzo, R.; Buob, D.; Pigeyre, M.; Verkindt, H.; Labreuche, J.; Raverdy, V.; Leteurtre, E.; Dharancy, S.; Louvet, A.; et al. Bariatric Surgery Reduces Features of Nonalcoholic Steatohepatitis in Morbidly Obese Patients. Gastroenterology 2015, 149, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Beer, N.L.; Tribble, N.D.; McCulloch, L.J.; Roos, C.; Johnson, P.R.; Orho-Melander, M.; Gloyn, A.L. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum. Mol. Genet. 2009, 18, 4081–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirazzi, C.; Adiels, M.; Burza, M.A.; Mancina, R.M.; Levin, M.; Ståhlman, M.; Taskinen, M.-R.; Orho-Melander, M.; Perman, J.; Pujia, A.; et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J. Hepatol. 2012, 57, 1276–1282. [Google Scholar] [CrossRef]
- Ma, Y.; Brown, P.M.; Lin, D.D.; Ma, J.; Feng, D.; Belyaeva, O.V.; Podszun, M.C.; Roszik, J.; Allen, J.N.; Umarova, R.; et al. 17-Beta Hydroxysteroid Dehydrogenase 13 Deficiency Does Not Protect Mice from Obesogenic Diet Injury. Hepatology 2021, 73, 1701–1716. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene Editing ofCCR5in Autologous CD4 T Cells of Persons Infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-X.; Zhang, Y.; Yin, H. Genome Editing with mRNA Encoding ZFN, TALEN, and Cas9. Mol. Ther. 2019, 27, 735–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltys, K.A.; Setoyama, K.; Tafaleng, E.N.; Gutiérrez, A.S.; Fong, J.; Fukumitsu, K.; Nishikawa, T.; Nagaya, M.; Sada, R.; Haberman, K.; et al. Host conditioning and rejection monitoring in hepatocyte transplantation in humans. J. Hepatol. 2017, 66, 987–1000. [Google Scholar] [CrossRef] [Green Version]
- Takeishi, K.; De L’Hortet, A.C.; Wang, Y.; Handa, K.; Guzman-Lepe, J.; Matsubara, K.; Morita, K.; Jang, S.; Haep, N.; Florentino, R.M.; et al. Assembly and Function of a Bioengineered Human Liver for Transplantation Generated Solely from Induced Pluripotent Stem Cells. Cell Rep. 2020, 31, 107711. [Google Scholar] [CrossRef]
- Collin de l’Hortet, A.; Takeishi, K.; Guzman-Lepe, J.; Morita, K.; Achreja, A.; Popovic, B.; Wang, Y.; Handa, K.; Mittal, A.; Meurs, N.; et al. Generation of Human Fatty Livers Using Custom-Engineered Induced Pluripotent Stem Cells with Modifiable SIRT1 Metabolism. Cell Metab. 2019, 30, 385–401.e9. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene | SNP | MAF | NAFLD Allelic OR (95% CI) | ALD Allelic OR (95% CI) | HCC Allelic OR (95% CI) | Assumed Molecular Mechanism of the Variant |
|---|---|---|---|---|---|---|
| PNPLA3 | rs738409 (C > G) | 0.14 (Africans)– 0.57 (Hispanics) | 1.91 (1.64–2.21) | 2.19 (1.97–2.43) | 5.9 (1.5–23.8) | Repressor of lipase activity in hepatocyte [16] and altered retinol metabolism in stellate cell |
| TM6SF2 | rs5842926 (G > A) rs58542926 (C > T) rs10401969 (T > C) | 0.03 (Hispanics)– 0.08 (Europeans) | 1.82 (1.59–2.08) | – | 1.72 (1.27–2.38) | Loss of function of secretion of VLDL particle, leading lipid accumulation in the liver [18] |
| GCKR | rs1260326 (C > T) rs780094 (T > C) | 0.5 (Asians)– 0.86 (Africans) | 1.38 (1.25–1.53) | – | 1.84 (1.23–2.75) | Loss of affinity for glucokinase, leading to increased lipogenesis [20] |
| MBOAT7 | rs641738 (C > T) | 0.24 (Asians)– 0.42 (Europeans) | 1.42 (1.07–1.91) | 1.35 (1.23–1.49) | 2.10 (1.33–3.31) | Loss of remodeling of phosphatidylinositol, resulting in increased TG synthesis [23] |
| LEPR | rs12077210 (C > T) | 0.01 (Asians)– 0.28 (Africans) | 1.48 (1.29—1.71) | – | – | Loss of leptin receptor function [26] |
| HSD17B13 | rs72613567 (T > TA) rs6834314 (A > G) rs9992651 (G > A) rs3923441 (C > G) | 0.06 (Africans)– 0.34 (Asians) | 0.70 (0.57–0.87) | 0.47 (0.23–0.97) | 0.72 (0.66–0.79) | Loss of retinol dehydrogenase activity [27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motomura, T.; Amirneni, S.; Diaz-Aragon, R.; Faccioli, L.A.P.; Malizio, M.R.; Coard, M.C.; Kocas-Kilicarslan, Z.N.; Frau, C.; Haep, N.; Ostrowska, A.; et al. Is HSD17B13 Genetic Variant a Protector for Liver Dysfunction? Future Perspective as a Potential Therapeutic Target. J. Pers. Med. 2021, 11, 619. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11070619
Motomura T, Amirneni S, Diaz-Aragon R, Faccioli LAP, Malizio MR, Coard MC, Kocas-Kilicarslan ZN, Frau C, Haep N, Ostrowska A, et al. Is HSD17B13 Genetic Variant a Protector for Liver Dysfunction? Future Perspective as a Potential Therapeutic Target. Journal of Personalized Medicine. 2021; 11(7):619. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11070619
Chicago/Turabian StyleMotomura, Takashi, Sriram Amirneni, Ricardo Diaz-Aragon, Lanuza A. P. Faccioli, Michelle R. Malizio, Michael C. Coard, Zehra N. Kocas-Kilicarslan, Carla Frau, Nils Haep, Alina Ostrowska, and et al. 2021. "Is HSD17B13 Genetic Variant a Protector for Liver Dysfunction? Future Perspective as a Potential Therapeutic Target" Journal of Personalized Medicine 11, no. 7: 619. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11070619