Possible Spillover of Pathogens between Bee Communities Foraging on the Same Floral Resource

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Bee Sampling
2.2. Virus Detection
2.2.1. RNA Extraction
2.2.2. RT-PCR
2.2.3. Prevalence and Sequencing
2.3. Barcoding
2.4. Data Analysis
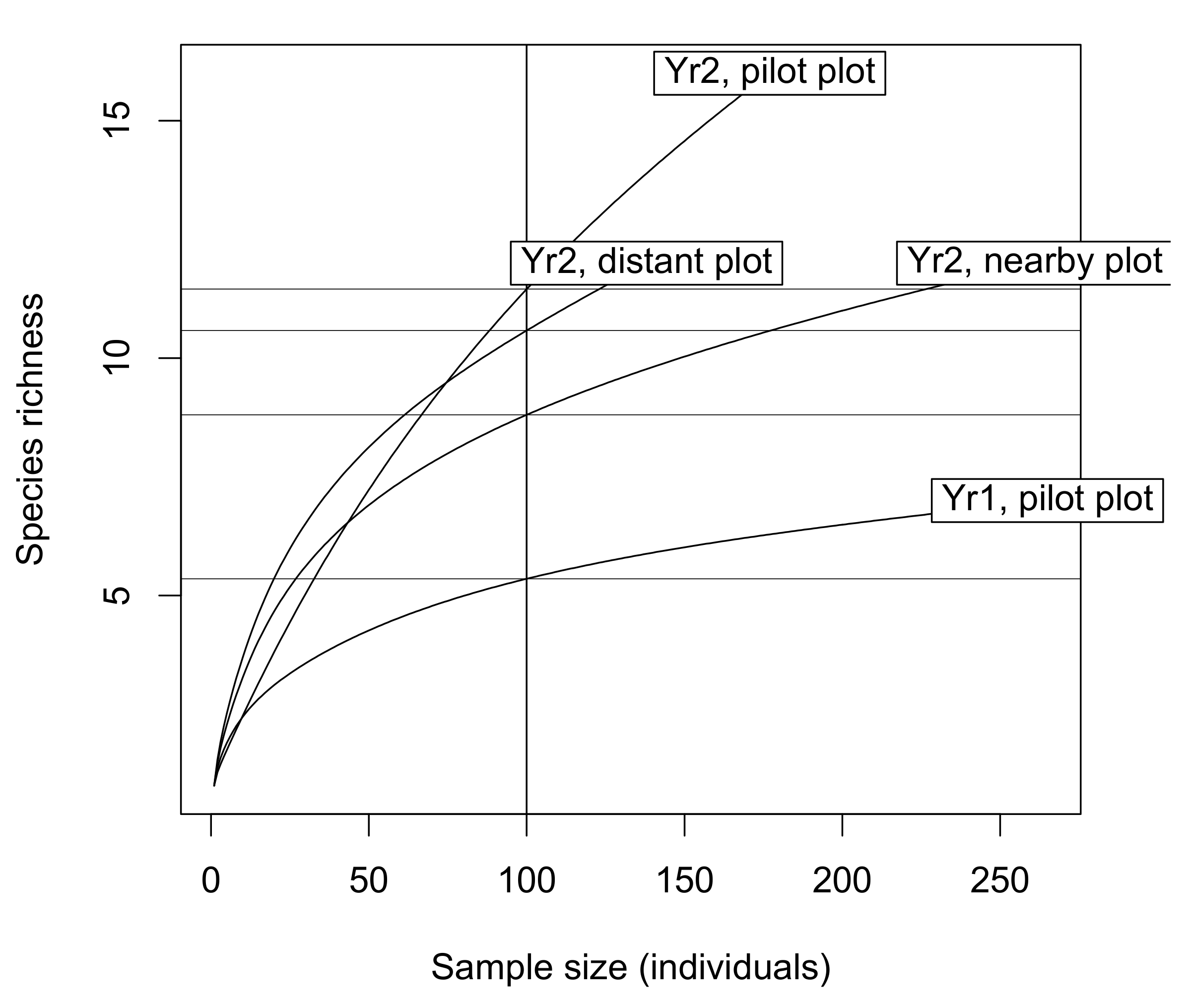
2.4.1. Characterizing the Sampled Wild Bee Communities
2.4.2. Virus Prevalence in Honey Bees and Wild Bees
- (i)
- Complete virus distribution model computation. We first fitted MGLMs for binomial presence-absence data with the traitglm function of the mvabund R package [84]. Complete models were computed, with all environmental variables, including: sampling plot identity and days since the onset of flowering, as well as taxonomic and functional assignations in the case of wild bee samples. The significance of the complete model was tested using a multivariate analysis of variance using 999 random permutations of samples among environmental variables.
- (ii)
- Backward stepwise model simplification. Whenever a significant environmental effect was detected, we further simplified the model by dropping non-significant environmental variables. We used the Akaike information criterion (AIC) to guide model simplification, considering a trade-off between model fit and complexity. Model simplification was iteratively pursued until only a subset of significant environmental variables was included. The resulting model was therefore viewed as the most parsimonious candidate model.
- (iii)
- Parsimonious model assessment. As a final model simplification step, we recomputed the most parsimonious model using the LASSO penalty function that automatically drops irrelevant virus × environment combinations explaining the overall variations of virus prevalence models. This algorithm permits further highlighting of the most salient virus prevalence patterns throughout the many possible combinations.
- (iv)
- Post-hoc univariate model confirmations. We are first and foremost interested in the potential rise of virus prevalence as time lapses since the onset of flowering. Whenever MGLMs detected such a temporal pattern, we performed posteriori univariate confirmatory analyses, focusing on each virus species of interest separately. To do so, we used a GLMM framework for binomial family date, specifying the number of days since the onset of flowering as a fixed variable, while controlling for the non-independency of samples proceeding from the same sampling plot and year using the plot identity as a random grouping variable.
3. Results
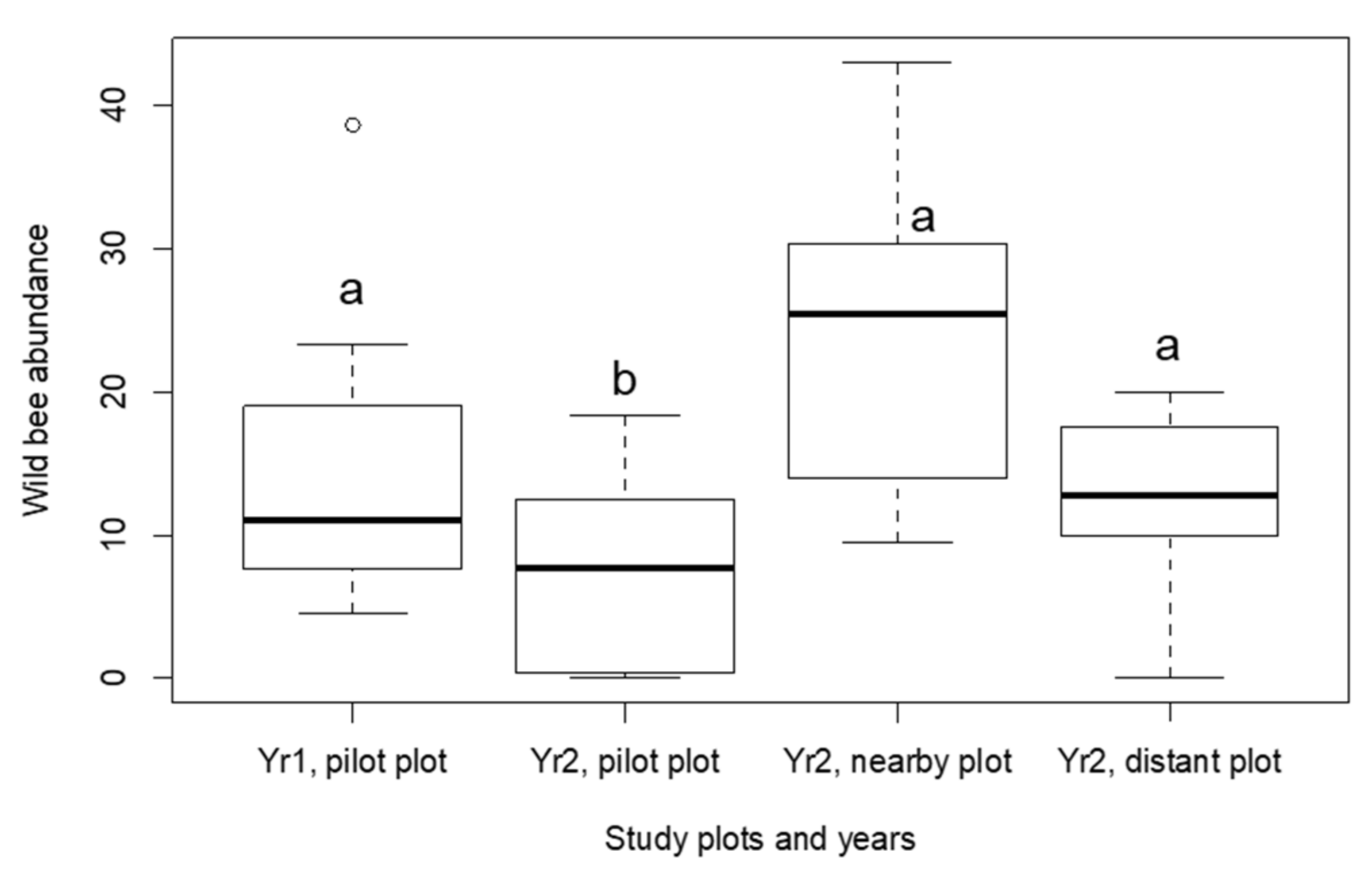
3.1. Characterising the Sampled Wild Bee Communities
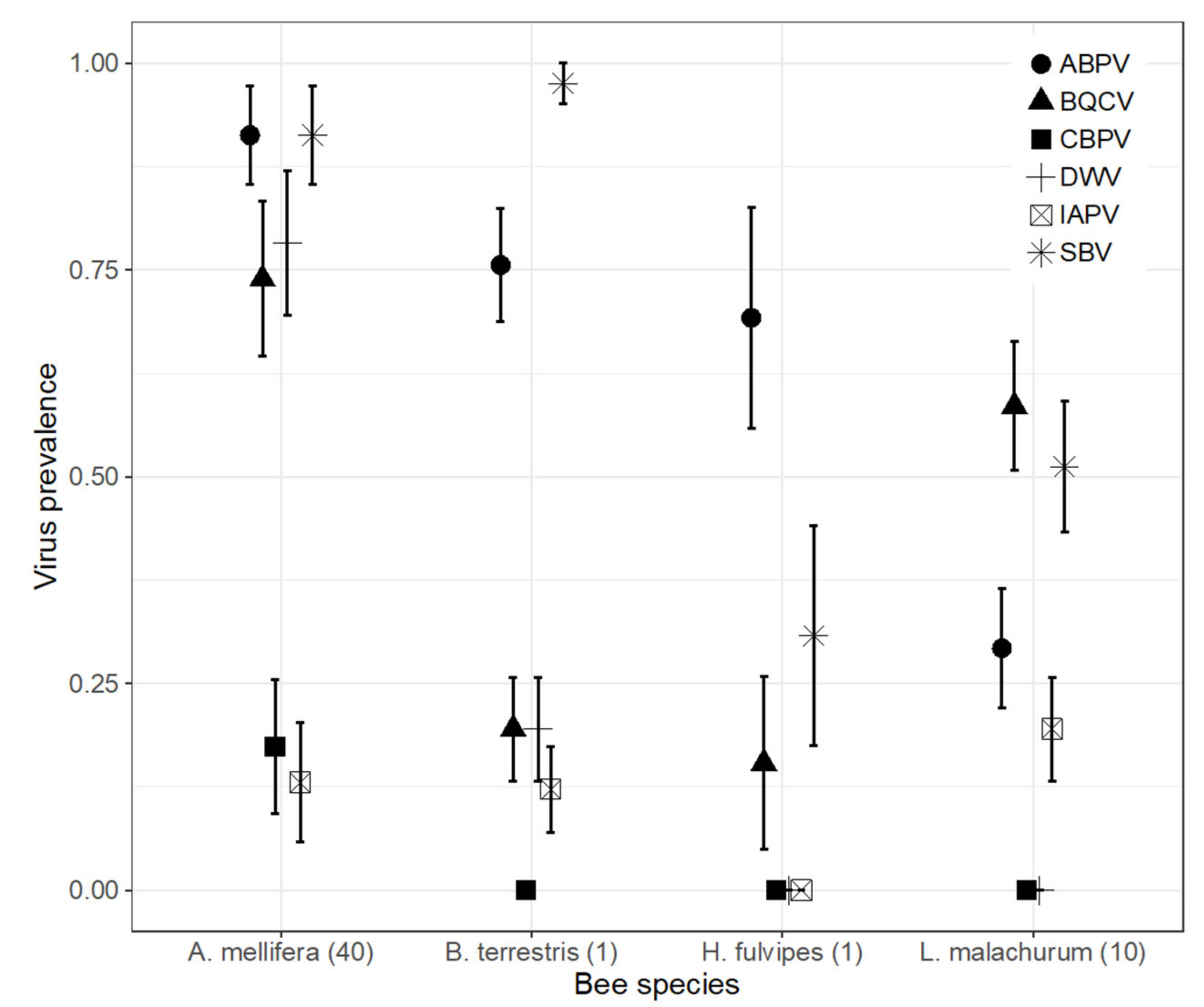
3.2. Virus Prevalence in Honey Bees and Wild Bees
3.2.1. SBV, BQCV, ABPV and IAPV Were Widespread in the Wild Bee Community
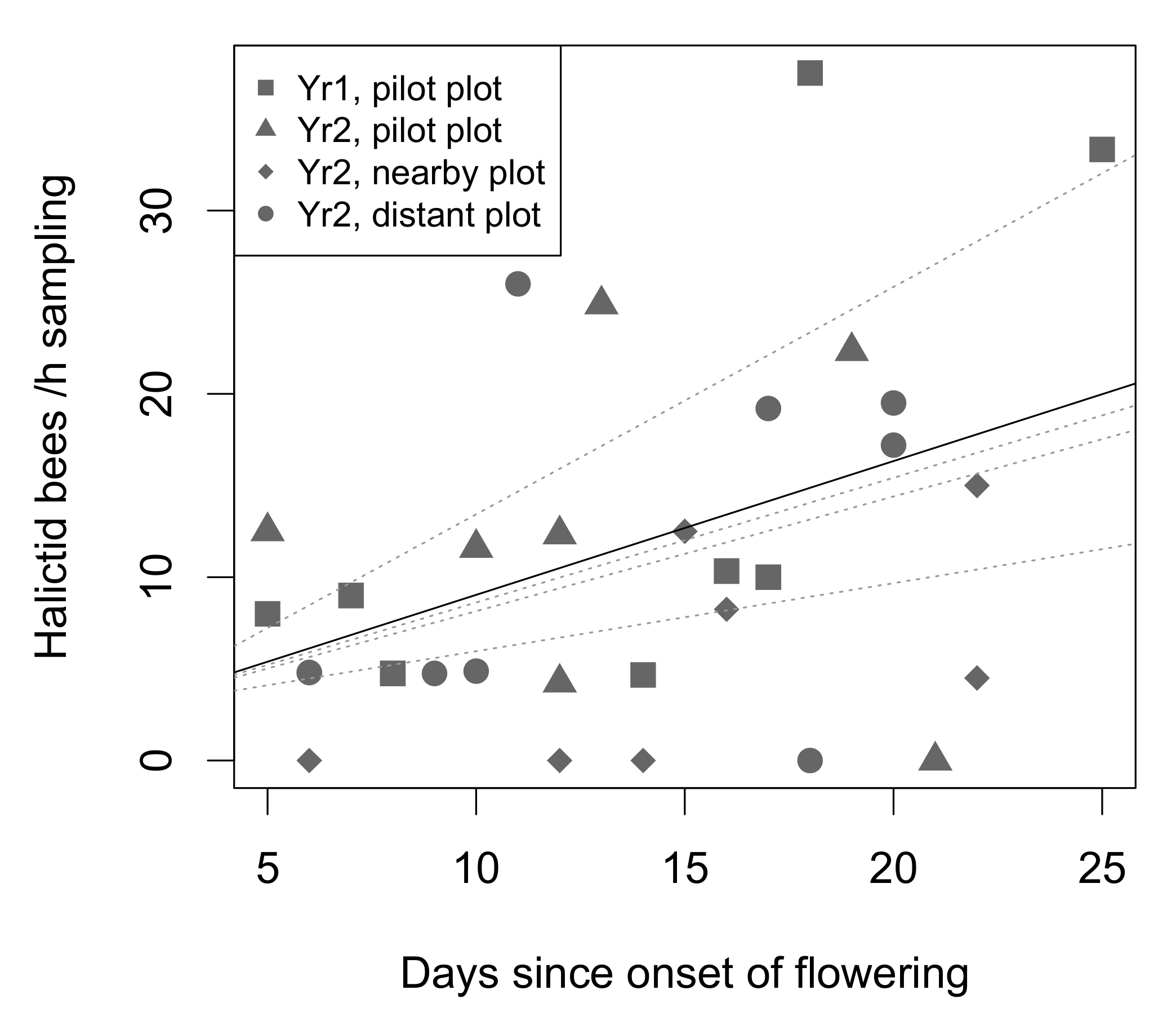
3.2.2. Drivers of Virus Prevalence in Honey Bees
3.2.3. Drivers of Virus Prevalence in Wild Bees
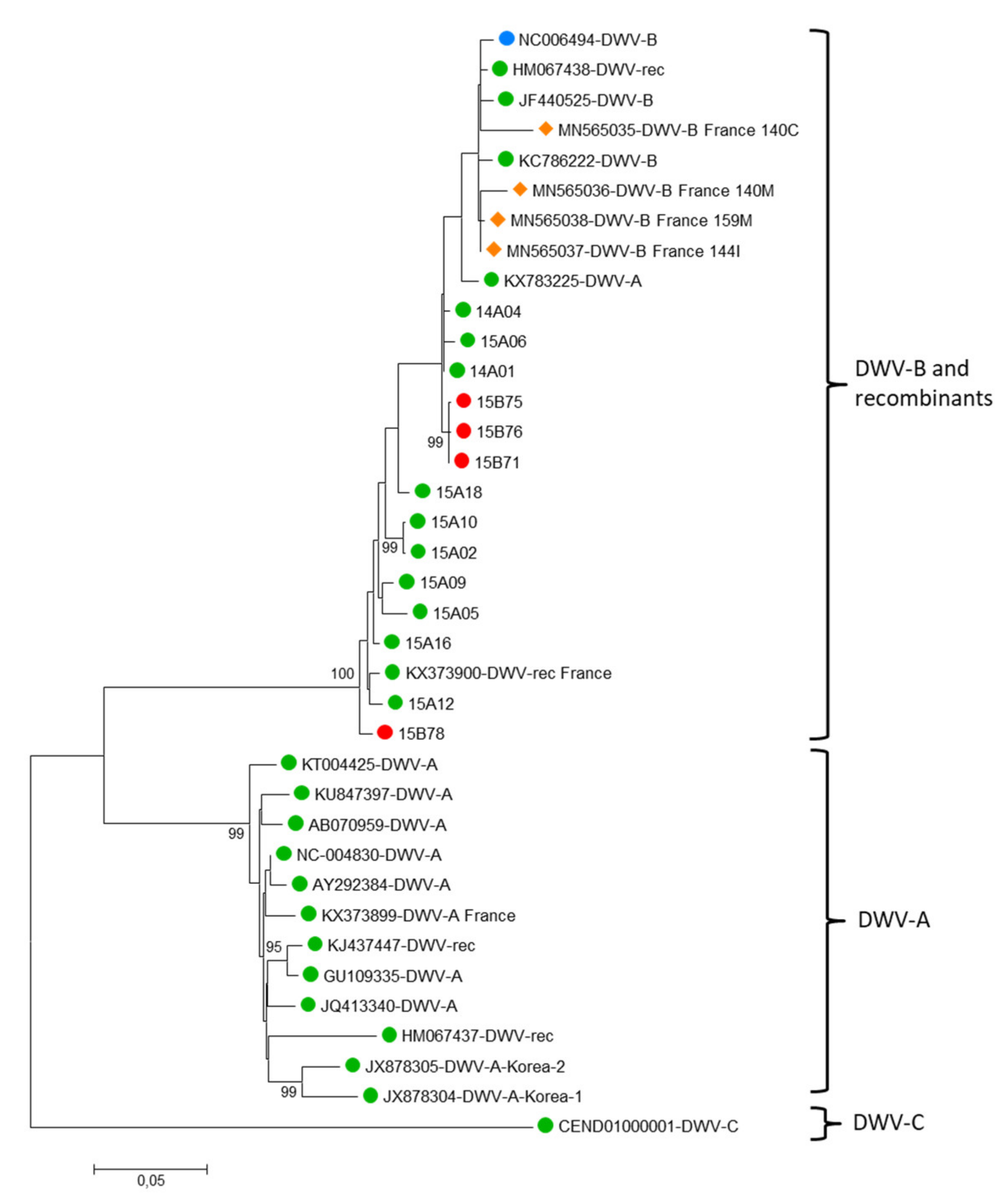
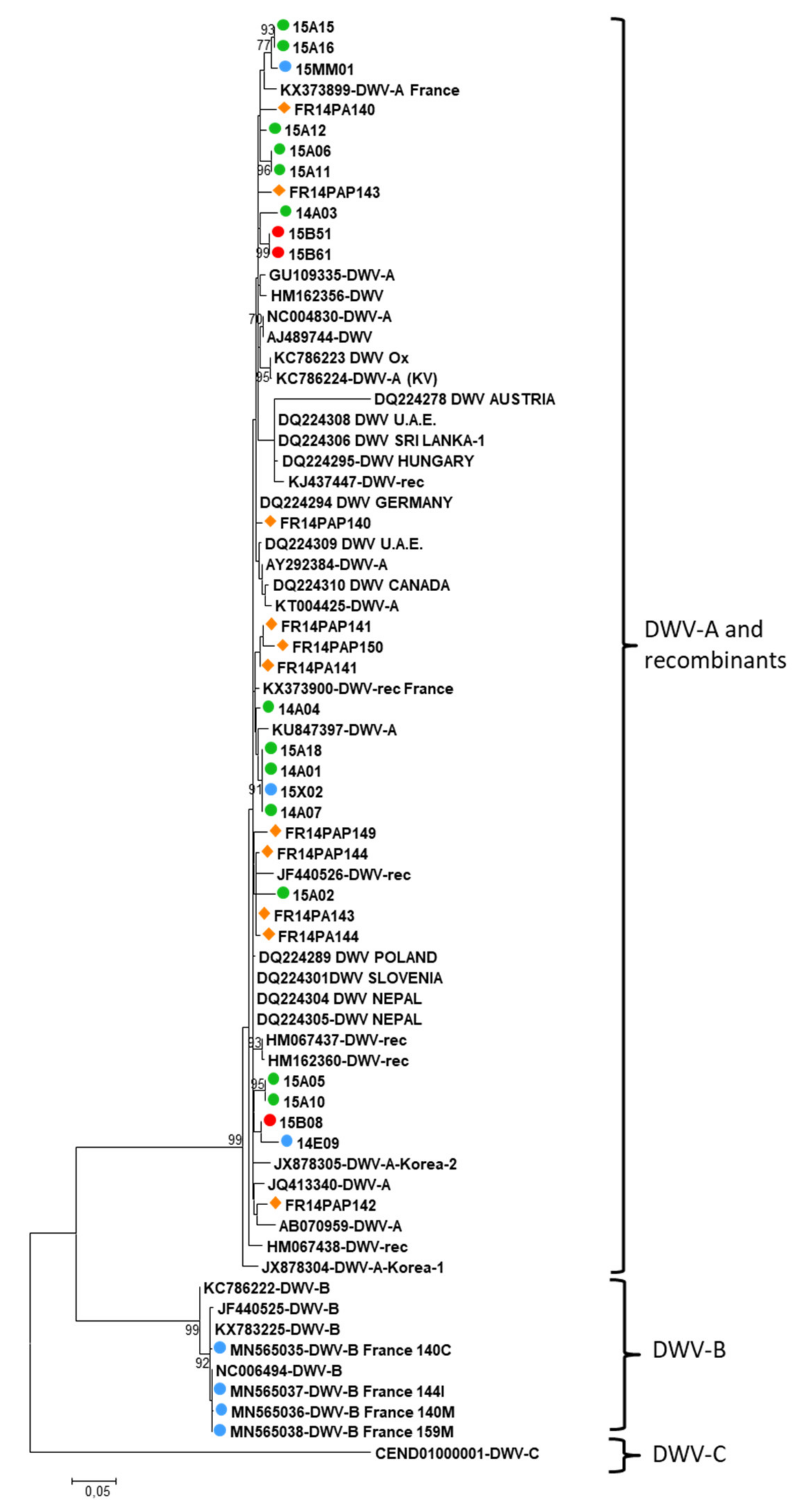
3.3. Virus Phylogenies
4. Discussion
4.1. Patterns of Virus Prevalence in Bees and Other Flower-Visiting Insects
4.2. Virus Prevalence Does not Provide Clear Insights on Possible Local Spillover Events
4.3. Virus Phylogenies Provide Insigths on Possible Spillover Events
4.4. Deficient Knowledge on Virus Replication, Transmission, and Pathogenicity in Wild Bees
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klein, A.-M.; Vaissière, B.E.; Cane, J.H.; Steffan-Dewenter, I.; Cunningham, S.A.; Kremen, C.; Tscharntke, T. Importance of pollinators in changing landscapes for world crops. Proc. R. Soc. B Biol. Sci. 2007, 274, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garibaldi, L.; Steffan-Dewenter, I.; Winfree, R.; Aizen, M.; Bommarco, R.; Cunningham, S.; Kremen, C.; Carvalheiro, L.; Harder, L.; Afik, O.; et al. Wild Pollinators Enhance Fruit Set of Crops Regardless of Honey Bee Abundance. Science (N. Y.) 2013, 339, 1608–1611. [Google Scholar] [CrossRef] [PubMed]
- Garibaldi, L.; Carvalheiro, L.; Vaissière, B.; Gemmill-Herren, B.; Hipólito, J.; Freitas, B.; Ngo, H.T.; Azzu, N.; Saez, A.; Åström, J.; et al. Mutually beneficial pollinator diversity and crop yield outcomes in small and large farms. Science 2016, 351, 388–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rader, R.; Bartomeus, I.; Garibaldi, L.A.; Garratt, M.P.D.; Howlett, B.G.; Winfree, R.; Cunningham, S.A.; Mayfield, M.M.; Arthur, A.D.; Andersson, G.K.S.; et al. Non-bee insects are important contributors to global crop pollination. Proc. Natl. Acad. Sci. USA 2016, 113, 146–151. [Google Scholar] [PubMed] [Green Version]
- Vanbergen, A.J.; Initiative, T.I.P. Threats to an ecosystem service: pressures on pollinators. Front. Ecol. Environ. 2013, 11, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Potts, S.G.; Roberts, S.P.M.; Dean, R.; Marris, G.; Brown, M.A.; Jones, R.; Neumann, P.; Settele, J. Declines of managed honey bees and beekeepers in Europe. J. Apic. Res. 2010, 49, 15–22. [Google Scholar] [CrossRef]
- Goulson, D.; Lye, G.C.; Darvill, B. Decline and Conservation of Bumble Bees. Annu. Rev. Entomol. 2008, 53, 191–208. [Google Scholar]
- Soroye, P.; Newbold, T.; Kerr, J. Climate change contributes to widespread declines among bumble bees across continents. Science 2020, 367, 685–688. [Google Scholar] [CrossRef]
- Cameron, S.A.; Lozier, J.D.; Strange, J.P.; Koch, J.B.; Cordes, N.; Solter, L.F.; Griswold, T.L. Patterns of widespread decline in North American bumble bees. Proc. Natl. Acad. Sci. USA 2011, 108, 662–667. [Google Scholar]
- Biesmeijer, J.C.; Roberts, S.P.; Reemer, M.; Ohlemüller, R.; Edwards, M.; Peeters, T.; Schaffers, A.P.; Potts, S.G.; Kleukers, R.; Thomas, C.D.; et al. Parallel declines in pollinators and insect-pollinated plants in Britain and the Netherlands. Science 2006, 313, 351–354. [Google Scholar] [CrossRef]
- Koh, I.; Lonsdorf, E.V.; Williams, N.M.; Brittain, C.; Isaacs, R.; Gibbs, J.; Ricketts, T.H. Modeling the status, trends, and impacts of wild bee abundance in the United States. Proc. Natl. Acad. Sci. USA 2016, 113, 140–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, J.T.; Pindar, A.; Galpern, P.; Packer, L.; Potts, S.G.; Roberts, S.M.; Rasmont, P.; Schweiger, O.; Colla, S.R.; Richardson, L.L.; et al. Climate change impacts on bumblebees converge across continents. Science 2015, 349, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Carreck, N.L. Honey bee colony losses. J. Apic. Res. 2010, 49, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Herrera, C.M. Gradual replacement of wild bees by honeybees in flowers of the Mediterranean Basin over the last 50 years. Proc. R. Soc. B: Biol. Sci. 2020, 287, 20192657. [Google Scholar] [CrossRef] [Green Version]
- Baldock, K.C.R. Opportunities and threats for pollinator conservation in global towns and cities. Curr. Opin. Insect Sci. 2020, 38, 63–71. [Google Scholar] [CrossRef]
- Fortel, L.; Henry, M.; Guilbaud, L.; Guirao, A.L.; Kuhlmann, M.; Mouret, H.; Rollin, O.; Vaissière, B.E. Decreasing Abundance, Increasing Diversity and Changing Structure of the Wild Bee Community (Hymenoptera: Anthophila) along an Urbanization Gradient. PLoS ONE 2014, 9, e104679. [Google Scholar] [CrossRef]
- Geslin, B.; Le Féon, V.; Folschweiller, M.; Flacher, F.; Carmignac, D.; Motard, E.; Perret, S.; Dajoz, I. The proportion of impervious surfaces at the landscape scale structures wild bee assemblages in a densely populated region. Ecol. Evol. 2016, 6, 6599–6615. [Google Scholar] [CrossRef]
- Poquet, Y.; Vidau, C.; Alaux, C. Modulation of pesticide response in honeybees. Apidologie 2016, 47, 412–426. [Google Scholar] [CrossRef] [Green Version]
- Whitehorn, P.R.; O’Connor, S.; Wackers, F.L.; Goulson, D. Neonicotinoid Pesticide Reduces Bumble Bee Colony Growth and Queen Production. Science 2012, 336, 351–352. [Google Scholar] [CrossRef] [Green Version]
- Rundlöf, M.; Andersson, G.K.S.; Bommarco, R.; Fries, I.; Hederström, V.; Herbertsson, L.; Jonsson, O.; Klatt, B.K.; Pedersen, T.R.; Yourstone, J.; et al. Seed coating with a neonicotinoid insecticide negatively affects wild bees. Nature 2015, 521, 77–80. [Google Scholar] [CrossRef]
- Beaurepaire, A.; Piot, N.; Doublet, V.; Antunez, K.; Campbell, E.; Chantawannakul, P.; Chejanovsky, N.; Gajda, A.; Heerman, M.; Panziera, D.; et al. Diversity and Global Distribution of Viruses of the Western Honey Bee, Apis mellifera. Insects 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Dainat, B.; Evans, J.D.; Chen, Y.P.; Gauthier, L.; Neumann, P. Dead or alive: deformed wing virus and Varroa destructor reduce the life span of winter honeybees. Appl. Environ. Microbiol. 2012, 78, 981–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, R.M.; Nielsen, S.L.; Kryger, P. Varroa-virus interaction in collapsing honey bee colonies. PLoS ONE 2013, 8, e57540. [Google Scholar] [CrossRef] [Green Version]
- Genersch, E.; Ohe, W.v.d.; Kaatz, H.; Schroeder, A.; Otten, C.; Buchler, R.; Berg, S.; Ritter, W.; Muhlen, W.; Gisder, S.; et al. The German bee monitoring project: A long term study to understand periodically high winter losses of honey bee colonies. Apidologie 2010, 41, 332–352. [Google Scholar] [CrossRef] [Green Version]
- Kielmanowicz, M.G.; Inberg, A.; Lerner, I.M.; Golani, Y.; Brown, N.; Turner, C.L.; Hayes, G.J.R.; Ballam, J.M. Prospective Large-Scale Field Study Generates Predictive Model Identifying Major Contributors to Colony Losses. PLoS Pathog. 2015, 11, e1004816. [Google Scholar] [CrossRef] [Green Version]
- Traynor, K.S.; Rennich, K.; Forsgren, E.; Rose, R.; Pettis, J.; Kunkel, G.; Madella, S.; Evans, J.; Lopez, D.; vanEngelsdorp, D. Multiyear survey targeting disease incidence in US honey bees. Apidologie 2016, 47, 325–347. [Google Scholar]
- Desai, S.D.; Currie, R.W. Effects of Wintering Environment and Parasite-Pathogen Interactions on Honey Bee Colony Loss in North Temperate Regions. PLoS ONE 2016, 11, e0159615. [Google Scholar] [CrossRef] [Green Version]
- Mordecai, G.J.; Wilfert, L.; Martin, S.J.; Jones, I.M.; Schroeder, D.C. Diversity in a honey bee pathogen: first report of a third master variant of the Deformed Wing Virus quasispecies. J. ISME 2016, 10, 1264–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, D.P.; Natsopoulou, M.E.; Doublet, V.; Fürst, M.; Weging, S.; Brown, M.J.F.; Gogol-Döring, A.; Paxton, R.J. Elevated virulence of an emerging viral genotype as a driver of honeybee loss. Proc. R. Soc. B: Biol. Sci. 2016, 283, 20160811. [Google Scholar]
- Gisder, S.; Möckel, N.; Eisenhardt, D.; Genersch, E. In vivo evolution of viral virulence: switching of deformed wing virus between hosts results in virulence changes and sequence shifts. Environ. Microbiol. 2018, 20, 4612–4628. [Google Scholar] [CrossRef] [Green Version]
- Tehel, A.; Streicher, T.; Tragust, S.; Paxton, R.J. Experimental infection of bumblebees with honeybee-associated viruses: no direct fitness costs but potential future threats to novel wild bee hosts. R. Soc. Open Sci. 2020, 7, 200480. [Google Scholar] [CrossRef] [PubMed]
- Ryabov, E.V.; Childers, A.K.; Chen, Y.; Madella, S.; Nessa, A.; vanEngelsdorp, D.; Evans, J.D. Recent spread of Varroa destructor virus-1, a honey bee pathogen, in the United States. Sci. Rep. 2017, 7, 17447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryabov, E.V.; Childers, A.K.; Lopez, D.; Grubbs, K.; Posada-Florez, F.; Weaver, D.; Girten, W.; van Engelsdorp, D.; Chen, Y.P.; Evans, J.D. Dynamic evolution in the key honey bee pathogen deformed wing virus: Novel insights into virulence and competition using reverse genetics. PLoS Biol. 2019, 17, e3000502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kevill, J.L.; de Souza, F.S.; Sharples, C.; Oliver, R.; Schroeder, D.C.; Martin, S.J. DWV-A Lethal to Honey Bees (Apis mellifera): A Colony Level Survey of DWV Variants (A, B, and C) in England, Wales, and 32 States across the US. Viruses 2019, 11, 426. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.; Jironkin, A.; Chandler, D.; Burroughs, N.; Evans, D.J.; Ryabov, E.V. Recombinants between Deformed wing virus and Varroa destructor virus-1 may prevail in Varroa destructor-infested honeybee colonies. J. Gen. Virol. 2011, 92, 156–161. [Google Scholar] [CrossRef]
- Ryabov, E.V.; Wood, G.R.; Fannon, J.M.; Moore, J.D.; Bull, J.C.; Chandler, D.; Mead, A.; Burroughs, N.; Evans, D.J. A Virulent Strain of Deformed Wing Virus (DWV) of Honeybees (Apis mellifera) Prevails after Varroa destructor-Mediated, or In Vitro, Transmission. PLoS Pathog. 2014, 10, e1004230. [Google Scholar] [CrossRef] [Green Version]
- Zioni, N.; Soroker, V.; Chejanovsky, N. Replication of Varroa destructor virus 1 (VDV-1) and a Varroa destructor virus 1-deformed wing virus recombinant (VDV-1-DWV) in the head of the honey bee. Virology 2011, 417, 106–112. [Google Scholar]
- Dalmon, A.; Desbiez, C.; Coulon, M.; Thomasson, M.; Conte, Y.L.; Alaux, C.; Vallon, J.; Moury, B. Evidence for positive selection and recombination hotspots in Deformed wing virus (DWV). Sci. Rep. 2017, 7, 41045. [Google Scholar] [CrossRef]
- Fei, D.; Guo, Y.; Fan, Q.; Wang, H.; Wu, J.; Li, M.; Ma, M. Phylogenetic and recombination analyses of two deformed wing virus strains from different honeybee species in China. PeerJ 2019, 7, e7214. [Google Scholar] [CrossRef]
- Manley, R.; Temperton, B.; Doyle, T.; Gates, D.; Hedges, S.; Boots, M.; Wilfert, L. Knock-on community impacts of a novel vector: spillover of emerging DWV-B from Varroa-infested honeybees to wild bumblebees. Ecol. Lett. 2019, 22, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- De Miranda, J.R.; Cordoni, G.; Budge, G. The Acute bee paralysis virus–Kashmir bee virus–Israeli acute paralysis virus complex. J. Invertebr. Pathol. 2010, 103, S30–S47. [Google Scholar] [CrossRef] [PubMed]
- Manley, R.; Boots, M.; Wilfert, L. Emerging viral disease risk to pollinating insects: ecological, evolutionary and anthropogenic factors. J. Appl. Ecol. 2015, 52, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Tehel, A.; Brown, M.J.F.; Paxton, R.J. Impact of managed honey bee viruses on wild bees. Curr. Opin. Virol. 2016, 19, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genersch, E.; Yue, C.; Fries, I.; de Miranda, J.R. Detection of Deformed wing virus, a honey bee viral pathogen, in bumble bees (Bombus terrestris and Bombus pascuorum) with wing deformities. J. Invertebr. Pathol. 2006, 91, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Dalmon, A.; Gayral, P.; Decante, D.; Klopp, C.; Bigot, D.; Thomasson, M.; Herniou, E.A.; Alaux, C.; Le Conte, Y. Viruses in the Invasive Hornet Vespa velutina. Viruses 2019, 11, 1041. [Google Scholar] [CrossRef] [Green Version]
- Levitt, A.L.; Singh, R.; Cox-Foster, D.L.; Rajotte, E.; Hoover, K.; Ostiguy, N.; Holmes, E.C. Cross-species transmission of honey bee viruses in associated arthropods. Virus Res. 2013, 176, 232–240. [Google Scholar] [CrossRef]
- Furst, M.A.; McMahon, D.P.; Osborne, J.L.; Paxton, R.J.; Brown, M.J.F. Disease associations between honeybees and bumblebees as a threat to wild pollinators. Nature 2014, 506, 364–366. [Google Scholar] [CrossRef]
- Singh, R.; Levitt, A.L.; Rajotte, E.G.; Holmes, E.C.; Ostiguy, N.; Vanengelsdorp, D.; Lipkin, W.I.; Depamphilis, C.W.; Toth, A.L.; Cox-Foster, D.L. RNA Viruses in Hymenopteran Pollinators: Evidence of Inter-Taxa Virus Transmission via Pollen and Potential Impact on Non-Apis Hymenopteran Species. PLoS ONE 2010, 5, e14357. [Google Scholar]
- Guzman-Novoa, E.; Hamiduzzaman, M.M.; Anguiano-Baez, R.; Correa-Benitez, A.; Castaneda-Cervantes, E.; Arnold, N.I. First detection of honey bee viruses in stingless bees in North America. J. Apic. Res. 2015, 54, 93–95. [Google Scholar] [CrossRef]
- Yañez, O.; Piot, N.; Dalmon, A.; de Miranda, J.R.; Chantawannakul, P.; Panziera, D.; Amiri, E.; Smagghe, G.; Schroeder, D.; Chejanovsky, N. Bee Viruses: Routes of Infection in Hymenoptera. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Gisder, S.; Genersch, E. Viruses of commercialized insect pollinators. J. Invertebr. Pathol. 2017, 147, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, M.; Carrozza, M.L.; Luisi, E.; Forzan, M.; Giusti, M.; Sagona, S.; Tolari, F.; Felicioli, A. Infectivity of DWV Associated to Flower Pollen: Experimental Evidence of a Horizontal Transmission Route. PLoS ONE 2014, 9, e113448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, R.S.; Kevill, J.L.; Correia-Aveira, M.E.; de Carvalho, C.A.L.; Martin, S.J. Occurrence of deformed wing virus variants in the stingless bee Melipona subnitida and honey bee Apis mellifera populations in Brazil. J. Gen. Virol. 2019, 100, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Meeus, I.; de Miranda, J.R.; de Graaf, D.C.; Wäckers, F.; Smagghe, G. Effect of oral infection with Kashmir bee virus and Israeli acute paralysis virus on bumblebee (Bombus terrestris) reproductive success. J. Invertebr. Pathol. 2014, 121, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Li, J.; Boncristiani, H.; Strange, J.P.; Hamilton, M.; Chen, Y. Host range expansion of honey bee Black Queen Cell Virus in the bumble bee, Bombus huntii. Apidologie 2011, 42, 650–658. [Google Scholar] [CrossRef] [Green Version]
- Ravoet, J.; De Smet, L.; Meeus, I.; Smagghe, G.; Wenseleers, T.; de Graaf, D.C. Widespread occurrence of honey bee pathogens in solitary bees. J. Invertebr. Pathol. 2014, 122, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Celle, O.; Blanchard, P.; Olivier, V.; Schuff, F.; Cougoule, N.; Faucon, J.P.; Ribiere, M. Detection of Chronic bee paralysis virus (CBPV) genome and its replicative RNA form in various hosts and possible ways of spread. Virus Res. 2008, 133, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Dobelmann, J.; Felden, A.; Lester, P.J. Genetic Strain Diversity of Multi-Host RNA Viruses that Infect a Wide Range of Pollinators and Associates is Shaped by Geographic Origins. Viruses-Basel 2020, 12, 358. [Google Scholar]
- Schlappi, D.; Chejanovsky, N.; Yanez, O.; Neumann, P. Foodborne Transmission and Clinical Symptoms of Honey Bee Viruses in Ants Lasius spp. Viruses-Basel 2020, 12, 321. [Google Scholar] [CrossRef] [Green Version]
- Schläppi, D.; Lattrell, P.; Yañez, O.; Chejanovsky, N.; Neumann, P. Foodborne Transmission of Deformed Wing Virus to Ants (Myrmica rubra). Insects 2019, 10, 394. [Google Scholar] [CrossRef] [Green Version]
- Alger, S.A.; Burnham, P.A.; Brody, A.K. Flowers as viral hot spots: Honey bees (Apis mellifera) unevenly deposit viruses across plant species. PLoS ONE 2019, 14, e0221800. [Google Scholar] [CrossRef] [PubMed]
- Graystock, P.; Goulson, D.; Hughes, W.O.H. Parasites in bloom: flowers aid dispersal and transmission of pollinator parasites within and between bee species. Proc. R. Soc. B Biol. Sci. 2015, 282, 20151371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazzi, F.; Le Conte, Y. Ecology of Varroa destructor, the Major Ectoparasite of the Western Honey Bee, Apis mellifera. Annu. Rev. Entomol. 2016, 61, 417–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribiere, M.; Lallemand, P.; Iscache, A.L.; Schurr, F.; Celle, O.; Blanchard, P.; Olivier, V.; Faucon, J.P. Spread of infectious chronic bee paralysis virus by honeybee (Apis mellifera L.) feces. Appl. Environ. Microbiol. 2007, 73, 7711–7716. [Google Scholar] [CrossRef] [Green Version]
- Whitton, J.L.; Cornell, C.T.; Feuer, R. Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 2005, 3, 765–776. [Google Scholar] [CrossRef]
- Chen, Y.; Evans, J.; Feldlaufer, M. Horizontal and vertical transmission of viruses in the honey bee, Apis mellifera. J. Invertebr. Pathol. 2006, 92, 152–159. [Google Scholar] [CrossRef]
- Schittny, D.; Yañez, O.; Neumann, P. Honey Bee Virus Transmission via Hive Products. Vet. Sci. 2020, 7, 96. [Google Scholar] [CrossRef]
- Yanez, O.; Zheng, H.Q.; Hu, F.L.; Neumann, P.; Dietemann, V. A scientific note on Israeli acute paralysis virus infection of Eastern honeybee Apis cerana and vespine predator Vespa velutina. Apidologie 2012, 43, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Mutien, G.; Bernard, T.; Noëmie El, A.; Daniel, C.; Gautier, G.; Marie, H.; Daniel, D.; Georges, D.; Annick, L.; Frédéric, F.; et al. Moku Virus in Invasive Asian Hornets, Belgium, 2016. J. Emerg. Infect. Dis. 2017, 23, 2109. [Google Scholar]
- Yang, S.; Gayral, P.; Zhao, H.; Wu, Y.; Jiang, X.; Bigot, D.; Wang, X.; Yang, D.; Herniou, E.; Deng, S.; et al. Occurrence and Molecular Phylogeny of Honey Bee Viruses in Vespids. Viruses 2020, 12, 6. [Google Scholar]
- Loope, K.J.; Baty, J.W.; Lester, P.J.; Rankin, E.E.W. Pathogen shifts in a honeybee predator following the arrival of the Varroa mite. Proc. R. Soc. B: Biol. Sci. 2019, 286, 20182499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, M.; Froechen, M.; Maillet-Mezeray, J.; Breyne, E.; Allier, F.; Odoux, J.-F.; Decourtye, A. Spatial autocorrelation in honeybee foraging activity reveals optimal focus scale for predicting agro-environmental scheme efficiency. Ecol. Model. 2012, 225, 103–114. [Google Scholar] [CrossRef]
- Ion, N.; Odoux, J.F.; Vaissière, B. Melliferous potential of weedy herbaceous plants in crop fields of Romania from 1949 to 2012. J. Apic. Sci. 2018, 62, 149–165. [Google Scholar] [CrossRef] [Green Version]
- Petanidou, T. Introducing plants for bee-keeping at any cost? – Assessment of Phacelia tanacetifolia as nectar source plant under xeric Mediterranean conditions. Plant Syst. Evol. 2003, 238, 155–168. [Google Scholar] [CrossRef]
- Williams, I.H.; Christian, D.G. Observations on Phacelia tanacetifolia Bentham (Hydrophyllaceae) as a food plant for honey bees and bumble bees. J. Apic. Res. 1991, 30, 3–12. [Google Scholar] [CrossRef]
- Westphal, C.; Bommarco, R.; Carré, G.; Lamborn, E.; Morison, N.; Petanidou, T.; Potts, S.G.; Roberts, S.P.M.; Szentgyörgyi, H.; Tscheulin, T.; et al. Measuring bee diversity in different european habitats and biogeographical regions. Ecol. Monogr. 2008, 78, 653–671. [Google Scholar]
- Sguazza, G.H.; Reynaldi, F.J.; Galosi, C.M.; Pecoraro, M.R. Simultaneous detection of bee viruses by multiplex PCR. J. Virol. Methods 2013, 194, 102–106. [Google Scholar] [CrossRef]
- Reynaldi, F.J.; Sguazza, G.H.; Albicoro, F.J.; Pecoraro, M.R.; Galosi, C.M. First molecular detection of co-infection of honey bee viruses in asymptomatic Bombus atratus in South America. Braz. J. Biol. 2014, 73, 797–800. [Google Scholar] [CrossRef]
- Berenyi, O.; Bakonyi, T.; Derakhshifar, I.; Koglberger, H.; Topolska, G.; Ritter, W.; Pechhacker, H.; Nowotny, N. Phylogenetic analysis of deformed wing virus genotypes from diverse geographic origins indicates recent global distribution of the virus. Appl. Environ. Microbiol. 2007, 73, 3605–3611. [Google Scholar] [CrossRef] [Green Version]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Community Ecology Package. R package version 2.5-6. 2019. Available online: http://sortie-admin.readyhosting.com/lme/R%20Packages/vegan.pdf (accessed on 13 July 2020).
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 48. [Google Scholar] [CrossRef]
- Ver Hoef, J.M.; Boveng, P.L. Quasi-Poisson vs. negative binomial regression: how should we model overdispersed count data? Ecology 2007, 88, 2766–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Naumann, U.; Eddelbuettel, D.; Wilshire, J.; Warton, D. mvabund: Statistical Methods for Analysing Multivariate Abundance Data. R package version 4.1.3. 2020. Available online: https://cran.r-project.org/web/packages/mvabund/mvabund.pdf (accessed on 13 July 2020).
- Colwell, R.K.C.J.A. Estimating terrestrial biodiversity through extrapolation. Phil. Trans. Roy. Soc. London B 345 1994, 345, 101–118. [Google Scholar]
- Geslin, B.; Gauzens, B.; Baude, M.; Dajoz, I.; Fontaine, C.; Henry, M. Chapter Four—Massively Introduced Managed Species and Their Consequences for Plant—Pollinator Interactions. In Advances in Ecological Research; Bohan, D.A., Dumbrell, A.J., Massol, F., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 147–199. [Google Scholar]
- Dolezal, A.G.; Carrillo-Tripp, J.; Judd, T.M.; Miller, W.A.; Bonning, B.C.; Toth, A.L. Interacting stressors matter: diet quality and virus infection in honeybee health. R. Soc. Open Sci. 2019, 6, 181803. [Google Scholar] [CrossRef] [Green Version]
- Dolezal, A.G.; Hendrix, S.D.; Scavo, N.A.; Carrillo-Tripp, J.; Harris, M.A.; Wheelock, M.J.; O’Neal, M.E.; Toth, A.L. Honey Bee Viruses in Wild Bees: Viral Prevalence, Loads, and Experimental Inoculation. PLOS ONE 2016, 11, e0166190. [Google Scholar] [CrossRef]
- Piot, N.; Meeus, I.; Kleijn, D.; Scheper, J.; Linders, T.; Smagghe, G. Establishment of wildflower fields in poor quality landscapes enhances micro-parasite prevalence in wild bumble bees. Oecologia 2019, 189, 149–158. [Google Scholar] [CrossRef]
- Spivak, M.; Cariveau, D.P. Flowers as parasite transmission hubs. Nat. Ecol. Evol. 2020, 4, 1298–1299. [Google Scholar] [CrossRef]
- Fontaine, C.; Dajoz, I.; Meriguet, J.; Loreau, M. Functional Diversity of Plant–Pollinator Interaction Webs Enhances the Persistence of Plant Communities. PLoS Biol. 2005, 4, e1. [Google Scholar] [CrossRef]
- Greenleaf, S.S.; Williams, N.M.; Winfree, R.; Kremen, C. Bee foraging ranges and their relationship to body size. Oecologia 2007, 153, 589–596. [Google Scholar] [CrossRef]
- McMahon, D.P.; Furst, M.A.; Caspar, J.; Theodorou, P.; Brown, M.J.F.; Paxton, R.J. A sting in the spit: widespread cross-infection of multiple RNA viruses across wild and managed bees. J. Anim. Ecol. 2015, 84, 615–624. [Google Scholar] [CrossRef]
- Schurr, F.; Tison, A.; Militano, L.; Cheviron, N.; Sircoulomb, F.; Riviere, M.P.; Ribiere-Chabert, M.; Thiery, R.; Dubois, E. Validation of quantitative real-time RT-PCR assays for the detection of six honeybee viruses. J. Virol. Methods 2019, 270, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Evison, S.E.F.; Roberts, K.E.; Laurenson, L.; Pietravalle, S.; Hui, J.; Biesmeijer, J.C.; Smith, J.E.; Budge, G.; Hughes, W.O.H. Pervasiveness of Parasites in Pollinators. PLoS ONE 2012, 7, e30641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forzan, M.; Felicioli, A.; Sagona, S.; Bandecchi, P.; Mazzei, M. Complete Genome Sequence of Deformed Wing Virus Isolated from Vespa crabro in Italy. Genome Announc. 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzei, M.; Forzan, M.; Cilia, G.; Sagona, S.; Bortolotti, L.; Felicioli, A. First detection of replicative deformed wing virus (DWV) in Vespa velutina nigrithorax. Bull. Insectol. 2018, 71, 211–216. [Google Scholar]
- Alger, S.A.; Burnham, P.A.; Boncristiani, H.F.; Brody, A.K. RNA virus spillover from managed honeybees (Apis mellifera) to wild bumblebees (Bombus spp.). PLoS ONE 2019, 14, e0217822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graystock, P.; Ng, W.H.; Parks, K.; Tripodi, A.D.; Muñiz, P.A.; Fersch, A.A.; Myers, C.R.; McFrederick, Q.S.; McArt, S.H. Dominant bee species and floral abundance drive parasite temporal dynamics in plant-pollinator communities. Nat. Ecol. Evol. 2020, 4, 1358–1367. [Google Scholar] [CrossRef]
- Sapir, Y.; Shmida, A.; Ne’eman, G. Pollination of Oncocyclus irises (Iris: Iridaceae) by Night-Sheltering Male Bees. Plant Biol. 2005, 7, 417–424. [Google Scholar] [CrossRef]
- Manley, R.; Temperton, B.; Boots, M.; Wilfert, L. Contrasting impacts of a novel specialist vector on multi-host viral pathogen epidemiology in wild and managed bees. Mol. Ecol. 2020, 29, 380–393. [Google Scholar] [CrossRef]
- Gusachenko, O.N.; Woodford, L.; Balbirnie-Cumming, K.; Ryabov, E.V.; Evans, D.J. Evidence for and against deformed wing virus spillover from honey bees to bumble bees: A reverse genetic analysis. Sci. Rep. 2020, 10, 16847. [Google Scholar] [CrossRef]
- Li, J.; Peng, W.; Wu, J.; Strange, J.P.; Boncristiani, H.; Chen, Y. Cross-species infection of deformed wing virus poses a new threat to pollinator conservation. J. Econ. Entomol. 2011, 104, 732–739. [Google Scholar] [CrossRef]
- Radzevičiūtė, R.; Theodorou, P.; Husemann, M.; Japoshvili, G.; Kirkitadze, G.; Zhusupbaeva, A.; Paxton, R.J. Replication of honey bee-associated RNA viruses across multiple bee species in apple orchards of Georgia, Germany and Kyrgyzstan. J. Invertebr. Pathol. 2017, 146, 14–23. [Google Scholar] [PubMed] [Green Version]
- Piot, N.; Snoeck, S.; Vanlede, M.; Smagghe, G.; Meeus, I. The Effect of Oral Administration of dsRNA on Viral Replication and Mortality in Bombus terrestris. Viruses 2015, 7, 3172–3185. [Google Scholar] [CrossRef]
- Niu, J.; Meeus, I.; De Coninck, D.I.; Deforce, D.; Etebari, K.; Asgari, S.; Smagghe, G. Infections of virulent and avirulent viruses differentially influenced the expression of dicer-1, ago-1, and microRNAs in Bombus terrestris. Sci. Rep. 2017, 7, 45620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzei, M.; Cilia, G.; Forzan, M.; Lavazza, A.; Mutinelli, F.; Felicioli, A. Detection of replicative Kashmir Bee Virus and Black Queen Cell Virus in Asian hornet Vespa velutina (Lepelieter 1836) in Italy. Sci. Rep. 2019, 9, 10091. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus/Species | Number of Samples | Number of Pooled Individuals | ABPV | IAPV | KBV | BQCV | SBV | DWV | CBPV |
|---|---|---|---|---|---|---|---|---|---|
| Apis mellifera | 23 | 920 | 91.3% | 13.0% | 0.0% | 73.9% | 91.3% | 78.3% | 17.4% |
| Andrenaspp. | 5 | 12 | 40.0% | 20.0% | 0.0% | 20.0% | 0.0% | 0.0% | 0.0% |
| Bombusspp. | 84 | 91 | 66.7% | 19.0% | 0.0% | 28.6% | 91.7% | 13.1% | 0.0% |
| Bombus terrestris | 43 | 45 | 31 | 7 | 0 | 9 | 41 | 8 | 0 |
| Bombus pascuorum | 7 | 7 | 5 | 1 | 0 | 1 | 7 | 0 | 0 |
| Bombus sp. | 29 | 34 | 17 | 8 | 0 | 12 | 24 | 2 | 0 |
| Euceraspp. | 9 | 55 | 22.2% | 77.8% | 0.0% | 66.7% | 55.6% | 11.1% | 0.0% |
| Halictusspp. | 25 | 56 | 56.0% | 8.0% | 0.0% | 24.0% | 28.0% | 8.0% | 0.0% |
| Halictus fulvipes | 14 | 17 | 10 | 0 | 0 | 2 | 5 | 0 | 0 |
| Halictus tectus | 4 | 15 | 0 | 0 | 0 | 2 | 1 | 0 | 0 |
| Halictidae sp. | 1 | 11 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hylaeusspp. | 5 | 10 | 20.0% | 40.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| Lasioglossumspp. | 77 | 676 | 26.0% | 31.2% | 0.0% | 54.5% | 41.6% | 1.3% | 0.0% |
| Lasioglossum malachurum | 65 | 633 | 17 | 22 | 0 | 35 | 28 | 0 | 0 |
| Lasioglossum pauperatum | 6 | 33 | 2 | 1 | 0 | 2 | 2 | 0 | 0 |
| undetermined Halictidae | 32 | 201 | 12.5% | 9.4% | 0.0% | 21.9% | 15.6% | 0.0% | 0.0% |
| Megachilespp. | 7 | 7 | 0.0% | 0.0% | 0.0% | 0.0% | 14.3% | 0.0% | 0.0% |
| Polistesspp. | 7 | 7 | 42.9% | 0.0% | 0.0% | 28.6% | 14.3% | 28.6% | 0.0% |
| Syrphidae spp. | 2 | 9 | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalmon, A.; Diévart, V.; Thomasson, M.; Fouque, R.; Vaissière, B.E.; Guilbaud, L.; Le Conte, Y.; Henry, M. Possible Spillover of Pathogens between Bee Communities Foraging on the Same Floral Resource. Insects 2021, 12, 122. https://0-doi-org.brum.beds.ac.uk/10.3390/insects12020122
Dalmon A, Diévart V, Thomasson M, Fouque R, Vaissière BE, Guilbaud L, Le Conte Y, Henry M. Possible Spillover of Pathogens between Bee Communities Foraging on the Same Floral Resource. Insects. 2021; 12(2):122. https://0-doi-org.brum.beds.ac.uk/10.3390/insects12020122
Chicago/Turabian StyleDalmon, Anne, Virgine Diévart, Maxime Thomasson, Romain Fouque, Bernard E. Vaissière, Laurent Guilbaud, Yves Le Conte, and Mickaël Henry. 2021. "Possible Spillover of Pathogens between Bee Communities Foraging on the Same Floral Resource" Insects 12, no. 2: 122. https://0-doi-org.brum.beds.ac.uk/10.3390/insects12020122