
Development of a Set of Microsatellite Markers to Investigate Sexually Antagonistic Selection in the Invasive Ant Nylanderia fulva


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
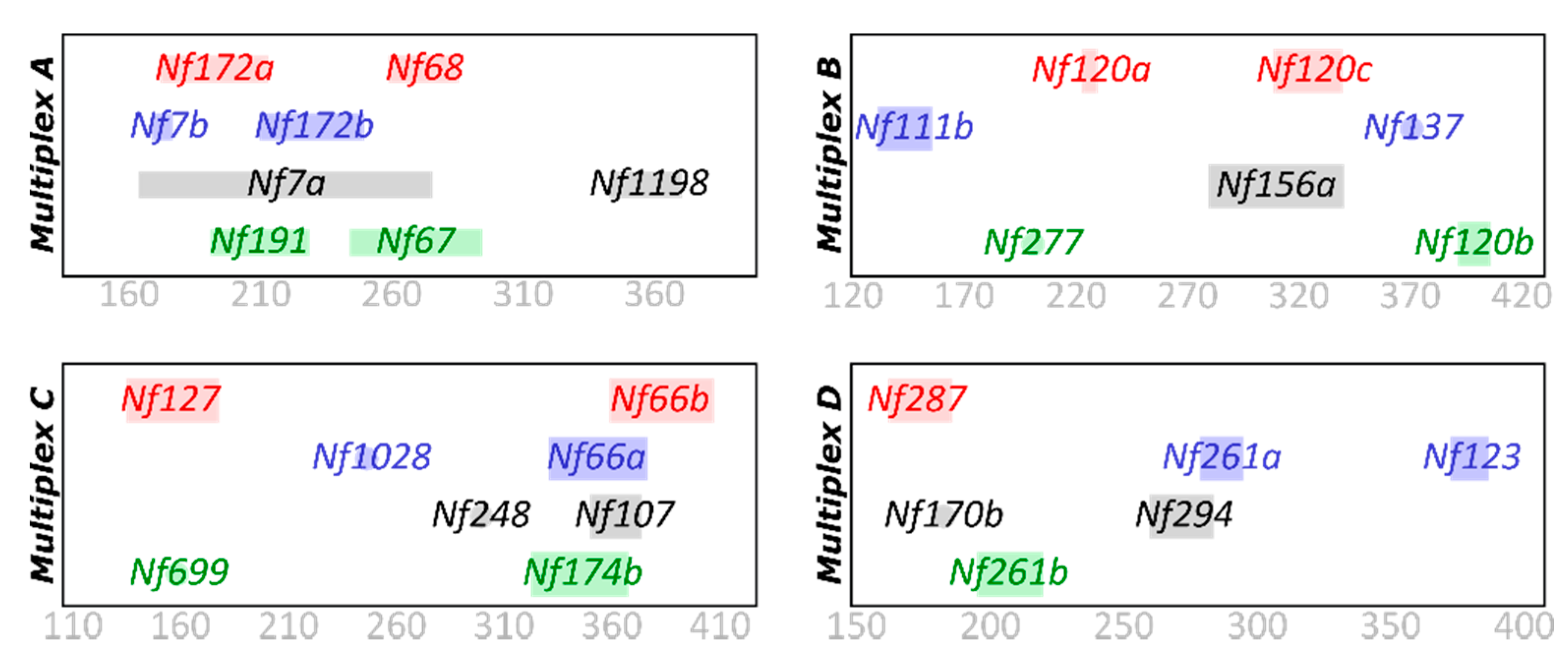
2.1. Microsatellite Primer Design
2.2. Genetic Procedures
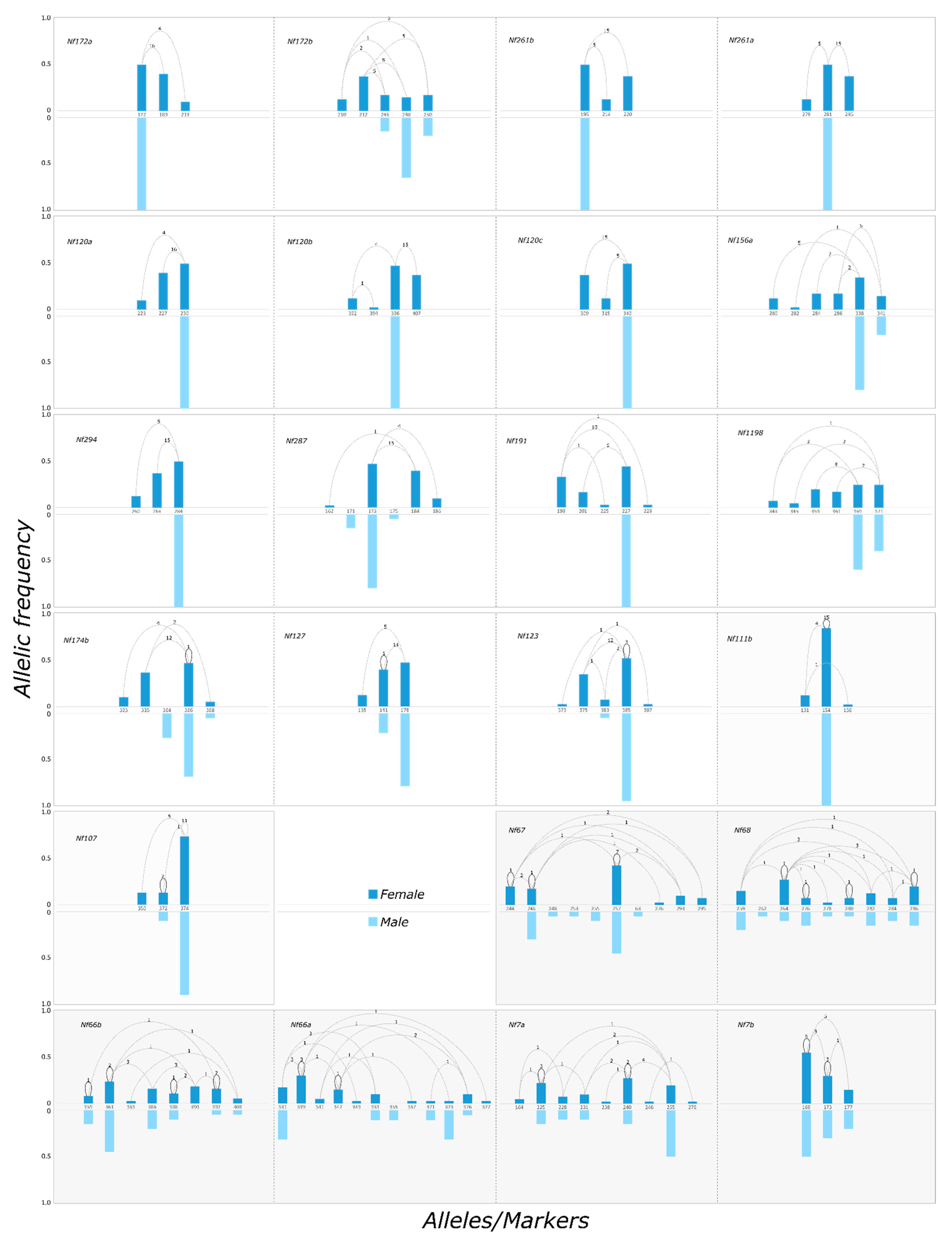
2.3. Confirming Inheritance Patterns of SAS and Non-SAS Markers
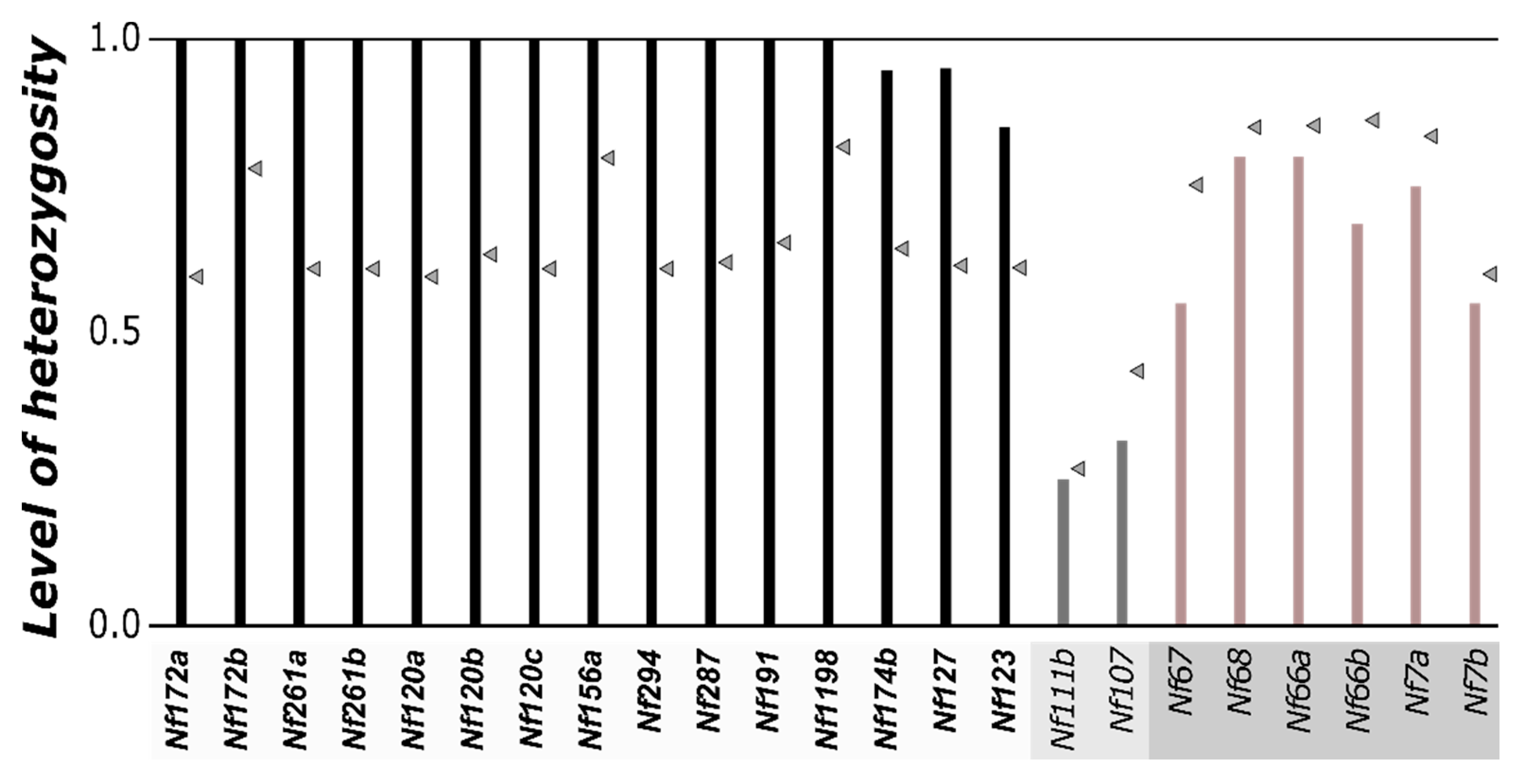
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Connallon, T.; Clark, A. Evolutionary inevitability of sexual antagonism. Proc. R. Soc. B Biol. Sci. 2014, 281, 20132123. [Google Scholar] [CrossRef]
- Bonduriansky, R.; Chenoweth, S. Intralocus sexual conflict. Trends Ecol. Evol. 2009, 24, 280–288. [Google Scholar] [CrossRef]
- Pennell, T.M.; Morrow, E.H. Two sexes, one genome: The evolutionary dynamics of intralocus sexual conflict. Ecol. Evol. 2013, 3, 1819–1834. [Google Scholar] [CrossRef]
- Griffin, R.M.; Dean, R.; Grace, J.L.; Rydén, P.; Friberg, U. The Shared Genome Is a Pervasive Constraint on the Evolution of Sex-Biased Gene Expression. Mol. Biol. Evol. 2013, 30, 2168–2176. [Google Scholar] [CrossRef] [Green Version]
- Rice, W.R. Sex chromosomes and the evolution of sexual dimorphism. Evolution 1984, 38, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B. The evolution of sex chromosomes. Science 1991, 251, 1030–1033. [Google Scholar] [CrossRef] [PubMed]
- Eyer, P.-A.; Blumenfeld, A.J.; Vargo, E.L. Sexually antagonistic selection promotes genetic divergence between males and females in an ant. Proc. Natl. Acad. Sci. USA 2019, 116, 24157–24163. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.; Crozier, R.H. Sex determination and population biology in the hymenoptera. Trends Ecol. Evol. 1995, 10, 281–286. [Google Scholar] [CrossRef]
- Eyer, P.-A.; McDowell, B.; Johnson, L.N.L.; Calcaterra, L.A.; Fernandez, M.B.; Shoemaker, D.; Puckett, R.T.; Vargo, E.L. Supercolonial structure of invasive populations of the tawny crazy ant Nylanderia fulva in the US. BMC Evol. Biol. 2018, 18, 209. [Google Scholar] [CrossRef]
- Gotzek, D.; Brady, S.G.; Kallal, R.J.; Lapolla, J.S. The Importance of Using Multiple Approaches for Identifying Emerging Invasive Species: The Case of the Rasberry Crazy Ant in the United States. PLoS ONE 2012, 7, e45314. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Moshman, L.; Kraus, E.; Wilson, B.E.; Acharya, N.; Diaz, R. A Review of the Tawny Crazy Ant, Nylanderia fulva, an Emergent Ant Invader in the Southern United States: Is Biological Control a Feasible Management Option? Insects 2016, 7, 77. [Google Scholar] [CrossRef]
- Butler, I.A.; Siletti, K.; Oxley, P.R.; Kronauer, D.J.C. Conserved Microsatellites in Ants Enable Population Genetic and Colony Pedigree Studies across a Wide Range of Species. PLoS ONE 2014, 9, e107334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamilo, P.; Gertsch, P.; Thorén, P.; Seppä, P. Molecular Population Genetics of Social Insects. Annu. Rev. Ecol. Syst. 1997, 28, 1–25. [Google Scholar] [CrossRef]
- Eyer, P.A.; Hefetz, A. Cytonuclear incongruences hamper species delimitation in the socially polymorphic desert ants of the Cataglyphis albicans group in Israel. J. Evol. Biol. 2018, 12, 1828–1842. [Google Scholar] [CrossRef]
- Eyer, P.A.; Vargo, E.L.; Peeters, C. One tree, many colonies: Colony structure, breeding system and colonization events of host trees in tunnelling Melissotarsus ants. Biol. J. Linn. Soc. 2021, 133, 237–248. [Google Scholar] [CrossRef]
- Schultner, E.; Saramäki, J.; Helanterä, H. Genetic structure of native ant supercolonies varies in space and time. Mol. Ecol. 2016, 25, 6196–6213. [Google Scholar] [CrossRef]
- Jacobs, S.; Heinze, J. Population and colony structure of an ant with territorial males, Cardiocondyla venustula. BMC Evol. Biol. 2019, 19, 115. [Google Scholar] [CrossRef]
- Lemos, A.S.M.; Azevedo-Silva, M.; Gonçalves-Neto, S.; Souza, A.P.; Oliveira, P.S. Microsatellites for the Neotropical Ant, Odontomachus chelifer (Hymenoptera: Formicidae). J. Insect Sci. 2020, 20, 117. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT (Version 1.2): A Computer Program to Calculate F-Statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Meglécz, E.; Pech, N.; Gilles, A.; Dubut, V.; Hingamp, P.; Trilles, A.; Grenier, R.; Martin, J.F. QDD version 3.1: A user-friendly computer program for microsatellite selection and primer design revisited: Ex-perimental validation of variables determining genotyping success rate. Mol. Ecol. Resour. 2014, 14, 1302–1313. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandström, M.; Ellegren, H. Genome-wide analysis of microsatellite polymorphism in chicken circumventing the ascer-tainment bias. BioTechniques 2001, 28, 6–24. [Google Scholar]
- Boutin-Ganache, I.; Raposo, M.; Raymond, M.; Deschepper, C. M13-Tailed Primers Improve the Readability and Usability of Microsatellite Analyses Performed with Two Different Allele-Sizing Methods. Biotechniques 2001, 31, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F. genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Mank, J.E. Population genetics of sexual conflict in the genomic era. Nat. Rev. Genet. 2017, 18, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Rowe, L.; Chenoweth, S.; Agrawal, A.F. The Genomics of Sexual Conflict. Am. Nat. 2018, 192, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Connallon, T.; Matthews, G. Cross-sex genetic correlations for fitness and fitness components: Connecting theoretical pre-dictions to empirical patterns. Evol. Lett. 2019, 3, 254–262. [Google Scholar] [CrossRef]
- Frank, S.A.; Patten, M.M. Sexual antagonism leads to a mosaic of X-autosome conflict. Evolution 2019, 74, 495–498. [Google Scholar] [CrossRef]
- Hitchcock, T.J.; Gardner, A. A gene’s-eye view of sexual antagonism. Proc. R. Soc. B Biol. Sci. 2020, 287, 20201633. [Google Scholar] [CrossRef]
- Kasimatis, K.R.; Ralph, P.L.; Phillips, P.C. Limits to Genomic Divergence Under Sexually Antagonistic Selection. G3 Genes Genomes Genet. 2019, 9, 3813–3824. [Google Scholar] [CrossRef] [Green Version]
- De la Filia, A.G.; Bain, S.; Ross, L. Haplodiploidy and the reproductive ecology of Arthropods. Curr. Opin. Insect Sci. 2015, 9, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraaijeveld, K. Male genes with nowhere to hide; sexual conflict in haplodiploids. Anim. Biol. 2009, 59, 403–415. [Google Scholar] [CrossRef]
- Patten, M.M. The X chromosome favors males under sexually antagonistic selection. Evolution 2019, 73, 84–91. [Google Scholar] [CrossRef] [Green Version]
- LaPolla, J.S.; Brady, S.G.; Shattuck, S.O. Phylogeny and taxonomy of the Prenolepis genus-group of ants (Hyme-noptera: Formicidae). Syst. Entomol. 2010, 35, 118–131. [Google Scholar] [CrossRef]
- Williams, J.L.; Zhang, Y.M.; Lloyd, M.W.; LaPolla, J.S.; Schultz, T.R.; Lucky, A. Global domination by crazy ants: Phylogenomics reveals biogeographical history and invasive species relationships in the genus Nylanderia (Hymenoptera: Formicidae). Syst. Èntomol. 2020, 45, 730–744. [Google Scholar] [CrossRef]
- Williams, J.L.; Lucky, A. Non-native and Invasive Nylanderia Crazy Ants (Hymenoptera: Formicidae) of the World: Integrating Genomics to Enhance Taxonomic Preparedness. Ann. Èntomol. Soc. Am. 2020, 113, 318–336. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Marker Name | Scaffold | FIS Scaffold | Scaffold FIS Value | MS Motif | # of Repeats | Left | Right | TM | Product Size | Peak Reading * | Color | Multiplex |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nf172a | S_172 | High | −0.900 | CT | 33 | TACAGCCGCGTTGTTTTCAC | GGCAACATATCAAGAACCCTGT | 57 | 172/213 | PET | A | |
| Nf172b | S_172 | High | −0.900 | TC | 27 | TTAATGAGGGGCCCGTTGAT | GCATGTATGAAAGAGCAGCGA | 57 | 210/250 | FAM | A | |
| Nf137 | S_137 | High | −0.900 | TA | 24 | ACGTGTGTTGTGTGTGTGTG | TGGTGCTTTTAATACAGTGGCT | 57 | 370 | monomorphic | FAM | B |
| Nf1028 | S_1028 | High | −0.900 | TA | 18 | GCAATGCCACTCAAGGTCAA | GCTCTGTTGGCCGATTTAAAA | 57 | 249 | monomorphic | FAM | C |
| Nf699 | S_699 | High | −0.900 | TA | 27 | AGTCAATTAAAACGAGTCCTGGA | TGTGTGAAAATACGTGTGTCTAC | 57 | 162 | monomorphic | VIC | C |
| Nf652 | S_652 | High | −0.900 | TCT | 17 | CGGAGATACAAGCGGTCAAA | AGGGAGGTGTGAGTGAAAGG | / | no amplification | |||
| Nf1198 | S_1198 | High | −0.878 | CATA | 12 | GAGACCACATACACCAAAAGGT | GCAGAAAATTAGTTGCGCGA | 57 | 343/371 | NED | A | |
| Nf191 | S_191 | High | −0.742 | AT | 24 | TGCGCGTTAATATCTCAAACTCT | TGGATGAAATGAGAGATGTGGG | 57 | 191/229 | VIC | A | |
| Nf248 | S_248 | High | −0.764 | AG | 28 | CTATGCACGCTCCTCACTCT | ACCGAGACCTTGTACACACT | 295 | monomorphic | NED | C | |
| Nf401a | S_401 | High | −0.667 | TA | 21 | ACGTGTGCATGTTGAGAGAG | TGCCCCTTTCGAAACGTAGA | 57 | [315] | inconsistent | ||
| Nf401b | S_401 | High | −0.667 | TA | 21 | CATACCTGCAGCATCCCCTA | TAAGATGCATGCACACACGC | 57 | [222] | inconsistent | ||
| Nf170a | S_170 | High | −0.609 | GAC | 52 | CCACAGATCTCGTTCCGTCT | TGAAGGTGCTGAGGAGGATG | / | no amplification | |||
| Nf170b | S_170 | High | −0.609 | AT | 24 | CGAGTGTCTTTATTTCGCGC | GTCCCAGAAATGAACACCGC | 60 | 172 | monomorphic | NED | D |
| Nf294 | S_294 | High | −0.900 | AT | 26 | GTTTGACGACATTTCTCTGTTCA | CGCAAGTGTAAACGCAATCT | 57 | 260/284 | NED | D | |
| Nf502 | S_502 | High | −0.900 | TC | 30 | GGTGGATGAGGGAGTTGGAA | TACCTTCCGCACATAACCCA | [336/367] | inconsistent | |||
| Nf174a | S_174 | High | −0.889 | TC | 63 | CCCGCTTCGAACATGACAAT | TCATGGAATATCGCATTGTCGT | / | no amplification | |||
| Nf174b | S_174 | High | −0.889 | TA | 30 | AGCTAACTGACTGACTGCGT | CGATATTCTGCTCGTCGTCAC | 57 | 323/368 | VIC | C | |
| Nf287 | S_287 | High | −0.900 | AG | 30 | CGAATTTTATGCTCGCCGGA | GATTTGATTCCAGAGCGCGA | 57 | 162/186 | PET | D | |
| Nf277 | S_277 | High | −0.900 | CT | 34 | GCGAGAGAGACGGTATCAC | AGAATTCGATGTACACCGGGT | 57 | 188 | monomorphic | VIC | B |
| Nf167a | S_167 | High | −0.629 | TA | 39 | AGCAGAGAGAAAGAAATGAGAGT | TTGTTAGGGATAGATCGGAGGA | / | no amplification | |||
| Nf167b | S_167 | High | −0.629 | TC | 28 | GGGGCTAACTTCATACAGGC | ACCTTCTGCGAATGGTAGCT | 57 | [281] | inconsistent | ||
| Nf107 | S_107 | Mod. | −0.125 | AG | 31 | ACAAGTCACTTCCGCTGAAAC | CGCAAGGATCAGGTACCGAT | 60 | 350/374 | NED | C | |
| Nf261a | S_261 | Mod. | −0.178 | CT | 32 | ATGCTCTTTGTCACGAGGGA | CGAGAGAAAGGGAAGGGTGA | 60 | 279/295 | FAM | D | |
| Nf261b | S_261 | Mod. | −0.178 | TCG | 21 | CATACTATCTGGGCGGGTGT | CACTGAGAAGATCGCGAGTG | 57 | 195/220 | VIC | D | |
| Nf66a | S_66 | Mod. | −0.439 | TA | 32 | AAACTACGCTCGCAATGCAA | ATGAGAGGGTGTGGAAGAGC | 57 | 331/377 | FAM | C | |
| Nf66b | S_66 | Mod. | −0.439 | TA | 31 | GTGCTCCACTCCAATAATGCT | TGGTCAGGAGTCACGGTAAA | 55 | 359/408 | PET | C | |
| Nf127 | S_127 | Mod. | −0.335 | AG | 31 | GCGGCTCGTTAGTGATTCTC | GAGACTCCATTTTGACGGCG | 57 | 135/178 | PET | C | |
| Nf123 | S_123 | Mod. | −0.279 | AG | 28 | TGAAAATGTACGCGCGACTT | CCAGCCTTTCAGTGATTCGA | 57 | 373/387 | FAM | D | |
| Nf57 | S_57 | Mod. | −0.540 | TA | 31 | GTGTGGAGGAGCAATTTGGG | AATAACGCACTGTCATCCGC | 57 | [208] | inconsistent | ||
| Nf120a | S_120 | SAS | −0.900 | GA | 28 | AGAAGCCGCCATCAAGAAGA | GGGAAGATGAGCGCGATCA | 60 | 223/230 | PET | B | |
| Nf120b | S_120 | SAS | −0.900 | TC | 26 | GCCTCTTTATTCGCGGAAGG | AGATTTTACAGCTACGCCGC | 57 | 392/407 | VIC | B | |
| Nf120c | S_120 | SAS | −0.900 | AC | 24 | AGATTGACATTTTCCGCTCTTCA | CCCCATTTGTTCGCTCGTAG | 57 | 309/340 | PET | B | |
| Nf111a | S_111 | SAS | −0.488 | GA | 32 | GGACAAGTTGGAACGGGATG | AACAGAGGAGAACGCGGTAA | 57 | [308] | inconsistent | ||
| Nf111b | S_111 | SAS | −0.488 | CA | 25 | GCGTGGATGCTCTTTTCACA | GAAAGTATCTTCTTGCGTgcg | 57 | 131/156 | FAM | B | |
| Nf111c | S_111 | SAS | −0.488 | TC | 24 | CACGCTAAACTGTCATCCGA | CGTGTTGAAGGGAGGGAAGA | 62 | [162/170] | inconsistent | ||
| Nf156a | S_156 | SAS | −0.875 | AT | 43 | ACACACTGTACTACTGCGGT | GCGAAATGAGAACGGTAGGT | 55 | 280/341 | NED | B | |
| Nf156b | S_156 | SAS | −0.875 | TA | 23 | CGTAACCTTCGAAATGGCTGT | GTTGCAGAGATCCGAACGAT | / | no amplification | |||
| Nf20 | S_20 | Low | 0.000 | AG | 73 | ACTCCTAACTGCTGCCTAATTT | GCACGAATTTACAATTGCGCA | / | no amplification | |||
| Nf68 | S_68 | Low | 0.007 | GA | 32 | TACCCACGCATAATCCACCC | CCTCCTTGTTTGTAACGGAAGA | 57 | 259/286 | PET | A | |
| Nf67 | S_67 | Low | 0.008 | TC | 36 | AAATCCCTGTTTTAAACTGCTGT | AGAACGTTCGAGTGTAGATAGGT | 57 | 244/295 | VIC | A | |
| Nf7a | S_7 | Low | 0.038 | CAT | 36 | ACGTGGTTGTTGGTGCATAC | AGCAAGAGAGAGACCGATGT | 55 | 164/276 | NED | A | |
| Nf7b | S_7 | Low | 0.038 | GGTA | 21 | TTAGTGGTGCAAAAGGGAAGA | GTTGTGGTGGCAAAGGGTG | 60 | 169/177 | FAM | A | |
| Nf53 | S_53 | Low | 0.024 | TA | 33 | CCTTGCATCATGTGTGGACC | TCACACGAGGAGACAAGAGG | / | no amplification |
| Marker Name | Number of Alleles | Heterozygosity | HW Sign. | FIS | M-F Diff. | Genomic Region | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Overall | Workers | Queens | Males | Obs. | Exp. | |||||
| Nf172a | 3 | 3 | 3 | 1 | 1.00 | 0.59 | *** | −0.712 | *** | SAS |
| Nf172b | 5 | 5 | 5 | 3 | 1.00 | 0.78 | ** | −0.293 | *** | SAS |
| Nf1198 | 6 | 6 | 6 | 2 | 1.00 | 0.82 | ** | −0.232 | *** | SAS |
| Nf191 | 5 | 4 | 4 | 1 | 1.00 | 0.68 | *** | −0.489 | *** | SAS |
| Nf294 | 3 | 3 | 3 | 1 | 1.00 | 0.61 | *** | −0.670 | *** | SAS |
| Nf174b | 5 | 4 | 3 | 3 | 0.95 | 0.64 | *** | −0.493 | *** | SAS |
| Nf287 | 6 | 3 | 4 | 3 | 1.00 | 0.62 | *** | −0.642 | *** | SAS |
| Nf120a | 3 | 3 | 3 | 1 | 1.00 | 0.59 | *** | −0.712 | *** | SAS |
| Nf120b | 4 | 4 | 3 | 1 | 1.00 | 0.63 | *** | −0.603 | *** | SAS |
| Nf120c | 3 | 3 | 3 | 1 | 1.00 | 0.61 | *** | −0.670 | *** | SAS |
| Nf156a | 6 | 5 | 6 | 2 | 1.00 | 0.80 | ** | −0.263 | *** | SAS |
| Nf261a | 3 | 3 | 3 | 1 | 1.00 | 0.61 | *** | −0.670 | *** | SAS |
| Nf261b | 3 | 3 | 3 | 1 | 1.00 | 0.61 | *** | −0.670 | *** | SAS |
| Nf127 | 3 | 3 | 3 | 2 | 0.95 | 0.61 | ** | −0.570 | *** | SAS |
| Nf123 | 5 | 4 | 3 | 2 | 0.85 | 0.61 | ** | −0.407 | *** | SAS |
| Nf66a | 12 | 7 | 9 | 6 | 0.80 | 0.85 | NS | 0.063 | *** | non-SAS |
| Nf66b | 8 | 7 | 7 | 6 | 0.68 | 0.86 | NS | 0.211 | * | non-SAS |
| Nf68 | 9 | 6 | 8 | 9 | 0.80 | 0.85 | NS | 0.060 | NS | non-SAS |
| Nf67 | 10 | 6 | 5 | 6 | 0.55 | 0.75 | NS | 0.273 | *** | non-SAS |
| Nf7a | 9 | 7 | 7 | 5 | 0.75 | 0.83 | NS | 0.104 | NS | non-SAS |
| Nf7b | 3 | 3 | 3 | 3 | 0.55 | 0.60 | NS | 0.085 | NS | non-SAS |
| Nf111b | 3 | 3 | 2 | 1 | 0.25 | 0.27 | NS | 0.069 | * | unclear |
| Nf107 | 3 | 3 | 3 | 2 | 0.32 | 0.43 | NS | 0.278 | * | unclear |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eyer, P.-A.; Moran, M.N.; Blumenfeld, A.J.; Vargo, E.L. Development of a Set of Microsatellite Markers to Investigate Sexually Antagonistic Selection in the Invasive Ant Nylanderia fulva. Insects 2021, 12, 643. https://0-doi-org.brum.beds.ac.uk/10.3390/insects12070643
Eyer P-A, Moran MN, Blumenfeld AJ, Vargo EL. Development of a Set of Microsatellite Markers to Investigate Sexually Antagonistic Selection in the Invasive Ant Nylanderia fulva. Insects. 2021; 12(7):643. https://0-doi-org.brum.beds.ac.uk/10.3390/insects12070643
Chicago/Turabian StyleEyer, Pierre-Andre, Megan N. Moran, Alexander J. Blumenfeld, and Edward L. Vargo. 2021. "Development of a Set of Microsatellite Markers to Investigate Sexually Antagonistic Selection in the Invasive Ant Nylanderia fulva" Insects 12, no. 7: 643. https://0-doi-org.brum.beds.ac.uk/10.3390/insects12070643