
Co-Expression Network and Time-Course Expression Analyses to Identify Silk Protein Regulatory Factors in Bombyx mori

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Constructing a Gene Co-Expression Network and Detecting Modules
2.2. Co-Expression Network Analysis
2.3. RNA Extraction
2.4. Gene Expression Analysis
3. Results
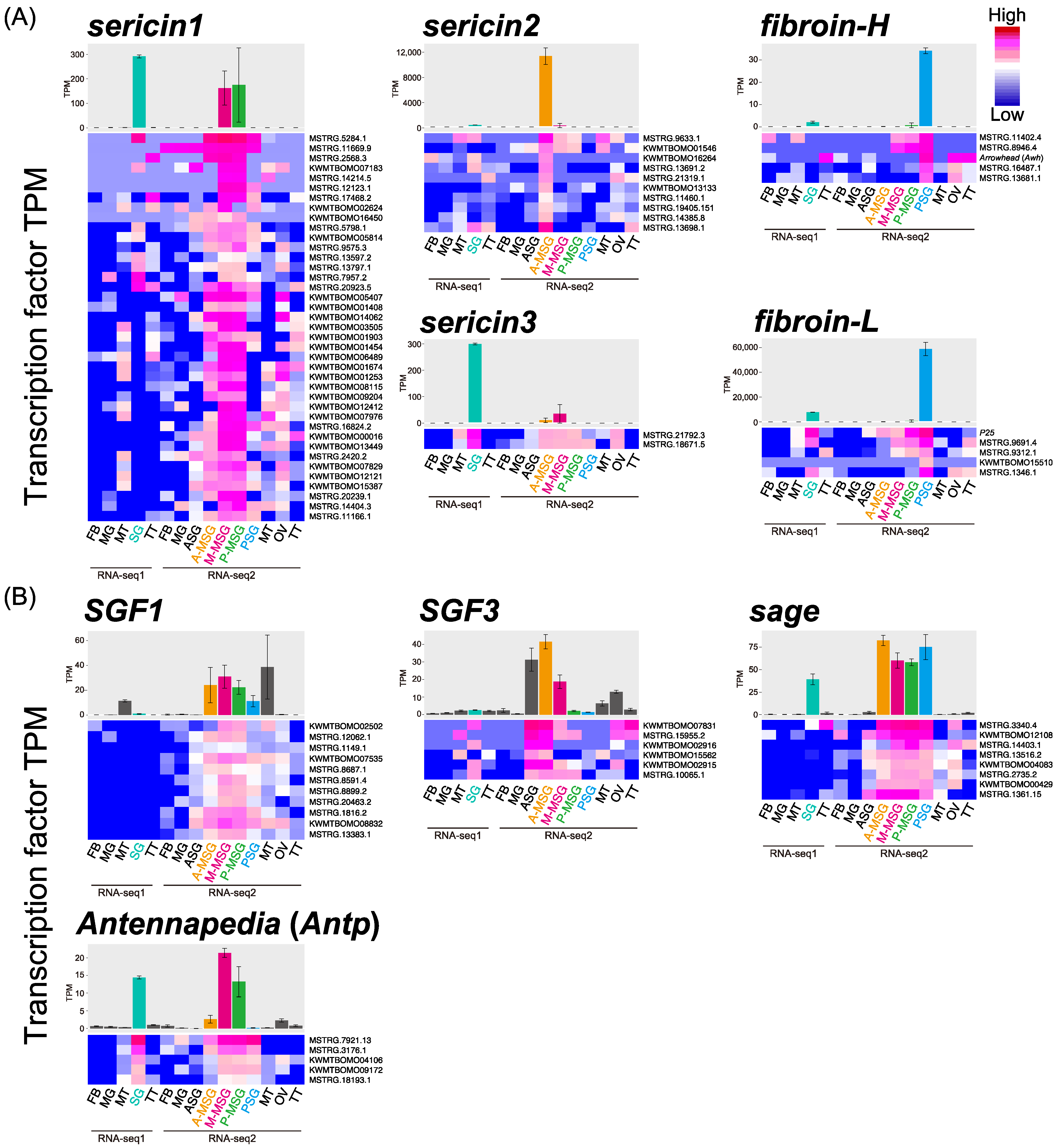
3.1. Co-Expression Network Analysis with Tissue Expression Data
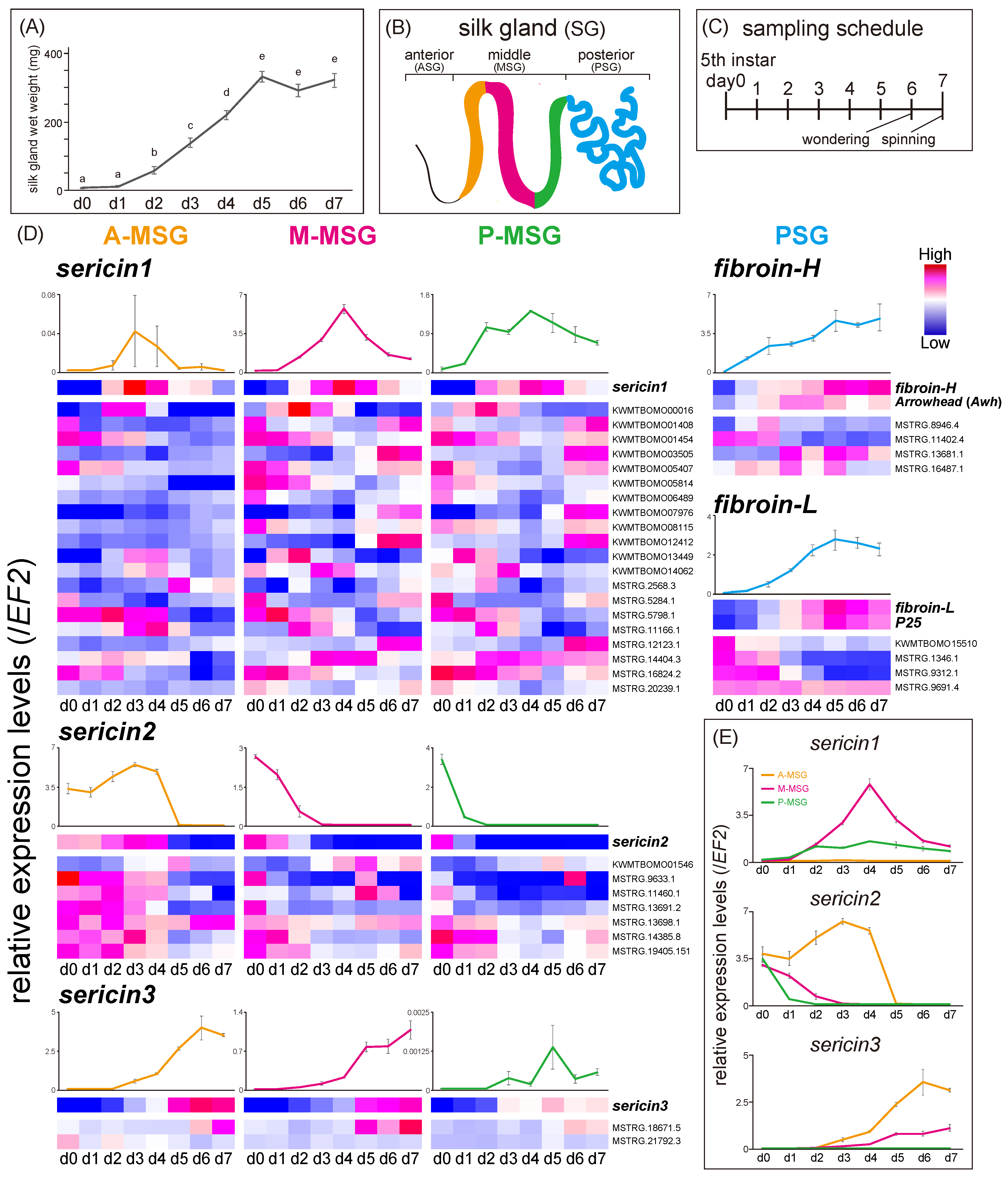
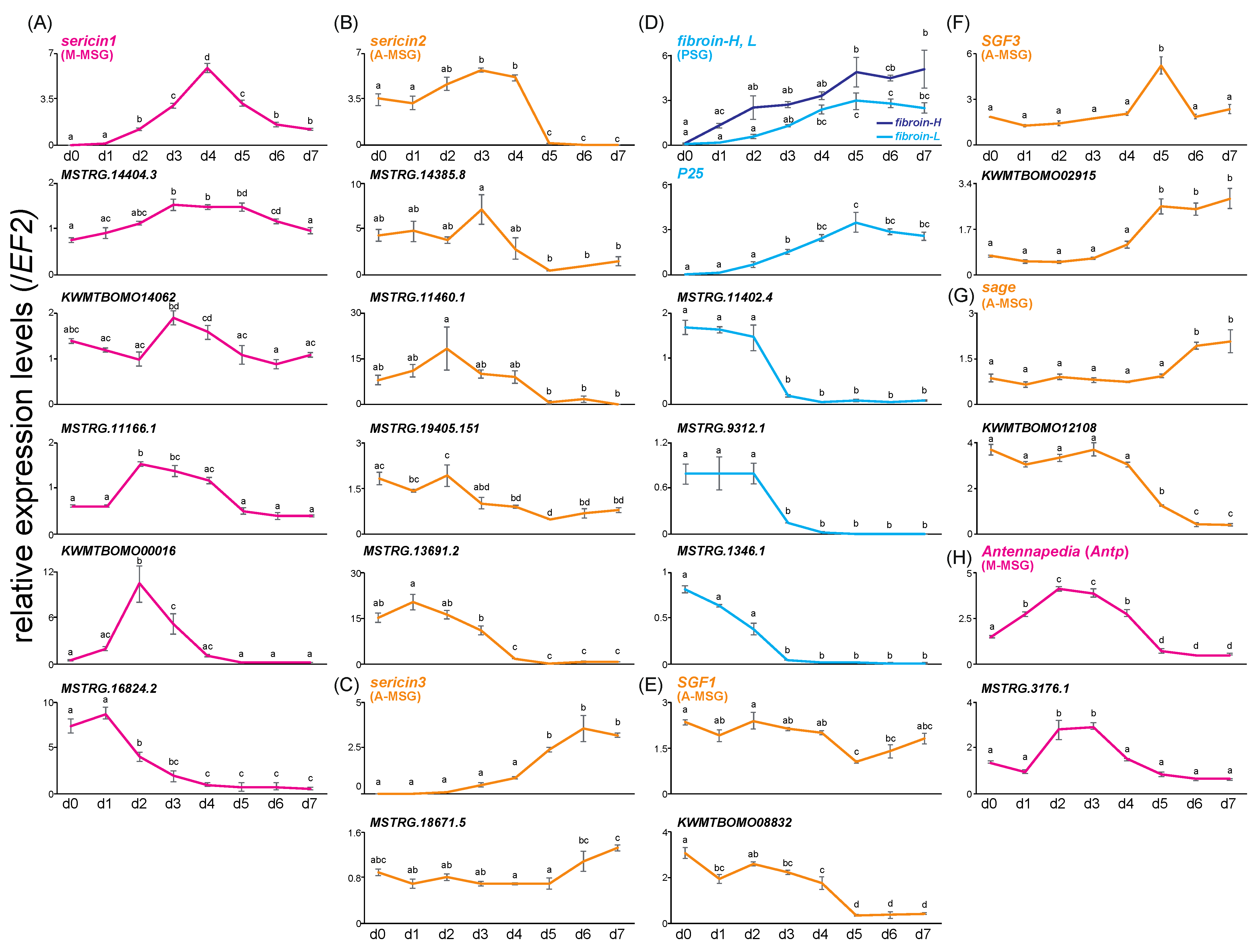
3.2. Time-Course Expression Analysis of the Four SG Regions during the Last Instar Period
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dhawan, S.; Gopinathan, K.P. Cell cycle events during the development of the silk glands in the mulberry silkworm Bombyx mori. Dev. Genes Evol. 2003, 213, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, M.; Yamaguchi, M.; Fujimoto, Y.; Takiya, S. Expression profiles of the genes for nine transcription factors and their isoforms in the posterior silk gland of the silkworm Bombyx mori during the last and penultimate instars. J. Insect Biotechnol. Sericology 2011, 79, 31–43. [Google Scholar]
- Takiya, S.; Tsubota, T.; Kimoto, M. Regulation of silk genes by hox and homeodomain proteins in the terminal differentiated silk gland of the silkworm Bombyx mori. J. Dev. Biol. 2016, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimoto, M.; Tsubota, T.; Uchino, K.; Sezutsu, H.; Takiya, S. Hox transcription factor Antp regulates sericin-1 gene expression in the terminal differentiated silk gland of Bombyx mori. Dev. Biol. 2014, 386, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Tsubota, T.; Tomita, S.; Uchino, K.; Kimoto, M.; Takiya, S.; Kajiwara, H.; Yamazaki, T.; Sezutsu, H. A hox gene, Antennapedia, regulates expression of multiple major silk protein genes in the silkworm Bombyx mori. J. Biol. Chem. 2016, 291, 7087–7096. [Google Scholar] [CrossRef] [Green Version]
- Ohno, K.; Sawada, J.; Takiya, S.; Kimoto, M.; Matsumoto, A.; Tsubota, T.; Uchino, K.; Hui, C.C.; Sezutsu, H.; Handa, H.; et al. Silk gland factor-2, involved in fibroin gene transcription, consists of LIM homeodomain, LIM-interacting, and single-stranded DNA-binding proteins. J. Biol. Chem. 2013, 288, 31581–31591. [Google Scholar] [CrossRef] [Green Version]
- Kimoto, M.; Tsubota, T.; Uchino, K.; Sezutsu, H.; Takiya, S. LIM-homeodomain transcription factor Awh is a key component activating all three fibroin genes, fibH, fibL and fhx, in the silk gland of the silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 2015, 56, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Mach, V.; Takiya, S.; Ohno, K.; Handa, H.; Imai, T.; Suzuki, Y. Silk gland factor-1 involved in the regulation of Bombyx sericin-1 gene contains fork head motif. J. Biol. Chem. 1995, 270, 9340–9346. [Google Scholar] [CrossRef] [Green Version]
- Matsuno, K.; Takiya, S.; Hui, C.C.; Suzuki, T.; Fakuta, M.; Ueno, K.; Suzuki, Y. Transcriptional stimulation via SG site of Bombyx sericin-1 gene through an interaction with a DNA binding protein SGF-3. Nucleic Acids Res. 1990, 18, 1853–1858. [Google Scholar] [CrossRef] [Green Version]
- Matsunami, K.; Kokubo, H.; Ohno, K.; Suzuki, Y. Expression pattern analysis of SGF-3/POU-M1 in relation to sericin-1 gene expression in the silk gland. Dev. Growth Differ. 1998, 40, 591–597. [Google Scholar] [CrossRef]
- Zhao, X.M.; Liu, C.; Li, Q.Y.; Hu, W.B.; Zhou, M.T.; Nie, H.Y.; Zhang, Y.X.; Peng, Z.C.; Zhao, P.; Xia, Q.Y. Basic helix-loop-helix transcription factor bmsage is involved in regulation of fibroin H-chain gene via interaction with SGF1 in Bombyx mori. PLoS ONE 2014, 9, e94091. [Google Scholar] [CrossRef] [PubMed]
- Dam, S.; Vosa, U.; Graaf, A.; Franke, L.; Magalhaes, J.P. Gene co-expression analysis for functional classification and gene-disease predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [PubMed]
- Tokimatsu, T.; Sakurai, N.; Suzuki, H.; Ohta, H.; Nishitani, K.; Koyama, T.; Umezawa, T.; Misawa, N.; Saito, K.; Shibata, D. KPPA-view. A web-based analysis tool for integration of transcript and metabolite data on plant metabolic pathway maps. Plant Physiol. 2005, 138, 1289–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, K.; Ogata, Y.; Shibata, D. Approaches for extracting practical information from gene co-expression networks in plant biology. Plant Cell Physiol. 2007, 48, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Amrine, K.C.H.; Blanco-Ulate, B.; Cantu, D. Discovery of core biotic stress responsive genes in Arabidopsis by weighted gene co-expression network analysis. PLoS ONE 2015, 10, e0118731. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Li, L.; Long, X.; You, W.; Zhong, Y.; Wang, M.; Tao, H.; Lin, S.; He, H. Construction and analysis of gene co-expression networks in Escherichia coli. Cells 2018, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.Q.; Li, Y.; Irwin, D.M.; Zhang, Y.P.; Wu, D.D. Integrative analysis of young genes, positively selected genes and IncRNAs in the development of Drosophila melanogaster. BMC Evol. Biol. 2014, 14, 241. [Google Scholar] [CrossRef] [Green Version]
- Behura, S.K.; Gomez-Machorro, C.; Harker, B.W.; deBruyn, B.; Lovin, D.D.; Hemme, R.R.; Mori, A.; Romero-Severson, J.; Severso, D.W. Global cross-talk of genes of the mosquito Aedes aegypti in response to dengue virus infection. PLoS Negl. Trop. Dis. 2011, 5, e1385. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Cheng, T.; Liu, C.; Liu, D.; Zhang, Q.; Long, R.; Zhao, P.; Xia, Q. Systematic identification and characterization of long non-cording RNAs in the silkworm, Bombyx mori. PLoS ONE 2016, 11, e0147147. [Google Scholar]
- Zhou, Q.Z.; Fu, P.; Li, S.S.; Zhang, C.J.; Yu, Q.Y.; Qiu, C.Z.; Zhang, H.B.; Zhang, Z. A comparison of co-expression networks in silk gland reveals the causes of silk yield increase during silkworm domestication. Front. Genet. 2020, 11, 225. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, A.; Nakazato, T.; Ito, K.; Nojima, Y.; Yokoyama, T.; Iwabuchi, K.; Bono, H.; Toyoda, A.; Fujiyama, A.; Sato, R. Identification of functional enolase genes of the silkworm bombyx mori from public databases with a combination of dry and wet bench processes. BMC Genom. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichino, F.; Bono, H.; Nakazato, T.; Toyoda, A.; Fujiyama, A.; Iwabuchi, K.; Sato, R.; Tabunoki, H. Construction of a simple evaluation system for the intestinal absorption of an orally administered medicine using Bombyx mori larvae. Drug Discov. Ther. 2018, 12, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, Y.; Nojima, Y.; Sakamoto, T.; Iwabuchi, K.; Nakazato, T.; Bono, H.; Toyoda, A.; Fujiyama, A.; Kanost, M.; Tabunoki, H. Comparative analysis of seven types of superoxide dismutases for their ability to respond to oxidative stress in Bombyx mori. Sci. Rep. 2019, 9, 2170. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, K.; Tsubota, T.; Jouraku, A.; Sezutsu, H.; Bono, H. Reference transcriptome data in silkworm Bombyx mori. Insects 2021, 12, 519. [Google Scholar] [CrossRef] [PubMed]
- Kruskal, J.B. On the shortest spanning subtree of a graph and the traveling salesman problem. Proc. Am. Math. Soc. 1956, 7, 48–50. [Google Scholar] [CrossRef]
- Campigotto, R.; Cespedes, P.C.; Guillaume, J.L. A generalized and adaptive method for community detection. arXiv 2014, arXiv:1406.2518. [Google Scholar]
- Koike, Y.; Mita, K.; Suzuki, M.G.; Maeda, S.; Abe, H.; Osoegawa, K.; deJong, P.J.; Shimada, T. Genomic sequence of a 320-kb segment of the Z chromosome of Bombyx mori containing a kettin ortholog. Mol. Genet. Genom. 2003, 269, 137–149. [Google Scholar] [CrossRef]
- Sakai, H.; Sumitani, M.; Chikami, Y.; Yahata, K.; Uchino, K.; Kiuchi, T.; Katsuma, S.; Aoki, F.; Sezutsu, H.; Suzuki, M.G. Transgenic expression of the piRNA-resistant Masculinizer gene induces female-specific lethality and partial female-to-male sex reversal in the silkworm, Bombyx mori. PLoS Genet. 2016, 12, e1006203. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.M.; Hu, B.L.; Zhou, Q.Z.; YU, Q.Y.; Zhang, Z. Comparative analysis of the silk gland transcriptomes between the domestic and wild silkworms. BMC Genom. 2015, 16, 60. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Cheng, T.; Wu, Y.; Hu, W.; Long, R.; Liu, C.; Zhao, P.; Xia, Q. Transcriptomic analysis of the anterior silk gland in the domestic silkworm (Bombyx mori)–insight into the mechanism of silk formation and spinning. PLoS ONE 2015, 10, e0139424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Sun, Q.; Huang, L.; Luo, Q.; Zeng, W.; Ou, Y.; Ma, J.; Xu, H. Genome-wide survey and characterization of transcription factors in the silk gland of the silkworm, Bombyx mori. PLoS ONE 2021, 16, e0259870. [Google Scholar] [CrossRef] [PubMed]
- Couble, P.; Michaille, J.J.; Garel, A.; Couble, M.L.; Prudhomme, J.C. Developmental switches of sericin mRNA splicing in individual cells of Bombyx mori silkgland. Dev. Biol. 1987, 124, 431–440. [Google Scholar] [CrossRef]
- Michaille, J.J.; Garel, A.; Prudhomme, J.C. Cloning and characterisation of the highly polymorphic Ser2 gene of Bombyx mori. Gene 1990, 86, 177–184. [Google Scholar] [CrossRef]
- Garel, A.; Deleage, G.; Prudhomme, J.C. Structure and organization of the Bombyx mori sericin1 gene and of the sericin1 deduced from the sequence of the ser 1B cDNA. Insect Biochem. Mol. Biol. 1997, 27, 469–477. [Google Scholar] [CrossRef]
- Kaneko, Y.; Takaki, K.; Iwami, M.; Sakurai, S. Developmental profile of annexin IX and its possible role in programmed cell death of the Bombyx mori anterior silk gland. Zool. Sci. 2006, 23, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, S.; Sagisaka, A.; Tanaka, H.; Ishibashi, J.; Kaneko, Y.; Yamaji, K.; Yamanaka, M. Molecular cloning and characterization of histone H2A.Z gene of the silkworm, Bombyx mori. J. Insect Biotechnol. Sericology 2007, 76, 121–127. [Google Scholar]
- Zhou, C.; Zha, X.; Shi, P.; Wei, S.; Wang, H.; Zheng, R.; Xia, Q. Multiprotein bridging factor 2 regulates the expression of the fibroin heavy chain gene by interacting with Bmdimmed in the silkworm Bombyx mori. Insect Mol. Biol. 2016, 25, 509–518. [Google Scholar] [CrossRef]
- Liu, Q.X.; Ueda, H.; Hirose, S. MBF2 is a tissue-and stage-specific coactivator that is regulated at the step of nuclear transport in the silkworm Bombyx mori. Dev. Biol. 2000, 225, 437–446. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Series | Tissue | Strain | SRA Run ID | Replicate | Reference |
|---|---|---|---|---|---|
| RNA-seq 1 | testis (TT) | o751 | DRR068893-068895 | 3 | Kikuchi et al., 2017 [21] |
| fat body (FB) | o751 | DRR095105-095107 | 3 | Kobayashi et al., 2019 [23] | |
| midgut (MG) | o751 | DRR095108-095110 | 3 | Ichino et al., 2018 [22] | |
| Malpighian tubule (MT) | o751 | DRR095111-095113 | 3 | Kobayashi et al., 2019 [23] | |
| whole silk gland (SG) | o751 | DRR095114-095116 | 3 | Kobayashi et al., 2019 [23] | |
| RNA-seq 2 | anterior SG (ASG) | p50T | DRR186474-186476 | 3 | Yokoi et al., 2021 [24] |
| anterior middle SG (A-MSG) | p50T | DRR186477-186479 | 3 | Yokoi et al., 2021 [24] | |
| middle middle SG (M-MSG) | p50T | DRR186480-186482 | 3 | Yokoi et al., 2021 [24] | |
| posterior middle SG (P-MSG) | p50T | DRR186483-186485 | 3 | Yokoi et al., 2021 [24] | |
| posterior SG (PSG) | p50T | DRR186486-186488 | 3 | Yokoi et al., 2021 [24] | |
| fat body (FB) | p50T | DRR186489-186491 | 3 | Yokoi et al., 2021 [24] | |
| midgut (MG) | p50T | DRR186492-186494 | 3 | Yokoi et al., 2021 [24] | |
| Malpighian tubule (MT) | p50T | DRR186495-186497 | 3 | Yokoi et al., 2021 [24] | |
| testis (TT) | p50T | DRR186498-186500 | 3 | Yokoi et al., 2021 [24] | |
| ovary (OV) | p50T | DRR186501-186503 | 3 | Yokoi et al., 2021 [24] |
| Target Gene | Modules | Total Transcripts | Transcription Factor |
|---|---|---|---|
| total | 1022 | ||
| sericin1 | 7 | 565 | 39 |
| sericin2 | 6 | 289 | 11 |
| sericin3 | 1 | 36 | 2 |
| fibroin-H | 1 | 42 | 5 (including Arrowhead) |
| fibroin-L | 1 | 80 (including P25) | 4 |
| SGF1 | 1 | 119 | 11 |
| SGF3 | 1 | 120 | 6 |
| sage | 1 | 114 | 8 |
| Antennapedia | 1 | 126 | 5 |
| Transcript ID | Module | Domain (PfamID) | Description (NCBI-nr) |
|---|---|---|---|
| KWMTBOMO00016 | sericin1 | zf-CCHC (PF00098), RT_RNaseH (PF17917), RVT_1 (PF00078), rve (PF00665) | unnamed protein product [Plutella xylostella] |
| KWMTBOMO14062 | sericin1 | zf-C2H2_4 (PF13894), PI-PLC-Y,X (PF00387, 00388), SH2 (PF00017), SH3_1 (PF00018), C2 (PF00168) | endonuclease-reverse transcriptase [Bombyx mori] |
| MSTRG.11166.1 | sericin1 | bZIP_1 (PF000170) | uncharacterized protein LOC101735428 isoform X2 [Bombyx mori] |
| MSTRG.14404.3 | sericin1 | Homeobox_KN (PF05920) | homeobox protein homothorax-like [Bombyx mori] |
| MSTRG.16824.2 | sericin1 | zf-C2HC_2 (PF13913) | homeobox protein 5 isoform X8 [Bombyx mori] |
| MSTRG.11460.1 | sericin2 | NCU-G1 (PF15065) | glycosylated lysosomal membrane protein B [Bombyx mori] |
| MSTRG.13691.2 | sericin2 | CENP-F_leu_zip (PF10473) | uncharacterized protein LOC114240082 [Bombyx mandarina] |
| MSTRG.14385.8 | sericin2 | Bromodomain (PF00439) | bromodomain adjacent to zinc finger domain protein 1A isoform X3 [Bombyx mori] |
| MSTRG.19405.151 | sericin2 | FLYWCH_zf (PF04500), BTB/POZ (PF00651) | Mod(mdg4)-heS00531 [Bombyx mori] |
| MSTRG.18671.5 | sericin3 | HSF_DNA-bind (PF00447) | heat shock factor-d isoform X4 [Bombyx mori] |
| MSTRG.11402.4 | fibroin-H | MBF2 (PF15868) | MBF2, partial [Bombyx mori] |
| MSTRG.1346.1 | fibroin-L | LIM (PF00412) | arrowhead PB [Bombyx mori] |
| MSTRG.9312.1 | fibroin-L | Myb_DNA-bind_7 (PF15963) | transcription factor TFIIIB component B’’ [Bombyx mori] |
| KWMTBOMO08832 | SGF1 | zf-CCHC (PF00098), rev (PF00665), Integrase_H2C2 (PF17921), Asp_protease_2 (PF13650) | uncharacterized protein LOC114250529 isoform X1 [Bombyx mandarina] |
| KWMTBOMO02915 | SGF3 | Forkhead (PF00250) | fork head domain-containing protein FD4 [Bombyx mori] |
| KWMTBOMO12108 | sage | Histone (PF00125), CBFD_NFYB_HMF (PF00808) | nuclear Y/CCAAT-box binding factor C subunit NF/YC isoform X1 [Bombyx mori] |
| MSTRG.3176.1 | Antennapedia | MTABC_N (PF16185) | transcriptional regulator ATRX homolog [Bombyx mandarina] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masuoka, Y.; Cao, W.; Jouraku, A.; Sakai, H.; Sezutsu, H.; Yokoi, K. Co-Expression Network and Time-Course Expression Analyses to Identify Silk Protein Regulatory Factors in Bombyx mori. Insects 2022, 13, 131. https://0-doi-org.brum.beds.ac.uk/10.3390/insects13020131
Masuoka Y, Cao W, Jouraku A, Sakai H, Sezutsu H, Yokoi K. Co-Expression Network and Time-Course Expression Analyses to Identify Silk Protein Regulatory Factors in Bombyx mori. Insects. 2022; 13(2):131. https://0-doi-org.brum.beds.ac.uk/10.3390/insects13020131
Chicago/Turabian StyleMasuoka, Yudai, Wei Cao, Akiya Jouraku, Hiroki Sakai, Hideki Sezutsu, and Kakeru Yokoi. 2022. "Co-Expression Network and Time-Course Expression Analyses to Identify Silk Protein Regulatory Factors in Bombyx mori" Insects 13, no. 2: 131. https://0-doi-org.brum.beds.ac.uk/10.3390/insects13020131