
Switch-Like Roles for Polycomb Proteins from Neurodevelopment to Neurodegeneration

Department of Translational Psychiatry, Max Planck Institute of Psychiatry, 80804 Munich, Germany
*
Author to whom correspondence should be addressed.
Epigenomes 2017, 1(3), 21; https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes1030021
Submission received: 16 October 2017
/
Revised: 13 November 2017
/
Accepted: 27 November 2017
/
Published: 1 December 2017
(This article belongs to the Special Issue Polycomb and Trithorax Group of Proteins in Development and Disease)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Polycomb Group (PcG) proteins are best-known for maintaining repressive or active chromatin states that are passed on across multiple cell divisions, and thus sustain long-term memory of gene expression. PcG proteins engage different, partly gene- and/or stage-specific, mechanisms to mediate spatiotemporal gene expression during central nervous system development. In the course of this, PcG proteins bind to various cis-regulatory sequences (e.g., promoters, enhancers or silencers) and coordinate, as well the interactions between distantly separated genomic regions to control chromatin function at different scales ranging from compaction of the linear chromatin to the formation of topological hubs. Recent findings show that PcG proteins are involved in switch-like changes in gene expression states of selected neural genes during the transition from multipotent to differentiating cells, and then to mature neurons. Beyond neurodevelopment, PcG proteins sustain mature neuronal function and viability, and prevent progressive neurodegeneration in mice. In support of this view, neuropathological findings from human neurodegenerative diseases point to altered PcG functions. Overall, improved insight into the multiplicity of PcG functions may advance our understanding of human neurodegenerative diseases and ultimately pave the way to new therapies.
1. Introduction
Polycomb Group (PcG) genes were originally discovered in Drosophila melanogaster (hereafter Drosophila) as a group of developmental regulators of the Hox gene cluster [1,2]. Hox genes, a subset of homeotic genes, comprise a group of related genes that control the embryonic body plan along the anterior-posterior axis. Since their original discovery, a large number of studies have established that PcG proteins maintain repressive chromatin structures at Hox and other (developmental) target genes that can be passed on across multiple cell divisions, thus adding to the long-term stability of gene expression states and the robustness of gene regulatory networks [3]. PcG protein mediated gene silencing is counteracted by the group of Thrithorax (TrxG) proteins that confer gene activation [4], and both repressive and activational states are equally important to normal development and beyond [4,5,6]. PcG and TrxG proteins are highly conserved in multicellular organisms [4,7] and share a central role in the (neuro-) development, differentiation, and maintenance of cell identity [8,9].
Developmental regulators are widely viewed to be expressed in a tightly controlled temporospatial pattern as a result of evolutionary constrained mechanisms that maintain robust changes in gene expression states governing patterning, proliferation, and differentiation [10]. In addition to this rigid role, recent findings show that PcG proteins are also involved in the dynamic and recurrent “On-Off” switches in gene regulatory activity [11] that contribute to distinct cell lineage decisions and the nascence of highly diverse cell types [10,12,13,14]. These regulatory transitions are guided by molecular epigenetic mechanisms that elsewise sustain a memory of cellular identity throughout development [3,15,16].
In the following, we take a snapshot of key features of PcG proteins comprising the assembly of core complexes, recruitment to target sites, chromatin marking, and the formation of topological hubs. Following this, we reconsider basic principles from mammalian neurodevelopment and current opportunities to model complex in vivo systems by means of pluripotent stem cells. This provides the opportunity to introduce the recent findings on dynamic switch-like roles of PcG proteins in cell lineages and cell fate transitions from early neural induction to neuronal maturation. Lastly, an unexpected role for PcG proteins in auto- and coregulatory transcriptional networks disrupting postmitotic neuronal identity is discussed.
2. A Snapshot of Polycomb Group Proteins
2.1. Composition of Polycomb Group Complexes
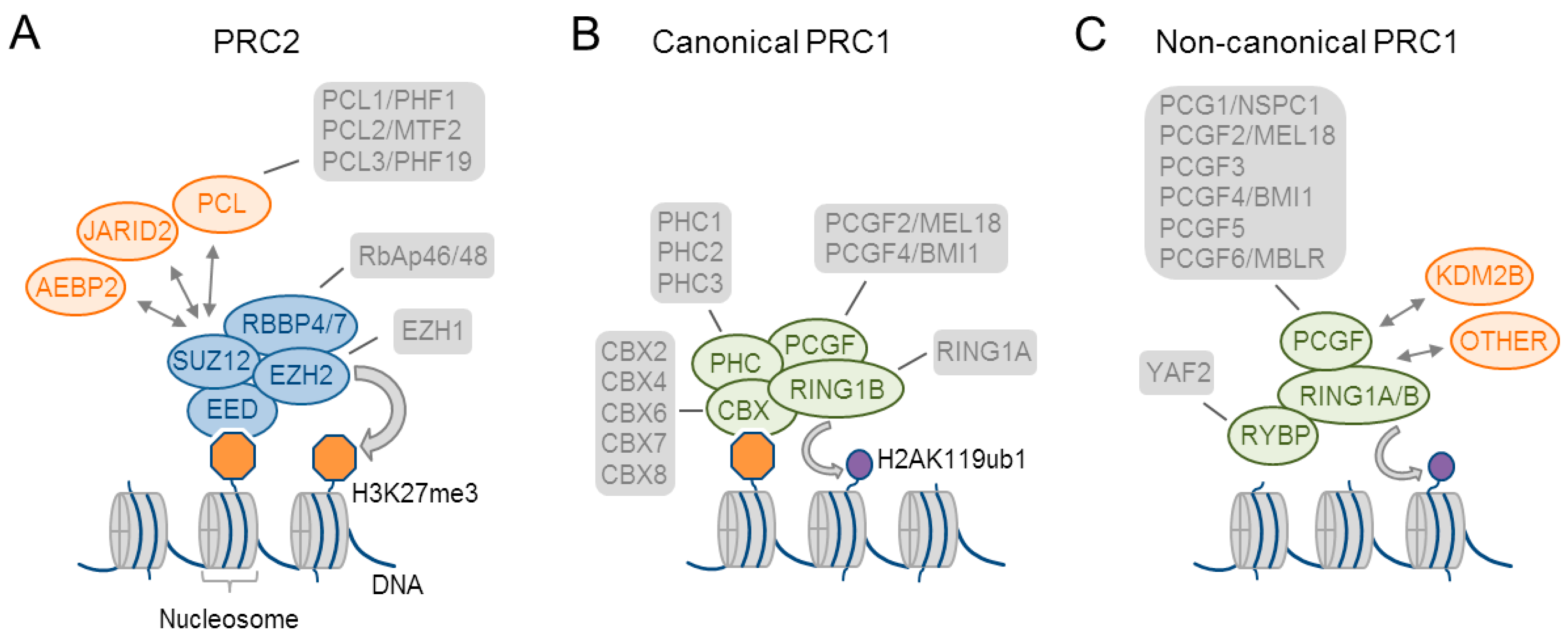
Two key PcG complexes have been identified by biochemical purification experiments: Polycomb repressive complex (PRC) 1 and PRC2 [7] (Figure 1A–C). PRC1 is distinguished into canonical (cPRC1) and non-canonical complexes (ncPCR1). All of the PRC1 complexes share common core subunits that comprise RING1 proteins (RING1A or RING1B), which catalyze ubiquitylation of histone H2A on lysine 119 (H2AK119ub) through their E3 ubiquitin ligase activity, and one of the six PcG ring-finger domain proteins (PCGF1–PCGF6). cPRC1 complexes (Figure 1B) organize around PCGF2/4 and contain as specific subunits: (i) a chromobox protein (CBX2, CBX4, and CBX6-CBX8), which recognizes trimethylation of histone 3 on lysine 27 (H3K27me3), and (ii) a Polyhomeotic (PH) homologous protein (PHC1–PHC3). PHCs contain a sterile alpha motif (SAM) domain that fulfills an important role in PcG-mediated gene repression. By contrast, ncPRC1 (Figure 1C) contains as specific subunit: (i) the zinc-finger domain and YY-1 binding protein (RYBP) or its paralog YY1-associated factor 2 (YAF2). These specific subunits associate with PCGF1, PCGF3/5, or PCGF6 to form ncPRC1.1, ncPRC1.3/PRC1.5, or ncPRC1.6, respectively (for a recent review see [17]). ncPCR1.1 also associates with the accessory regulatory subunit KDM2B, a H3K36-specific histone demethylase with a possible role in DNA binding (see Section 2.2).
The functional core of mammalian PRC2 consists of one of the SET-domain-containing histone methyltransferases enhancer of zeste homologue 1 or 2 (EZH1 or 2), embryonic ectoderm development (EED), suppressor of zeste (SUZ12), and the CAF1 histone-binding proteins RBBP4 and RBBP7 (Figure 1A). In mammals, EZH1 or 2 have markedly different expression patterns (see Section 4.6) and activities: (i) EZH2 catalyzes preferentially di- and tri-methylation of histone H3 on lysine 27 (H3K27me2 and 3) in embryonic stem cells (ESCs) and proliferating cells. By contrast, EZH1 replaces EZH2 in distinct differentiating, and post-mitotic cells and may be less active [18], or even promote, transcriptional activation [19,20].
PRC2 associates additionally with accessory regulatory subunits (Figure 1A) that modulate its enzymatic activities and the recruitment to chromatin sites. This diversity may reflect an increased need for specification of cell identity during development and differentiation in metazoans. Two alternate complexes consisting of the PRC2 core complex and accessory proteins [21] have been described: (i) PRC2.1 is characterized by the mutually exclusive binding of one of the three Polycomb-like homologs (PCLs) PHF1, PHF19, or MTF2. Within this complex, PHF1 enhances EZH2’s catalytic activity toward the H3K27me2 substrate. (ii) PRC2.2 is characterized by the association with the zinc-finger proteins AEBP2 (Adipocyte Enhancer-Binding Protein 2) and JARID2 (Jumonji, AT-Rich Interactive Domain 2 protein), which stimulate EZH2’s catalytic activity and regulate chromatin binding (Figure 1A).
Taken together, mammalian PcG proteins assemble in various multifaceted complexes that are endowed with distinct functional properties. Further studies are necessary to translate this diversity into defined biological functions and to elucidate the functional importance of individual subunits involved.
2.2. Recruitment of Polycomb Group Complexes
The binding of PcG complexes is best understood in Drosophila that harbors in its genome so-called PcG response elements (PREs). These cis-regulatory sequences (i) recruit PcG and TrxG proteins; (ii) maintain a silent or active state dependent on the inputs from their associated promoter and enhancer; (iii) may secure a stable epigenetic memory of transcriptional states; and (iv) show an inherent flexibility enabling the switches or modulation of their output in response to developmental or environmental cues. By contrast, the foundations of mammalian PcG binding sites are less well understood, and different mechanisms have been suggested (for recent review [17,22]). The hypothesis that CpG islands (CGIs, regions above a certain threshold of C+G and CpG dinucleotides) are the long-sought vertebrate PREs has been supported by the finding that PRC1, PRC2, and H3K27me3 localize at approximately 30% of promoter CGIs featuring the active mark H3K4me3 in ESCs [23,24]. However, genome-wide profiling showed that one-quarter to one-third of the PRC1 and PRC2 binding sites do not localize to annotated promoters and map to intergenic sites devoid of canonical CGIs [25,26]. Conversely, insertion of GC-rich sequences at ectopic sites or the reduction of endogenous DNA methylation has been shown to trigger H3K4me3/H3K27me3 marking and wide-spread redistribution of both PRC1 and PRC2 to GC-rich sites.
A plausible mechanism for a direct DNA based mechanism for PcG complex binding is provided by the accessory subunit KDM2B, a component of ncPRC1.1 (Figure 1C), that binds specifically to non-methylated CpG dinucleotides via its CxxC domain. Yet, KDM2B associates genome-wide with CGIs, whereas PRC1 components are detected at only 15 to 30% of these sites [27,28,29]. Furthermore, ChIP (chromatin immunoprecipitation) overlaps were assessed by RING1B protein, which is a component of multiple PRC1s, raising the question to which degrees RING1B bound sites are actually shared by KDM2B. Several models have been advanced to explain the puzzling finding that mammalian PcG proteins do not bind to all CGIs, nor to all KDM2B bound sites. Briefly, a “chromatin sampling” model suggests that PcG proteins interact only transiently with all of the potential sites since transcriptional activity precludes PcG from potential sites with high binding capacity [30]. Consistent with this scenario, global inhibition of transcription triggers a significant colonization of these silenced sites by PRC2 [31] Still, 30 to 40% of CGIs do not recruit PRC2 under this condition and 10 to 20% of the active genes show PcG binding in ESCs. Alternative models suggest that sequence-specific DNA-binding proteins “instruct” the recruitment of PcG and/or TrxG proteins to specific sites that are reminiscent of Drosophila [30]. If yes, then these factors still do not associate with PcG globally (for review see [22]) except JARID2 (Figure 1A) that is globally required for PRC2 binding [32,33,34,35]. JARID2 directly binds DNA in vitro though with low sequence specificity. This promiscuity questions a self-standing role in chromatin targeting of PRC2. Instead, recent findings suggest that PRC2 containing JARID2 and AEBP2 (Figure 1A) can bind to ubiquitinated histone H2A and that this event enhances additionally PRC2’s catalytic activity [36]. Moreover, PRC2 recognizes H3K27me3 by its core component EED (Figure 1A) that can recruit PRC2 to sites of pre-existing H3K27me3. At the same time, H3K27me3 binding stimulates PRC2’s catalytic activity and may self-reinforce the propagation of repressive chromatin [37,38].
Lastly, many PcG target genes from Drosophila and vertebrates are transcribed in non-coding RNAs (ncRNA), and several PcG and TrxG proteins can bind to RNA in vitro. These findings have prompted the hypothesis that interactions of PcG and TrxG proteins with specific ncRNAs can guide PcG complexes to specific sites in vivo [39,40]. Consistent with this concept, several ncRNAs support PcG and TrxG function in vivo with high specificity (for review [41]). Still, this model raises the question of how a generic PcG complex can recognize specifically hundreds of targeting RNAs among thousands of highly expressed unrelated RNA species.
Altogether, chromatin recruitment of mammalian PcG complexes builds on multiple, mutually non-exclusive, mechanisms that are likely to depend to a varying degree on sequence content, cell-type, and the developmental stage. Metaphorically speaking, “many roads lead to Rome”.
2.3. Polycomb Group Complexes Regulate Chromatin at Different Scales
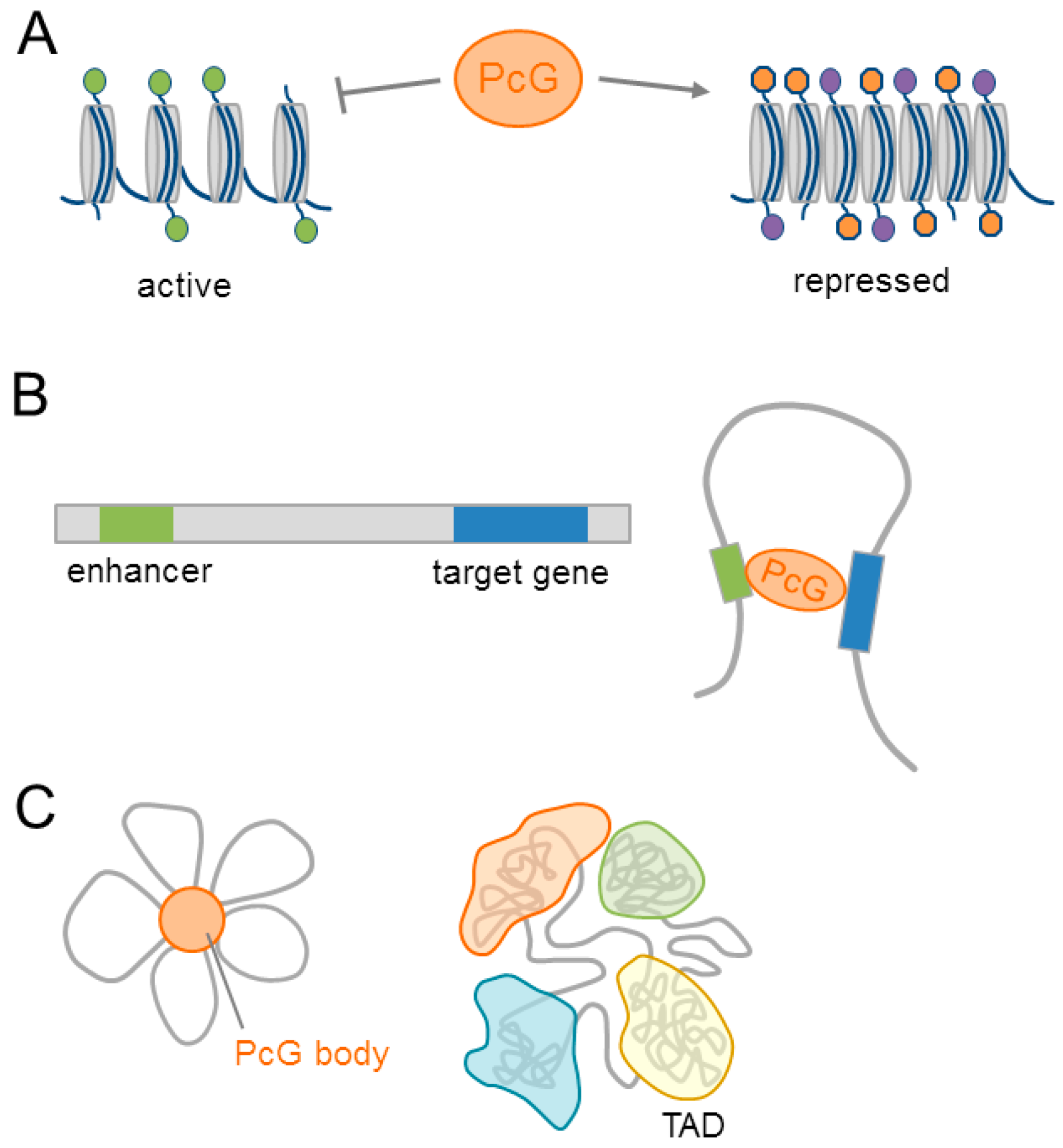
PcG mediated H3K27me3 prevents the acetylation of histone H3 on lysine 27 (H3K27ac), an activating mark, and the subsequent recruitment of Pol II (DNA polymerase II) [42]. In addition, PcG proteins bind directly to the acetyltransferase CREB-binding protein (CBP), and inhibit its catalytic activity towards H3K27 [43]. Together, these events maintain local chromatin compaction precluding the transcription of target genes (Figure 2A).
Beyond the linear chromatin, PcG proteins mediate interactions between distant DNA regulatory elements (e.g., promoters and enhancers) via chromatin looping (Figure 2B). Originally detected in Drosophila [44], increasing evidence supports PcG dependent chromatin looping in mammals (for a role in neurodevelopment see Section 4.1). Moreover, PcG-bound DNA regions assemble in discrete subnuclear structures termed PcG bodies [45] (Figure 2C). The relevance of these regulatory structures to neurodevelopment will be discussed in Section 4.1 and Section 4.3.
A potential layer in PcG regulated gene expression is the modulation of global nuclear architecture. Linear chromatin can fold into distinct three-dimensional (3D) structures, called topological associated domains (TAD), via strong genomic interactions within a domain. By contrast, the segments that are localized between the respective TAD borders show little interactions (Figure 2C). TADs are highly conserved across different species and cell types [46,47], and segregate dependent on their association with the nuclear lamina, replication timing, histone signatures, and the transcriptional activity of the genes residing within a TAD [48,49].
In Drosophila, the PRC1 component PHC polymerizes via its SAM domain and assembles PcG complexes into Polycomb bodies [50], with an essential role in gene repression [51]. The loss of PHC function abolishes the more compact and higher inter-TAD interactions of polycomb associated TADs when compared to other domains, and it indicates that PHC could influence global chromatin packaging [52].
This finding raises the question of whether mammalian SAM domains fulfill a similar role, and whether different PcG complexes (Figure 1A–C) could differ in their effects on 3D genome organization (see also Section 4.1). In this respect, the loss of PRC2 function disrupts long range interactions [53,54], but does not affect mammalian TAD formation.
Conclusively, mammalian PcG proteins bind to different regulatory sites in the genome to mediate chromatin compaction, chromatin looping, and the formation of PcG bodies. Together, these processes are key to maintaining gene expression states that are already established by master regulators during development and beyond.
3. Embryonic and Adult Neurogenesis
The central nervous system of mammals consists of highly specialized neuronal and glial cells that closely interact with each other. All of these cells are derived from common neural precursor cells (NPCs), which are also known as neural stem cells (NSCs) that reside in delimited anatomical regions during distinct developmental time windows and beyond [55]. Neurogenesis begins at embryonic day 8 (E8) in mice, and reaches a plateau around E14. NPCs originate from neuroepithelial cells, which line the spinal canal and forebrain ventricles at embryonic stages of development. At E8-E10, neuroepithelial cells develop into astroglial-like cells, termed radial glial cells (RGCs) that show both neuroepithelial and glial properties [56]. Epithelial features are thought to sustain the apical-basal polarity of RGCs that contributes to the migration of nascent neurons. RGCs represent fate-restricted progenitors, which generate neuronal and glial cell types in a tightly controlled sequential manner. Early RGCs directly produce nascent neurons or intermediate neuronal progenitor cells that differentiate in turn into neurons through symmetrical mitosis. At the same time, RGCs sustain self-renewal by undergoing asymmetrical cell divisions [57]. Following this early “neurogenic” phase, the late RGCs participate also in gliogenesis through the production of intermediate progenitor cell types that further develop into astrocytes or oligodendrocytes. During this stage, self-renewing RGCs are anchored on both the pial and ventricular surfaces and contact blood vessels for signaling transduction and nutrient supply. Concomitantly, such spatially aligned RGCs serve as a scaffold for the continuous migration of newly born neurons and glial cells towards the cortex, thickening of the neocortex, and the formation of an interneuron network. With the end of embryonic neurogenesis around postnatal day one, RGCs finally detach from the ventricular site, migrate towards the cortical plate, and transform into astrocytes. Only a small number of residual RGCs subsists quiescently as so-called B cells in the subventricular zone (SVZ) of the lateral ventricles, and contributes to adult neurogenesis [56,57]. B cells firstly produce transient amplifying cells (also known as C cells) that correspond to intermediate neuronal progenitor cells and subsequently differentiate into neuroblasts (A cells). These immature neurons travel via the rostral migratory stream through the olfactory bulb and finally differentiate into different subtypes of local interneurons [55].
Also, the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) participates in adult neurogenesis [55,58] and shares with the SVZ many features of embryonic neurogenesis. Similar to the SVZ, radial astrocytes in the SGZ (termed type I progenitors) act as primary NSCs that produce intermediate neuronal progenitor cells (termed type II progenitors) that migrate into the inner granular cell layer of the DG to integrate in the existing circuitry.
In brief, embryonic NSCs correspond to RGCs that share many features with adult NSCs comprising subventricular B cells and subgranular type I progenitors [55]. This commonality suggests that many developmental factors and pathways from embryonic neurogenesis may also operate in SVZ- and SGZ-derived neurogenesis.
4. Polycomb Group Complexes Guide Dynamic Transitions in Neurodevelopment
4.1. Polycomb Group Complexes in Embryonic Stem Cells Restrain Neural Differentiation
The chromatin of ESCs is highly plastic and contributes to their developmental pluripotency: it is decondensed, histone proteins are only loosely bound, and exchanged dynamically [59]. With the onset of differentiation these features are reversed and heterochromatic foci form and spread across the genome. Although the ESC state depends on a core network of sequence-specific TFs, namely Nanog (homeobox TF Nanog), Sox2 (SRY-related HMG-Box Gene2), and Oct4 (POU Domain, class 5, TF1; POU5F1) [60], increasing evidence suggests that the chromatin landscape constitutes an additional layer of gene regulation. Genome-wide mapping of chromatin modifications has uncovered, among common regulatory marks, also distinct chromatin signatures that appear to be more specific to ESCs. These include the activating mark H3K4me3 and the repressive PcG associated mark H3K27me3. In a pioneering study, Bernstein et al. [23] conducted ChIP experiments with antibodies against specific histones marks and hybridized the enriched DNA fragments to tilling arrays. The mark H3K4me3 and H3K27me3 mapped to highly conserved noncoding regions preferentially localized at developmental genes, such as the Hox gene cluster. Interestingly, ≈75% of the H3K27me3 sites localized to transcriptional start sites (TSS) that also carried the mark H3K4me3. Additional sequential ChIP sequencing experiments confirmed that both marks co-occurred mostly at genes encoding developmental TFs. Genes featuring these so-called bivalent domains were expressed at low levels in ESCs irrespective of the activating mark. By contrast, some of them rapidly lost their repressive mark and were robustly induced upon neural differentiation, while others lost the activating, but not the repressive mark, and became silenced. In agreement with these findings, EED deficient undifferentiated ESCs showed an upregulation of some bivalently marked genes, leading to premature expression of lineage specific genes. Together, these studies suggest to PRC2 a role in maintaining the undifferentiated state of ESCs [61].
Soon after the initial report [23], bivalent domains were genome-wide mapped through next-generation sequencing of ChIP enriched DNA (ChIP-seq) from mouse ESCs [62]. Almost all of the promoters covered by a CGI carried the activating mark H3K4me3, while only ≈25% featured also the repressive mark H3K27me3. The corresponding genes were weakly expressed and encoded mostly developmental TF, morphogens, and surface molecules. Bivalent domains were additionally identified in different kind of pluripotent cells [63,64,65,66,67], which is consistent with the idea that bivalent domains keep major developmental genes in a silent, though poised state.
Bivalent domains from mouse are well-conserved in human ESCs [68,69] and rapidly resolve upon (neural) differentiation. Conversely, reprogramming of terminal differentiated cells into induced pluripotent stem cells [70] reinstates a chromatin landscape and bivalent domains closely resembling those from ESCs [71,72,73], and suggests that chromatin bivalency evolves during the genesis of ESCs.
Beyond linear chromatin marking, PcG proteins assemble promoters and enhancers in 3D networks [74]. Enhancers are regulatory regions that can locate on the linear genome quite far from their target genes and still contact them via chromatin looping that joins target genes and enhancers in 3D space [75] through the action of structural protein complexes comprising CTCF (CCCTC-binding factor), cohesin, and Mediator [76]. Similar to promoters, many enhancers operate in a cell-type specific and spatiotemporal manner during development. Active enhancers confer strong gene expression, are enriched in the chromatin marks H3K4me1 and H3K27ac, and recruit the histone acetyltransferase EP300 (E1A-binding protein). By contrast, primed enhancers are devoid of H3K27ac although they retain EP300 and H3K4me1, and support basal levels of gene expression. Finally, poised enhancers likewise feature H3K4me1 and EP300, but also carry the repressive PcG mediated mark H3K27me3 that prevents the accumulation of H3K27ac and spurious activation by EP300 [77]. Poised enhancers associate with developmental loci in pluripotent mouse and human ESCs, in which they bookmark a limited set of regulatory sequences to prime timely and lineage-specific activation once the appropriate differentiation cue is received [78].
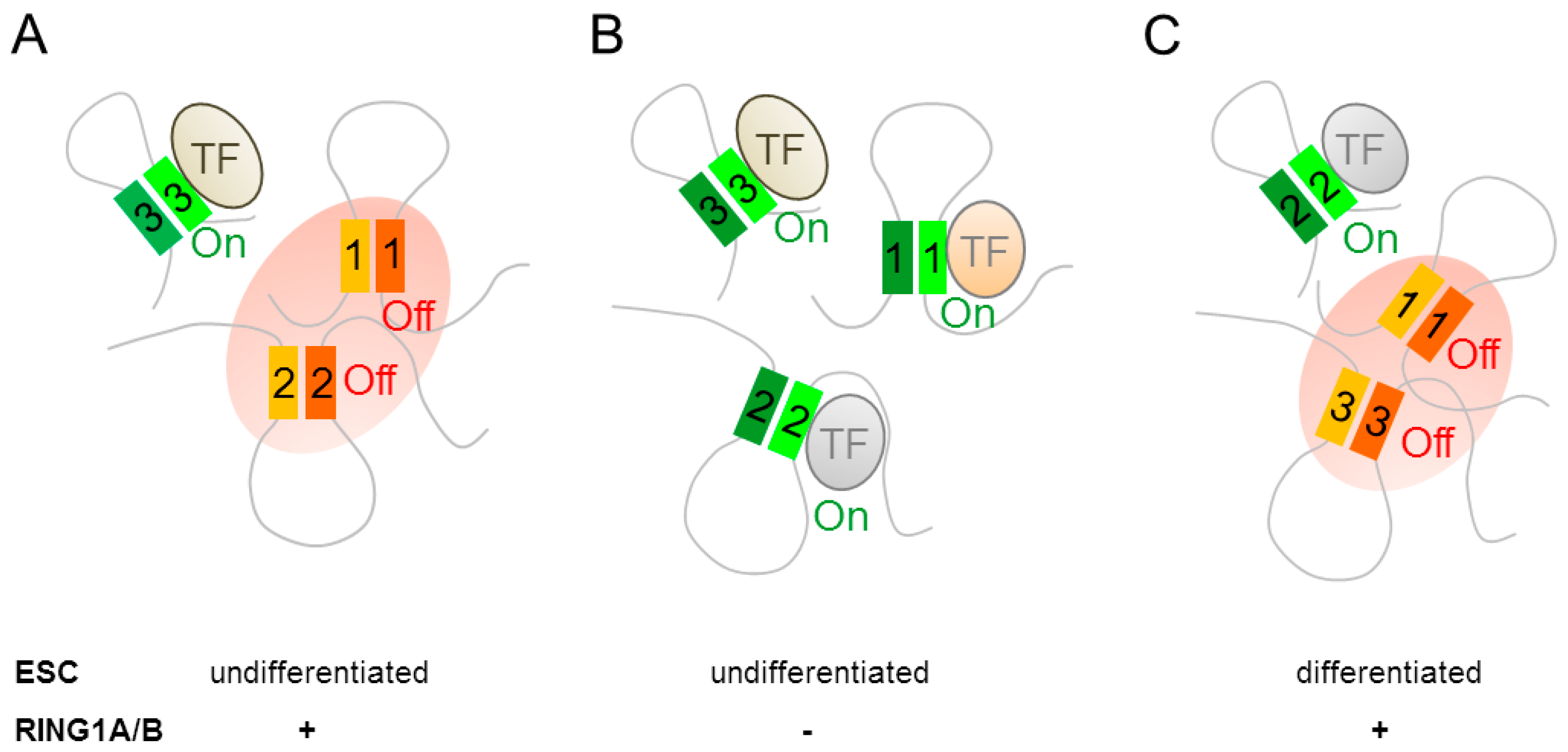
A role for PcG proteins in 3D organization of mammalian chromatin was inferred by Schoenfelder et al. [74] from high-throughput chromosome conformation capture assays (i.e., promoter capture Hi-C) on undifferentiated mouse ESCs. Genes that were bound by the PRC1 component RING1B formed distinct contact networks; among these, the strongest consisted of the 4 Hox gene clusters and 66 other key developmental regulators. PRC1 mediated contacts localized to the promoters and distal enhancers of the respective target genes (Figure 3A), and clearly separated from a network of genes that were recognized by pluripotency factors. Knock out experiments showed that loss of RING1A alone reduced network connectivity, whereas additional loss of RING1B disrupted the highly centralized promoter interactome of the Hox gene network and induced gene expression (Figure 3B).
Within this network, RING1B contacted a group of poised enhancers that were marked by H3K4me1 and H3K27me3. Although the loss of RING1B disrupted promoter-promoter contacts in the Hox gene network, promoter-enhancer contacts persisted since they were not mediated by RING1B (Figure 3B). Concomitantly, poised network enhancers acquired active chromatin signatures (replacement of H3K27me3 by H3K27ac) and stimulated expression of the associated genes. Notably, Hox network genes contacting poised enhancers in double-knock out cells showed the most significant transcriptional activation. By contrast, the small number of RING1B regulated promoters that establishing de novo contact to active enhances showed only weak transcriptional activation. These results indicate that PRC1 dependent assembly of promoters and poised enhancers in 3D space mediates silencing and that preformed, PRC1 independent, contacts between promoters and poised enhancers confer robust transcription once the PRC1 complex disengages during neural differentiation (Figure 3C).
Moving forward, Kundu et al. [79] showed by 5C and super-resolution microscopy that the chromatin of PRC1 bound developmentally controlled genes assembled in compacted structures that depended on cPRC1 and on interactions solely between PRC1 bound regions. By contrast, ncPRC1 and H2A ubiquitylation were neither necessary nor sufficient to domain formation. PRC1 domains encompassed entire or multiple genes and surpassed in size those formed by PRC1 mediated nucleosome compaction in vitro. For example, PRC1 domains spanned sizes, ranging from 140 (HoxA) to 20 kb (Pax6), although these are considerably smaller than those that were formed by TADs, straddling several hundreds of Kb to 1–2 Mb in size. Activation of PRC1 target genes during neural cell-fate specification triggered de-compaction of PRC1 bound domains and stimulated interactions within mapped TAD domains. This finding could indicate a potential co-regulation of TAD’s long-range interactions via PRC1.
Collectively, PcG proteins regulate chromatin configuration at different scales in neural gene expression. At the linear chromatin, PcG proteins map to bivalently marked domains that poise target genes for rapid, switch-like, induction in response to differentiation signals. Additionally, PRC1 assembles (neuro-) developmental genes, and their poised enhancers in a silenced, though preactivated, 3D network, whose disassembly initiates early differentiation. In this respect, PRC1 bodies may act as chromatin hubs with distinct structural features that maintain the repression or activation of early (neuro-) developmental genes.
4.2. Polycomb Group Complexes in Neural Progenitor Cells
As early as 2007, Mikkelsen et al. [62] observed that some 8% of the bivalent domains from mouse ESCs subsisted following neural differentiation. Moreover, Mohn et al. [14] noted that ≈675 bivalent domains that were resolved during the progression of pluripotent mouse ESCs to RGC-like neural progenitors, whereas, ≈575 bivalent domains emerged anew. Following terminal differentiation into glutamatergic pyramidal neurons, ≈1000 bivalent domains were resolved concomitantly to the formation of ≈340 new bivalent domains. These patterns suggest that bivalent domains form and resolve dynamically during successive neurodevelopmental stages to regulate diverse sets of target genes.
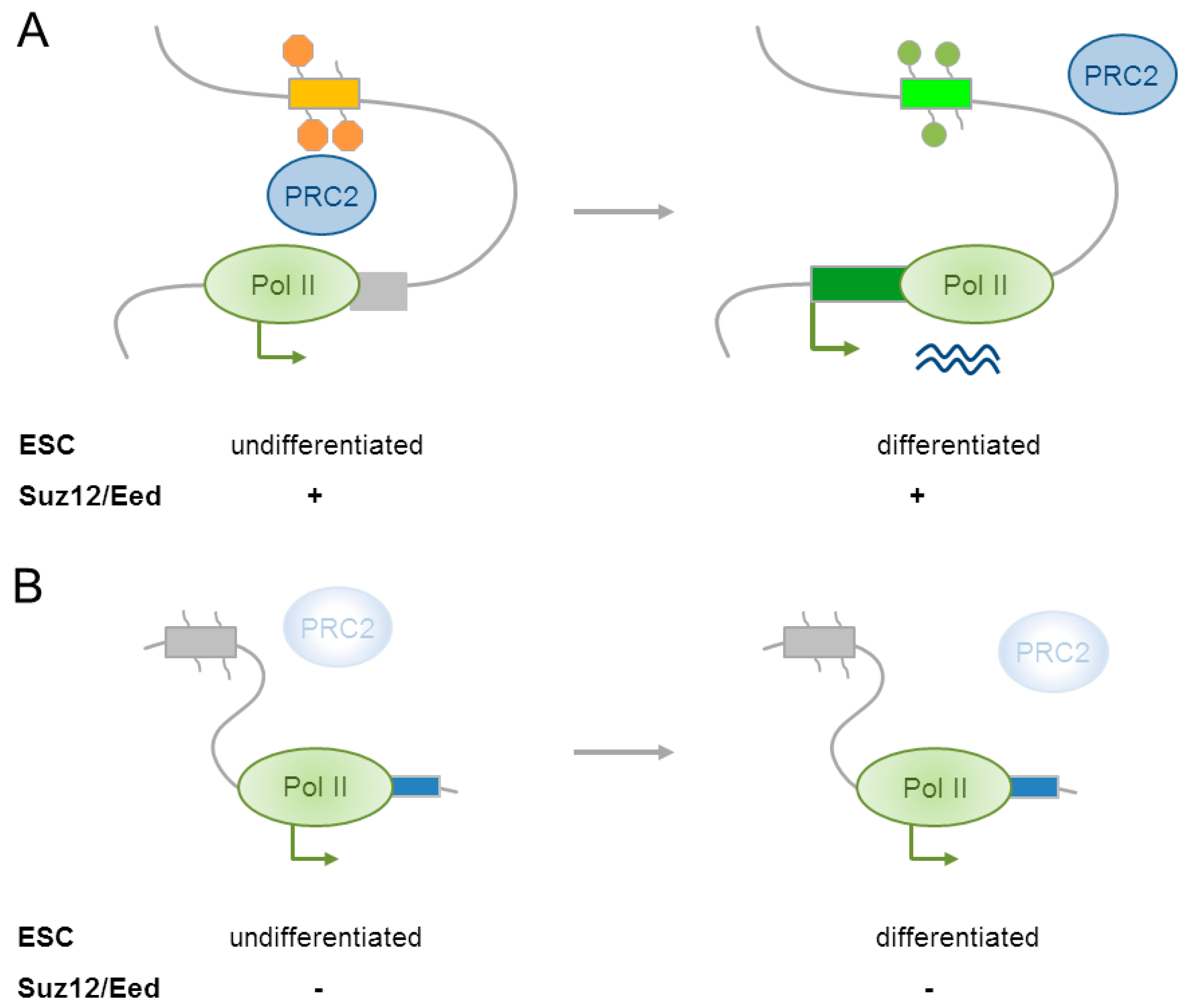
Moving beyond the linear chromatin landscape, most recently Cruz-Molina et al. [12] showed that PRC2 mediated contacts with poised enhancers (see Section 4.1) and promoters in ESCs established a permissive environment that preconditioned the induction of major anterior neural genes (e.g., Lhx5, Six3, Sox1, Wnt8b, Sox21) in NPCs. Genome-wide mapping of EP300 and H3K27me3 detected ≈1000 poised enhancers that were enriched in genes that were necessary for the development of the anterior neural tube. Furthermore, these enhancers aligned preferentially with active enhancers (H3K27ac) from the embryonic forebrain. About ≈20% of the poised enhancers acquired H3K27ac following directed differentiation of mouse ESCs into anterior neural progenitors and almost all of these associated with genes that were regulating early brain development. Interestingly, poised enhancers physically contacted their target promoters already in the silenced state in ESCs as evidenced by 4C-seq (high resolution circularized chromosome conformation capture sequencing) preceding their subsequent activation in anterior NPCs (Figure 4A). To assess the functional relevance of poised enhancers, a selected subset was deleted by the CRISPR/Cas9 technique in mouse ESCs. This experiment showed that poised enhancers were not required for maintaining the inactive state of target genes in ESCs, arguing against a role as silencers. By contrast, following neural differentiation, the induction of genes devoid of poised enhancers was severely reduced. This result shows that poised enhancers are necessary for the induction of major anterior neural genes.
As evidenced by 4C-seq experiments, a knockout of the core subunits Suz12 and Eed disrupted enhancer-promoter contacts and supported a role of PRC2 in their mediation (Figure 4B). By contrast, other structural proteins that were involved in chromatin looping (e.g., CTCF, cohesin, and Mediator) were undetectable at the base of the contacting loops. Following differentiation of Suz12/Eed knockout cells into anterior NPCs, poised enhancers did not acquire H3K27ac even though they lacked H3K27me3 from the very beginning (Figure 4B).
Taken together, PcG proteins are recurrently recruited to neural genes to guide dynamic transitions in gene regulation from ESCs to NPCs to neurons. Beyond the prevention of premature activation of early developmental regulators, PRC2 presets also future activation of major anterior neural genes. PRC2 mediated contacts with poised enhancers, and their associated promoters can establish a permissive regulatory topology that enables pluripotent cells to timely respond to neural differentiation signals.
4.3. Polycomb Group Complexes in Differentiating Neural Cell Types
The histone methyltransferase Ezh2 is highly expressed in neural progenitors, but rapidly declines as cortical neurons differentiate [80]. Conditional deletion of Ezh2 from E9.5, the onset of the neurogenic period in mice, caused a persistent loss of H3K27me3 accompanied by a substantial upregulation of gene expression. Functionally, loss of Ezh2 accelerated neurogenesis and neuronal differentiation. This event cumulated in an exhaustion of apical and basal progenitor cells and, consequently, a thinned cortex [80]. Beyond neurogenic progenitors, loss of Ezh2 affected also glial lineages and resulted in a strongly advanced production of mature astrocytes in the E16 ventricular zone. Even though Ezh2 deficiency constricted the neurogenic period for progenitor cells and their neuronal production, the temporal order of the developmental fate switch remained largely intact.
Beyond neural progenitors, PcG complexes also regulate the switch from neurogenesis to astrogliogenesis [81]. Knockout of the Ring1B, Ezh2, or Eed genes at later stages of cortical development (E13.5) prolonged the neurogenic phase of NPCs at the cost of a postponed, rather than advanced, astrogliogenesis. Neurogenin 1 (Ngn1) and Ngn2, which are two basic helix-loop-helix (bHLH) TFs, are expressed solely during the neurogenic, but not in the astrocytic, phase of neocortical development and appear critical to phase transition in NPCs [82]. In support of this hypothesis, PcG proteins are recruited to the promoters of Ngn1 and Ngn2 as corticogenesis proceeds and maintain their repressed state [81]. Ngn1 inhibits the astrocytic differentiation of NPCs in part by sequestration of the p300/CBP-Smad1 complex from STAT3 (signal transducer and activator of transcription), which promotes astrogenesis [83]. In brief, PcG protein mediated silencing of Ngn1 turns off the neurogenic phase and primes the neurogenic-to-astrogenic fate switch.
Polycomb’s role in developmental fate switches extends further to later cortical gliogenesis when oligodendrogenesis displaces the production of astrocytes. In the ventral telencephalon, neural progenitors can generate gamma-aminobutyric acid (GABA)-ergic neurons and oligodendrocytes through incompletely understood mechanisms. Petryniak et al. [84] showed that distal-less homeobox transcription factors (Dlx) were necessary for GABA-ergic interneuron production and the repression of oligodendrocyte precursor cell (OPC) formation by acting on a common progenitor to define neuronal versus oligodendroglial cell fate acquisition. In the embryonic cortex, the loss of Ring1B de-repressed Dlx2 [81] and promoted neurogenesis. This finding indicates that the repression of neurogenic Dlx1/2 activity was necessary for the developmental fate switch to oligodendrogenesis.
Collectively, PcG complexes play a critical role during successive cortical fate switches by restraining the neurogenic, gliogenic, and astrogenic competence of neuronal progenitors.
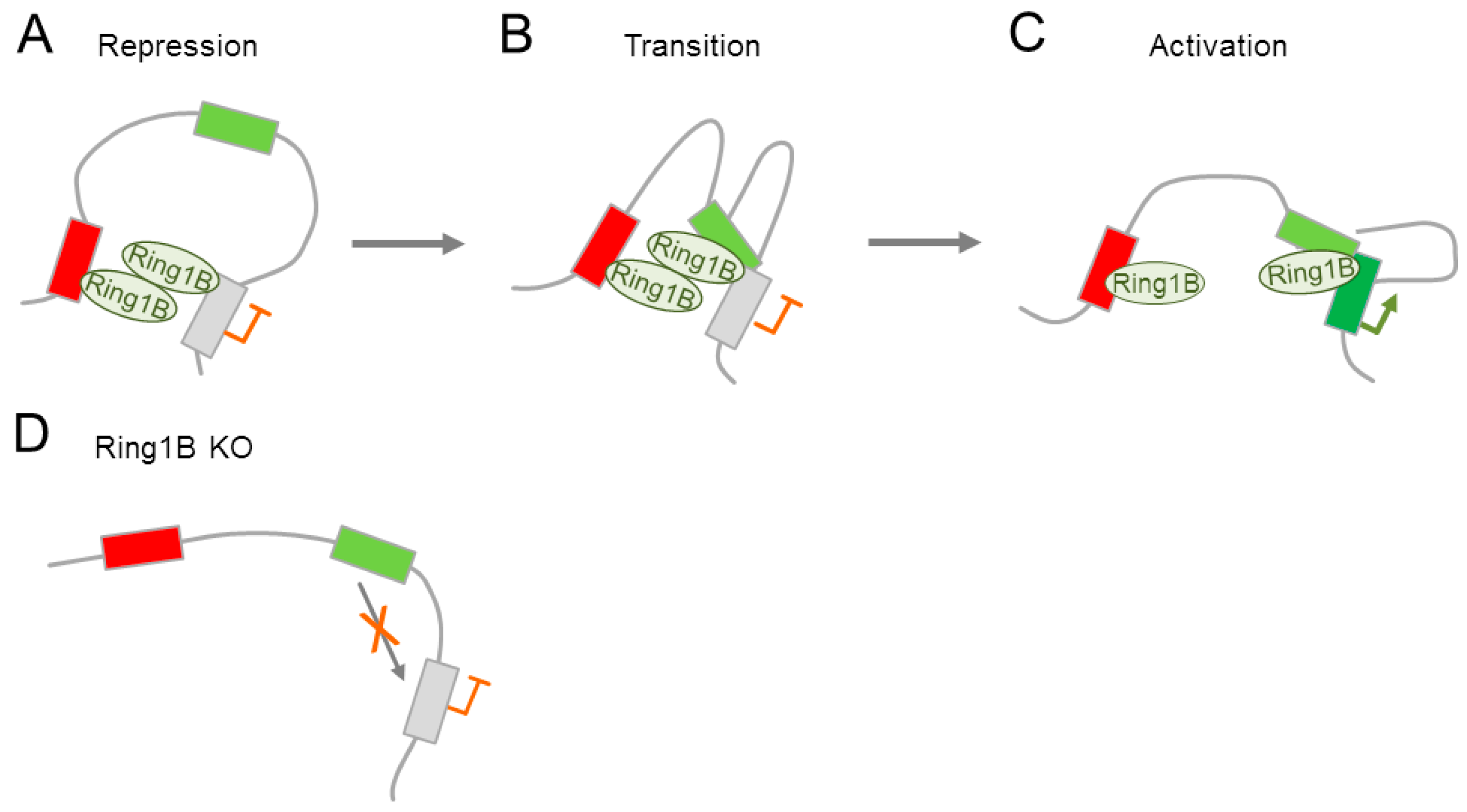
The ability of PcG proteins to mediate 3D interactions (see Section 4.2) could also apply to differentiated neural cells. Indeed, Kondo et al. [13] showed that PcG proteins regulate the mouse homeobox gene Meis2 in the embryonic midbrain and forebrain by mediating an interaction with its promoter and a tissue specific enhancer. In the preactivational state, Ring1B established contacts with the Meis2 promoter and a putative silencer region downstream of the poly-A site (Figure 5A). By contrast, late transcriptional activation in the embryonic midbrain was associated with promoter-enhancer interactions and the gradual dissociation of Ring1B from the promoter (Figure 5B,C). Progression from the repressive to the activated state is associated with a transient Ring1B dependent tripartite interaction among promoter, enhancer, and silencer in vivo (Figure 5B). Conversely, knock out of Ring1A or Ring1B prevented this tripartite interaction (Figure 5D), and led to a downregulation of Meis2 expression in the midbrain and forebrain, in which it is usually expressed. Concurrently, Meis2 expression was induced in many tissues in which it is usually not expressed Hence, PcG proteins were necessary for the formation of Meis2 enhancer-promoter interactions and subsequent transcriptional activation.
Collectively, PcG proteins control tissue-specifically both Meis2 activation and repression by mediating Ring1B dependent dynamic promoter-enhancer interactions during neurodevelopment. Furthermore, Ring1B and/or associated PcG proteins may provide via their binding to selected silencers a platform for future (tissue-) specific enhancer associations. Since previous reports suggested that promoter-enhancer contacts are largely stable during development and precede gene activation [85,86], those promoter-enhancer interactions may be independent of PcG proteins in some cells or developmental stages.
4.4. Polycomb Group Complexes in Adult NSCs
Subventricular NSCs resemble RGCs and show a sustained expression of Ezh2 [87], pointing to additional tasks. In support of this view, Ezh2 sustains self-renewal of subventricular NSCs by direct repression of the cyclin-dependent kinase inhibitor (CDKI) 2a (hereafter Ink4a/Arf), which generates through the use of shared coding regions and alternative reading frames the two major proteins p16ink4a and p19arf. Mechanistically, p16ink4a induces G1 cell cycle arrest by inhibiting the phosphorylation of the retinoblastoma protein through the cyclin-dependent kinases (CDK) 4 and 6. Moreover, the expression of Ezh2 in type C and A cells suggests an additional role in lineage specification. Consistent with this hypothesis, Ezh2 enhanced the neurogenic competence of adult subventricular NSCs by the direct repression of Olig2 (encoding oligodendrocyte lineage transcription factor, a bHLH protein). At the same time, Ezh2 prevented the aberrant activation of many homeodomain containing transcriptional regulators specifying non-SVZ neuronal subtypes [87].
To address a potential role for PcG mediated ubiquitination in NSCs, Román-Trufero et al. [88] inactivated Ring1B in embryonic NSCs from the olfactory bulb. This led to an impaired neural stem/progenitor cell proliferation, with single cells showing a premature neuronal differentiation in vitro; an effect that was further enhanced by the simultaneous deletion of Ring1A. Gene expression profiling on Ring1B knockout cells showed an upregulation of neuronal differentiation-related TFs, particularly of Neurod1, a potential direct Ring1B target, concomitantly to downregulation of the Notch signaling pathway, a known inhibitor of neuronal differentiation. Furthermore, the loss of Ring1B resulted in a substantial induction of the CDKI p21cip1 in the absence of direct promoter binding.
In addition to the enzymatic core subunits, BMI1 (leukemia viral homolog), a component of cPRC1 (Figure 1B), likewise affects adult NSC proliferation and self-renewal. Bmi1 knockout mice died by early adulthood with signs of hematopoietic failure and neurological abnormalities [89]. Although they showed initially normal proliferation and differentiation of NSCs from the central and peripheral nervous system [90], the rate of proliferation in the SVZ was reduced by postnatal day 30. Moreover, cerebellar development was impaired, partly because Bmi1 was required for the proliferation of granule precursor cells [91]. Consistent with Bmi1’s role in the repression of the Ink4a-Arf locus [92,93], the deletion of Ink4a-Arf partially reversed the self-renewal defect in Bmi1 deficient NSCs [90]. In general, Arf is repressed by Bmi1 in lymphoid cells, NSCs, and NPCs, while Ink4a is preferentially repressed by Bmi1 in NSCs [94]. These results indicate cell type-specific roles for Ink4a and Arf in Bmi1 dependent cell proliferation.
Moreover, Bmi1 prevents p21cip1 upregulation via the forebrain specific TF Foxg1 [95], which is expressed in forebrain progenitor cells throughout development and into adulthood. An acute knock down of Foxg1 in embryonic cortical cells caused de-repression of p21cip1 and led to the depletion of the stem/progenitor pool concomitant to neural differentiation [96]. By contrast, the overexpression of Bmi1 in adult SVZ cells prolonged the production of neurons, even beyond one month, while neuropotency rapidly declined under the control condition. Sustained NSC multipotency and self-renewal associated with an early and lasting increase in Foxg1. In support of this finding, the depletion of Foxg1 blocked the Bmi1 mediated an increase in NSC self-renewal [95].
Taken together, PcG’s catalytic and accessory subunits critically regulate adult NSC proliferation, self-renewal, and differentiation through distinct cell cycle inhibitors, TFs, and signaling pathways during distinct neurodevelopmental stages.
4.5. Polycomb Group Complexes in Fate Restricted Neuronal Precursors and Differentiating Neurons
During mammalian brain development, some neural precursor populations travel from their birth place to their final destination by radial or tangential migration.
The precerebellar pontine nuclei (PNs) transmit cortical motor and sensory information to the cerebellum. Neural progenitor compartments of the developing hindbrain are rostrocaudally segregated in rhombomeres (r1 to r8), which are defined by nested Hox gene expression [97]. PN neurons originate from lower rhombic lip progenitors (r6 to r8) that migrate via a long-distance caudorostral tangential path to settle beside the ventral midline. A role of PcG proteins in this process has been recently reported by Di Meglio et al. [98]. Ezh2 repressed Netrin1, a guidance cue in the dorsal hindbrain by counteracting the sonic hedgehog pathway, and thus curtailed pontine neuron migration. By contrast, ectopic Netrin1 expression in Ezh2 knockout mice resulted in abnormal migration and supernumerary nuclei integrating in brain circuitry. Moreover, Ezh2 retained throughout migration intrinsic topographic organization of pontine nuclei, according to rostrocaudal progenitor origin and correlated with patterned cortical input. Thereby Ezh2 sustained spatially restricted Hox gene expression and differential expression of Unc5b (i.e., a member of the family of netrin-1 receptors thought to mediate chemorepulsive effects) in migrating neurons. Consequently, subsets of migratory neurons responded differentially to the developmental guidance cue Netrin1.
A role for PcG proteins in neuronal differentiation is further suggested by the inactivation of Kdm6b (Jumonji Domain Containing Protein 3, which demethylates specifically trimethylation of histone 3 on lysine 27) [99]. Loss of Kdm6b resulted in perinatal lethality due to the complete and selective disruption of the neuronal pre-Bötzinger complex (PBC), the pacemaker of the respiratory rhythm generator. Although electrophysiological and immunohistochemical studies on Kdm6b knockout embryos showed that early stages of PBC development proceeded normally, additional expression profiling and ChIP experiments revealed that PBC specific genes were selectively targeted by Kdm6b during the acquisition of glutamatergic neuronal fate.
In sum, these findings suggest that Ezh1 or 2 associates with diverse functions in neurodevelopment ranging from tangential migration of differentiating hindbrain neurons to late structuring and function of respiratory neuronal networks.
4.6. Polycomb Group Complexes in Mature Neurons and Neurodegenerative Disease
Recent findings [100] suggest to PcG proteins an additional role in neuronal maturation. CDYL (chromodomain on Y-like protein), which is a component of the repressor complex CtBP1 (C-terminal-binding protein 1) [101], recognizes histone lysine methylation, including H3K27me3, through its chromodomain [102], and recruits subsequently PRC2 [103]. Qi and coworkers [100] showed through a series of gain- and loss-of-function experiments that CDYL was a negative regulator of dendrite morphogenesis in rat/mouse hippocampal neurons both in vitro and in vivo, and repressed a number of genes that were relevant to synaptic transmission, signaling, and neuronal differentiation. For example, CDYL repressed Bdnf (brain derived neurotrophic factor, a key regulator of dendrite development) by directly interacting with Ezh2 and recruiting it to the promoter. By contrast, the onset of neuronal activity in maturating neurons rapidly degraded CDYL through the proteasome and caused de-repression of Bdnf triggering in turn dendrite growth.
Collectively, this study links dynamic changes in neuronal activity to the availability of CDYL, its interaction with Ezh2, and dendrite morphogenesis during neuronal maturation.
Expression of the PCR2 subunits Ezh1, Ezh2, Suz12, and Jarid2b, persists in adult neurons, and points to a role in gene regulation even after completion of neuronal differentiation [104]. Likewise, the H3K27me3 specific demethylases Kdm6b and Utx (ubiquitously transcribed tetratricopeptide repeat, X alias Kdm6a) continue to be expressed in adult neurons. The activity of H3K27me3 specific demethylases is regulated by intracellular levels of α-ketoglutarate that fluctuate in response to neuronal activation [105]. Additionally, neuronal activation can directly phosphorylate histone H3 at Ser28 (H3S28p), leading to the displacement of PRC2 [106].
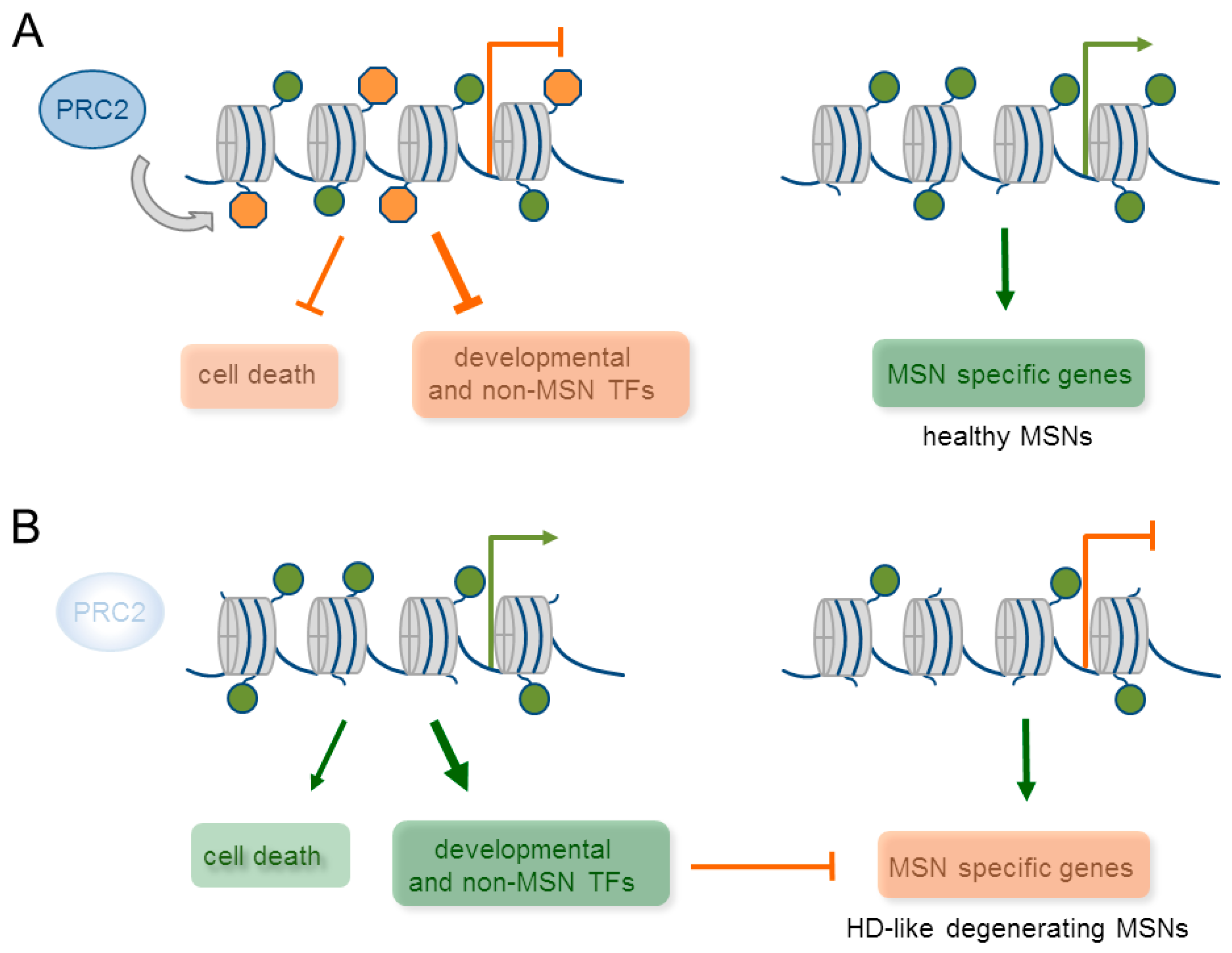
To obtain more direct evidence for PRC2’s function in adult neurons, von Schimmelmann et al. [104] investigated mice with null mutations in Ezh1 and 2 in medium spiny neurons (MSNs, a highly specialized population of striatal cells) and cerebellar Purkinje cells. Both cell types fulfil an important role in motor control and are commonly affected in various neurodegenerative diseases. Particularly, MSCs are the major projecting neurons in the basal ganglia that coordinate motor behaviors, habit and reward learning, and cognition. Although Ezh2 is largely replaced by Ezh1 during neuronal development and maturation, neither knock-out alone changed overall H3K27me3 levels. By contrast, a combined knockout resulted in a loss of H3K27me3 in MSNs around six weeks of age, approximately three–four weeks after inactivation. This delayed effect made developmental effects on mature MSN function rather unlikely. ChIP-sequencing of highly-purified, ex vivo-isolated MSNs nuclei from adult wild-type mice detected over 2000 silenced potential PRC2 target genes that were strongly enriched in H3K27me3 at their TSS. Interestingly, 835 of these genes featured also the activating mark H3K4me3, as evidenced by sequential ChIP-sequencing and matched the criterion of chromatin bivalency. Of note, about two-third of the bivalent domains in MSNs were identical to those from ESC, while one-third was specific to MSNs. Furthermore, bivalent domains were enriched in genes encoding transcriptional regulators thought to impact on non-MSNs and non-neuronal cells (Figure 6A). This unexpected result raised the possibility that terminal differentiated neurons are more plastic than commonly thought. However, the majority of putative PRC2 target genes was unaffected by Ezh1and 2 deficiency and only a selected subset (<200)—half of which carried the bivalent mark—were progressively upregulated in MSNs. These responders encoded predominantly TFs that were normally expressed during development, particularly, Hox genes that are switched off during early MSN development or non-MSN and non-neuronal cell lineage expressed genes together with several cell death promoting genes (Figure 6B). About one-quarter of these transcriptional regulators belonged to self-regulatory transcriptional networks that could underpin their robust up-regulation in the absence of Ezh1 and 2 [104]. Surprisingly though, this up-regulation failed in inducing neuronal programs that were unrelated to MSNs. Instead, it repressed in a cell-intrinsic manner highly expressed genes specific to MSNs consisting of neurotransmitter receptors, signaling proteins, and TFs (Figure 6B). Since MSN specific genes featured H3K4me3 in the absence of H3K27me3, their de-regulation appeared to result primarily from de-repression of PRC2 target genes. Although the downregulation of MSN specific genes was only partial with 38 out of 502 such genes, Ezh1 and 2 deficient mice manifested with a progressive and severe neurodegeneration of MSNs. Molecular, cellular, and behavioral analyses suggested that the underlying neuropathology closely resembled the one from mouse models of Huntington disease (HD): enhanced MSN intrinsic excitability together with increased input resistance, enhanced presence of γH2ax (a DNA damage histone mark), dark degenerating neurons, brain atrophy, and striatal neuronal loss. Importantly, the relevance of this data was supported in Purkinje cell-specific Ezh1 and 2 knock-out mice, in which ectopic TFs and cell-death promoting genes were similarly induced concomitantly to the downregulation of cell type specific genes.
Taken together, Ezh1 and 2 jointly safeguard bivalent domains in mature neurons, and prevent the switch-on of transcriptional circuits that repress progressively cell-type specific genes. Conversely, loss of cell type specific gene expression patterns triggers severe neurodegeneration.
A role of PcG proteins in maintaining mature neuron function is further supported by the findings from human neurodegenerative diseases. For example, symptoms from a genetic defect in ATM (ataxia-telangiectasia mutated gene,) encoding a member of the phosphatidylinositol 3-kinase family include progressive neurodegeneration concomitant with an increase in H3K27me3 [107]. Functionally, this kinase was found to phosphorylate EZH2 on Ser734, leading to its enhanced degradation. In agreement with this finding, lentiviral mediated knockdown of Ezh2 rescued Purkinje cell degeneration and behavioral abnormalities in Atm deficient mice [107]. Furthermore, in a mouse model of Parkinson disease, acute administration of the dopamine precursor L-DOPA induced a robust increase in H3K27me3S28 phosphorylation that is specific to MSNs [108]. Genome-wide ChIP experiments showed that the induction of H3K27me3S28p was followed by reduced PcG protein binding to a subset of target genes, and resulted in their derepression. Lastly, several recent reports implicate altered PRC2 function in HD [109,110]. Huntingtin protein can directly interact with PRC2 and stimulate its catalytic activity, leading to increased H3K27me3 marking at mice Hox genes [110]. By contrast, expression of mutated Huntingtin protein in mouse ESCs or NPCs distorts genome-wide patterns of H3K27me3 together with a reduced presence of H3K4me3 at active loci [111]. The potential link between PRC2 and H3K4me3 is further supported by a significant depletion of H3K4me3 in HD-enriched peaks from ChIP-sequencing of neuronal chromatin from prefrontal cortices [109] and the upregulation of some inflammatory and developmental PRC2 target genes, particularly of certain Hox genes, in HD brains [112,113].
Taken together, PRC2 is necessary to maintain adult neuronal specification, function, and survival by the repression of bivalently marked genes controlling cell death and auto- or coregulatory networks. Conversely, derepression of these networks induces progressive neuronal degeneration. In support of this view, PRC2 function appears compromised through specific mechanisms in different neurodegenerative disorders.
5. Concluding Remarks
PcG proteins participate in dynamic changes in gene expression in the developing brain that underpin robust transitions from multipotency to cell lineage to cell fate decisions. Furthermore, PcG proteins contribute to cell differentiation and maturation, and finally, the prevention of neurodegeneration. These transitions frequently comprise switch-like, rather than graded, changes in the expression state of selected bivalently marked genes. Bivalency is characterized by a built-in state of balanced inhibition enabling rapid “On-Off” decisions once the balance is tilted in response to differentiation cues. During neurodevelopment, chromatin bivalency evolves and resolves recurrently across distinct stages that are extending from ESCs to postmitotic neurons that are compatible with a general regulatory principle.
Although bivalent domains are common during neurodevelopment and in the mature brain, switch-like transitions in gene expression states are reserved to selected genes and associate with different, partly gene- and/or stage specific, mechanisms. PcG proteins can bind to different kinds of regulatory regions, such as promoters or enhancers, and/or mediate the interactions between distantly separated genomic regions to regulate chromatin conformation at different scales. In ESCs, PcG proteins maintain bivalent chromatin domains that poise target genes for rapid, switch-like, induction in response to neural differentiation [23,62]. Additionally, PcG proteins mediate interactions with neurodevelopmental genes and their poised enhancers that maintain a silenced, albeit preactivated, state whose dissolution induces rapid neural differentiation of ESCs [74]. Thereby, PcG dependent interactions may form 3D chromatin hubs that coordinate repression and the activation of early (neuro-) developmental regulators (Figure 3). Beyond preventing precocious activation of early neural genes, PcG proteins can also preset future activation of major anterior neural genes once the differentiation signal has been received [12]. In this scenario, PcG proteins endow poised enhancers with a permissive regulatory topology that enables pluripotent cells to respond effectively to neural induction signals (Figure 4). Through this mechanism, PcG proteins may constitute an important component of the classical “neural default” model [114], which posits that epiblast cells in vivo and ESCs in vitro are fated by default (i.e., in the absence of extrinsic signals) toward the neural lineage. Interestingly, the resulting neural progenitors exhibit initially marked anterior features (i.e., forebrain). The preferential association of poised enhancers with major regulators of anterior neural identity and the essential role of poised enhancers during the induction of these genes indicates that poised enhancers are an important molecular element of this model. Lastly, PcG proteins also control in vivo tissue-specifically activation and the repression of a midbrain TF [13] through the coordination of tripartite promoter-silencer-enhancer interactions during embryonic development (Figure 5).
Conclusively, PcG complexes regulate neurodevelopmental gene expression through local chromatin conformation and through 3D long-range interaction between regulatory elements. On a temporal scale, PcG dependent linear chromatin modifications mediate immediate “On-Off” switches in gene expression. By contrast, PcG dependent 3D chromatin hubs may also preset future “On-Off” decisions. Hence, PcG dependent gene regulation provides spatial and temporal control of gene expression during neurodevelopment.
Beyond early cell-lineage decisions, PcG proteins are also critical regulatory determinants in developmental fate switches by restraining the neurogenic, gliogenic, and astrogenic competence of NPCs in the developing cortex. A more limited role applies to PcG proteins in adult NSCs from the SVZ and SGZ, in which they regulate proliferation, self-renewal, and differentiation through distinct cell cycle inhibitors, TFs, and signaling pathways.
In addition to cell-lineage and cell-fate decisions, PcG proteins contribute to neuronal differentiation and maturation, including tangential migration of hindbrain neurons [98], dendrite morphogenesis of hippocampal neurons [101], and late structuring and function of respiratory neuronal networks [99]. Remarkably, PcG proteins safeguard bivalent domains also in mature neurons and prevent the switch-on of self-reinforcing transcriptional circuits that repress cell-type specific genes. De-repression of these networks in the absence of PCR2’s catalytic subunits triggers in turn the progressive neurodegeneration of MSN and Purkinje cells [104] (Figure 6). By contrast to chromatin bivalency in ESC and NPCs, PcG binding in adult MSNs does not position target genes for rapid switch-like expression, but maintains constitutive silencing in order to prevent neurodegeneration. Consistent with this possibility, human neurodegenerative diseases show specific changes in PRC2 activity together with alterations in H3K27me3 levels and distribution.
Overall, the multiplicity of PcG functions in the nervous system is readily apparent and extends from self-renewal and cell lineage decisions to the timing of cell fate switches and differentiation, and finally, the prevention of neurodegeneration. Further insight into these manifold functions of PcG proteins will benefit our understanding of neuropathological alterations in various progressive neurodegenerative diseases and eventually pave the way to new therapies. Hopefully, these may also include targeted manipulations to improve the efficiency of NPC differentiation in therapeutically relevant cell types and thus advance cell replacement in neurodegenerative diseases.
Acknowledgments
We are thankful to members of our groups for thoughtful comments and advice. Michael Ziller is supported by BMBF grant 01ZX1504.
Author Contributions
Anke Hoffmann, Vincenza Sportelli, Michael Ziller and Dietmar Spengler wrote jointly this manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Duncan, I.M. Polycomblike: A gene that appears to be required for the normal expression of the bithorax and antennapedia gene complexes of Drosophila melanogaster. Genetics 1982, 102, 49–70. [Google Scholar] [PubMed]
- Lewis, E.B. A gene complex controlling segmentation in Drosophila. Nature 1978, 276, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Ringrose, L.; Paro, R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu. Rev. Genet. 2004, 38, 413–443. [Google Scholar] [CrossRef] [PubMed]
- Capecchi, M.R. Hox genes and mammalian development. Cold Spring Harb. Symp. Quant. Biol. 1997, 62, 273–281. [Google Scholar] [PubMed]
- Nüsslein-Volhard, C. Of flies and fishes. Science 1994, 266, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Corley, M.; Kroll, K.L. The roles and regulation of Polycomb complexes in neural development. Cell Tissue Res. 2015, 359, 65–85. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, A.; Magnuson, T. Murine Polycomb- and trithorax-group genes regulate homeotic pathways and beyond. Trends Genet. 1997, 13, 167–170. [Google Scholar] [CrossRef]
- Zakany, J.; Duboule, D. The role of Hox genes during vertebrate limb development. Curr. Opin. Genet. Dev. 2007, 17, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Tanay, A.; O’Donnell, A.H.; Damelin, M.; Bestor, T.H. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc. Natl. Acad. Sci. USA 2007, 104, 5521–5526. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Molina, S.; Respuela, P.; Tebartz, C.; Kolovos, P.; Nikolic, M.; Fueyo, R.; van Ijcken, W.F.J.; Grosveld, F.; Frommolt, P.; Bazzi, H.; et al. PRC2 Facilitates the Regulatory Topology Required for Poised Enhancer Function during Pluripotent Stem Cell Differentiation. Cell Stem Cell 2017, 20, 689–705.e9. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Isono, K.; Kondo, K.; Endo, T.A.; Itohara, S.; Vidal, M.; Koseki, H. Polycomb potentiates Meis2 activation in midbrain by mediating interaction of the promoter with a tissue-specific enhancer. Dev. Cell 2014, 28, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schübeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Zimmermann, C.A.; Spengler, D. Molecular epigenetic switches in neurodevelopment in health and disease. Front. Behav. Neurosci. 2015, 9, 120. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Bourbon, H.-M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shao, Z.; Li, D.; Xie, H.; Kim, W.; Huang, J.; Taylor, J.E.; Pinello, L.; Glass, K.; Jaffe, J.D.; et al. Developmental control of polycomb subunit composition by GATA factors mediates a switch to non-canonical functions. Mol. Cell 2015, 57, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, K.; Zare, H.; Wang, A.H.; Sartorelli, V. Polycomb protein Ezh1 promotes RNA polymerase II elongation. Mol. Cell 2012, 45, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Hauri, S.; Comoglio, F.; Seimiya, M.; Gerstung, M.; Glatter, T.; Hansen, K.; Aebersold, R.; Paro, R.; Gstaiger, M.; Beisel, C. A High-Density Map for Navigating the Human Polycomb Complexome. Cell Rep. 2016, 17, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Trupke, J.; Ringrose, L. The quest for mammalian Polycomb response elements: Are we there yet? Chromosoma 2016, 125, 471–496. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Ku, M.; Koche, R.P.; Rheinbay, E.; Mendenhall, E.M.; Endoh, M.; Mikkelsen, T.S.; Presser, A.; Nusbaum, C.; Xie, X.; Chi, A.S.; et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008, 4, e1000242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, N.; Lerdrup, M.; Landt, E.; Agrawal-Singh, S.; Bak, M.; Tommerup, N.; Rappsilber, J.; Södersten, E.; Hansen, K. REST-mediated recruitment of polycomb repressor complexes in mammalian cells. PLoS Genet. 2012, 8, e1002494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hekimoglu-Balkan, B.; Aszodi, A.; Heinen, R.; Jaritz, M.; Ringrose, L. Intergenic Polycomb target sites are dynamically marked by non-coding transcription during lineage commitment. RNA Biol. 2012, 9, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Farcas, A.M.; Blackledge, N.P.; Sudbery, I.; Long, H.K.; McGouran, J.F.; Rose, N.R.; Lee, S.; Sims, D.; Cerase, A.; Sheahan, T.W.; et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. eLife 2012, 1, e00205. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Johansen, J.V.; Helin, K. Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol. Cell 2013, 49, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Shen, L.; Wan, M.; Taranova, O.; Wu, H.; Zhang, Y. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat. Cell Biol. 2013, 15, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Cooper, S.; Farcas, A.M.; Blackledge, N.P.; Brockdorff, N. Chromatin sampling—An emerging perspective on targeting polycomb repressor proteins. PLoS Genet. 2013, 9, e1003717. [Google Scholar] [CrossRef] [PubMed]
- Riising, E.M.; Comet, I.; Leblanc, B.; Wu, X.; Johansen, J.V.; Helin, K. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol. Cell 2014, 55, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Margueron, R.; Ku, M.; Chambon, P.; Bernstein, B.E.; Reinberg, D. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 2010, 24, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Landeira, D.; Sauer, S.; Poot, R.; Dvorkina, M.; Mazzarella, L.; Jørgensen, H.F.; Pereira, C.F.; Leleu, M.; Piccolo, F.M.; Spivakov, M.; et al. Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA Polymerase II to developmental regulators. Nat. Cell Biol. 2010, 12, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Cloos, P.A.C.; Walfridsson, J.; Olsson, L.; Bukowski, J.-P.; Johansen, J.V.; Bak, M.; Tommerup, N.; Rappsilber, J.; Helin, K. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 2010, 464, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.C.; Valouev, A.; Swigut, T.; Zhang, J.; Zhao, Y.; Sidow, A.; Wysocka, J. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell 2009, 139, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.T.C.; Müller, C.W.; Vermeulen, M.; Müller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.H.; Bracken, A.P.; Pasini, D.; Dietrich, N.; Gehani, S.S.; Monrad, A.; Rappsilber, J.; Lerdrup, M.; Helin, K. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 2008, 10, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Justin, N.; Ohno, K.; Sharpe, M.L.; Son, J.; Drury, W.J.; Voigt, P.; Martin, S.R.; Taylor, W.R.; De Marco, V.; et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 2009, 461, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Rojas, C.; Hernandez, A.J.; Sarma, K.; Lee, J.T. Regulatory interactions between RNA and polycomb repressive complex. Mol. Cell 2014, 55, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Son, J.; Bonasio, R.; Shen, S.S.; Reinberg, D. Nascent RNA interaction keeps PRC2 activity poised and in check. Genes Dev. 2014, 28, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N. Noncoding RNA and Polycomb recruitment. RNA 2013, 19, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Chopra, V.S.; Hendrix, D.A.; Core, L.J.; Tsui, C.; Lis, J.T.; Levine, M. The polycomb group mutant esc leads to augmented levels of paused Pol II in the Drosophila embryo. Mol. Cell 2011, 42, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Tie, F.; Banerjee, R.; Fu, C.; Stratton, C.A.; Fang, M.; Harte, P.J. Polycomb inhibits histone acetylation by CBP by binding directly to its catalytic domain. Proc. Natl. Acad. Sci. USA 2016, 113, E744–E753. [Google Scholar] [CrossRef] [PubMed]
- Lanzuolo, C.; Roure, V.; Dekker, J.; Bantignies, F.; Orlando, V. Polycomb response elements mediate the formation of chromosome higher-order structures in the bithorax complex. Nat. Cell Biol. 2007, 9, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Cheutin, T.; Cavalli, G. Polycomb silencing: from linear chromatin domains to 3D chromosome folding. Curr. Opin. Genet. Dev. 2014, 25, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Vietri Rudan, M.; Barrington, C.; Henderson, S.; Ernst, C.; Odom, D.T.; Tanay, A.; Hadjur, S. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015, 10, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N.; Weintraub, A.S.; Schuijers, J.; Lee, T.I.; Zhao, K.; et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Isono, K.; Endo, T.A.; Ku, M.; Yamada, D.; Suzuki, R.; Sharif, J.; Ishikura, T.; Toyoda, T.; Bernstein, B.E.; Koseki, H. SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev. Cell 2013, 26, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Gambetta, M.C.; Müller, J. O-GlcNAcylation prevents aggregation of the Polycomb group repressor polyhomeotic. Dev. Cell 2014, 31, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.; Zhuang, X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Denholtz, M.; Bonora, G.; Chronis, C.; Splinter, E.; de Laat, W.; Ernst, J.; Pellegrini, M.; Plath, K. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell 2013, 13, 602–616. [Google Scholar] [CrossRef] [PubMed]
- Joshi, O.; Wang, S.-Y.; Kuznetsova, T.; Atlasi, Y.; Peng, T.; Fabre, P.J.; Habibi, E.; Shaik, J.; Saeed, S.; Handoko, L.; et al. Dynamic Reorganization of Extremely Long-Range Promoter-Promoter Interactions between Two States of Pluripotency. Cell Stem Cell 2015, 17, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.-L.; Song, H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [PubMed]
- Götz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H.; Temple, S. Neural stem cells: generating and regenerating the brain. Neuron 2013, 80, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Meshorer, E.; Yellajoshula, D.; George, E.; Scambler, P.J.; Brown, D.T.; Misteli, T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell 2006, 10, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Young, R.A. Control of the embryonic stem cell state. Cell 2011, 144, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Azuara, V.; Perry, P.; Sauer, S.; Spivakov, M.; Jørgensen, H.F.; John, R.M.; Gouti, M.; Casanova, M.; Warnes, G.; Merkenschlager, M.; et al. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 2006, 8, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Alder, O.; Lavial, F.; Helness, A.; Brookes, E.; Pinho, S.; Chandrashekran, A.; Arnaud, P.; Pombo, A.; O’Neill, L.; Azuara, V. Ring1B and Suv39h1 delineate distinct chromatin states at bivalent genes during early mouse lineage commitment. Development 2010, 137, 2483–2492. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Zang, C.; Roh, T.-Y.; Schones, D.E.; Childs, R.W.; Peng, W.; Zhao, K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 2009, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J.A.; Reiner, A.H.; Klungland, A.; Wakayama, T.; Collas, P. Histone H3 lysine 27 methylation asymmetry on developmentally-regulated promoters distinguish the first two lineages in mouse preimplantation embryos. PLoS ONE 2010, 5, e9150. [Google Scholar] [CrossRef] [PubMed]
- Rugg-Gunn, P.J.; Cox, B.J.; Ralston, A.; Rossant, J. Distinct histone modifications in stem cell lines and tissue lineages from the early mouse embryo. Proc. Natl. Acad. Sci. USA 2010, 107, 10783–10790. [Google Scholar] [CrossRef] [PubMed]
- Vastenhouw, N.L.; Zhang, Y.; Woods, I.G.; Imam, F.; Regev, A.; Liu, X.S.; Rinn, J.; Schier, A.F. Chromatin signature of embryonic pluripotency is established during genome activation. Nature 2010, 464, 922–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, G.; Tian, S.; Nie, J.; Yang, C.; Ruotti, V.; Wei, H.; Jonsdottir, G.A.; Stewart, R.; Thomson, J.A. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 2007, 1, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.D.; Han, X.; Chew, J.L.; Liu, J.; Chiu, K.P.; Choo, A.; Orlov, Y.L.; Sung, W.-K.; Shahab, A.; Kuznetsov, V.A.; et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 2007, 1, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Zeltner, N.; Studer, L. Pluripotent stem cell-based disease modeling: current hurdles and future promise. Curr. Opin. Cell Biol. 2015, 37, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Guenther, M.G.; Frampton, G.M.; Soldner, F.; Hockemeyer, D.; Mitalipova, M.; Jaenisch, R.; Young, R.A. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell Stem Cell 2010, 7, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Maherali, N.; Sridharan, R.; Xie, W.; Utikal, J.; Eminli, S.; Arnold, K.; Stadtfeld, M.; Yachechko, R.; Tchieu, J.; Jaenisch, R.; et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 2007, 1, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Hanna, J.; Zhang, X.; Ku, M.; Wernig, M.; Schorderet, P.; Bernstein, B.E.; Jaenisch, R.; Lander, E.S.; Meissner, A. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008, 454, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Schoenfelder, S.; Sugar, R.; Dimond, A.; Javierre, B.-M.; Armstrong, H.; Mifsud, B.; Dimitrova, E.; Matheson, L.; Tavares-Cadete, F.; Furlan-Magaril, M.; et al. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat. Genet. 2015, 47, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Beagrie, R.A.; Pombo, A. Gene activation by metazoan enhancers: Diverse mechanisms stimulate distinct steps of transcription. Bioessays 2016, 38, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yu, N.-K.; Kaang, B.-K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 2015, 47, e166. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: what, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Ji, F.; Sunwoo, H.; Jain, G.; Lee, J.T.; Sadreyev, R.I.; Dekker, J.; Kingston, R.E. Polycomb Repressive Complex 1 Generates Discrete Compacted Domains that Change during Differentiation. Mol. Cell 2017, 65, 432–446.e5. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.D.; Sansom, S.N.; Smith, J.; Dobenecker, M.-W.; Tarakhovsky, A.; Livesey, F.J. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 15957–15962. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, Y.; Suzki, N.; Tsuboi, M.; Endo, T.A.; Toyoda, T.; Shinga, J.; Koseki, H.; Vidal, M.; Gotoh, Y. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 2009, 63, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, F. Spatial and temporal specification of neural fates by transcription factor codes. Development 2007, 134, 3771–3780. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ge, W.; Martinowich, K.; Becker-Catania, S.; Coskun, V.; Zhu, W.; Wu, H.; Castro, D.; Guillemot, F.; Fan, G.; et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat. Neurosci. 2005, 8, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Petryniak, M.A.; Potter, G.B.; Rowitch, D.H.; Rubenstein, J.L. R. Dlx1 and Dlx2 control neuronal versus oligodendroglial cell fate acquisition in the developing forebrain. Neuron 2007, 55, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Ghavi-Helm, Y.; Klein, F.A.; Pakozdi, T.; Ciglar, L.; Noordermeer, D.; Huber, W.; Furlong, E.E.M. Enhancer loops appear stable during development and are associated with paused polymerase. Nature 2014, 512, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Lonfat, N.; Montavon, T.; Darbellay, F.; Gitto, S.; Duboule, D. Convergent evolution of complex regulatory landscapes and pleiotropy at Hox loci. Science 2014, 346, 1004–1006. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.W.; Salinas, R.D.; Siu, J.J.; Kelley, K.W.; Delgado, R.N.; Paredes, M.F.; Alvarez-Buylla, A.; Oldham, M.C.; Lim, D.A. Distinct and separable roles for EZH2 in neurogenic astroglia. eLife 2014, 3, e02439. [Google Scholar] [CrossRef] [PubMed]
- Román-Trufero, M.; Méndez-Gómez, H.R.; Pérez, C.; Hijikata, A.; Fujimura, Y.; Endo, T.; Koseki, H.; Vicario-Abejón, C.; Vidal, M. Maintenance of undifferentiated state and self-renewal of embryonic neural stem cells by Polycomb protein Ring1B. Stem Cells 2009, 27, 1559–1570. [Google Scholar] [CrossRef] [PubMed]
- Van der Lugt, N.M.; Domen, J.; Linders, K.; van Roon, M.; Robanus-Maandag, E.; te Riele, H.; van der Valk, M.; Deschamps, J.; Sofroniew, M.; van Lohuizen, M. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 1994, 8, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.-K.; Clarke, M.F.; Morrison, S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 2003, 425, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Lingbeek, M.; Shakhova, O.; Liu, J.; Tanger, E.; Saremaslani, P.; Van Lohuizen, M.; Marino, S. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 2004, 428, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Scheijen, B.; Voncken, J.W.; Kieboom, K.; Berns, A.; van Lohuizen, M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999, 13, 2678–2690. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, S.W.M.; Valk-Lingbeek, M.E.; van der Stoop, P.P.M.; Jacobs, J.J.L.; Kieboom, K.; Tanger, E.; Hulsman, D.; Leung, C.; Arsenijevic, Y.; Marino, S.; et al. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 2005, 19, 1438–1443. [Google Scholar] [CrossRef] [PubMed]
- Fasano, C.A.; Phoenix, T.N.; Kokovay, E.; Lowry, N.; Elkabetz, Y.; Dimos, J.T.; Lemischka, I.R.; Studer, L.; Temple, S. Bmi-1 cooperates with Foxg1 to maintain neural stem cell self-renewal in the forebrain. Genes Dev. 2009, 23, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Wang, Y.; Dimos, J.T.; Fasano, C.A.; Phoenix, T.N.; Lemischka, I.R.; Ivanova, N.B.; Stifani, S.; Morrisey, E.E.; Temple, S. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat. Neurosci. 2006, 9, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Tümpel, S.; Wiedemann, L.M.; Krumlauf, R. Hox genes and segmentation of the vertebrate hindbrain. Curr. Top. Dev. Biol. 2009, 88, 103–137. [Google Scholar] [CrossRef] [PubMed]
- Di Meglio, T.; Kratochwil, C.F.; Vilain, N.; Loche, A.; Vitobello, A.; Yonehara, K.; Hrycaj, S.M.; Roska, B.; Peters, A.H.F.M.; Eichmann, A.; et al. Ezh2 orchestrates topographic migration and connectivity of mouse precerebellar neurons. Science 2013, 339, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Burgold, T.; Voituron, N.; Caganova, M.; Tripathi, P.P.; Menuet, C.; Tusi, B.K.; Spreafico, F.; Bévengut, M.; Gestreau, C.; Buontempo, S.; et al. The H3K27 demethylase JMJD3 is required for maintenance of the embryonic respiratory neuronal network, neonatal breathing, and survival. Cell Rep. 2012, 2, 1244–1258. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Liu, S.; Qin, R.; Zhang, Y.; Wang, G.; Shang, Y.; Wang, Y.; Liang, J. Coordinated regulation of dendrite arborization by epigenetic factors CDYL and EZH. J. Neurosci. 2014, 34, 4494–4508. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Sawada, J.; Sui, G.; Affar, E.B.; Whetstine, J.R.; Lan, F.; Ogawa, H.; Luke, M.P.-S.; Nakatani, Y.; Shi, Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 2003, 422, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, M.; Eberl, H.C.; Matarese, F.; Marks, H.; Denissov, S.; Butter, F.; Lee, K.K.; Olsen, J.V.; Hyman, A.A.; Stunnenberg, H.G.; et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 2010, 142, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, X.; Gui, B.; Xie, G.; Zhang, D.; Shang, Y.; Liang, J. Corepressor protein CDYL functions as a molecular bridge between polycomb repressor complex 2 and repressive chromatin mark trimethylated histone lysine 27. J. Biol. Chem. 2011, 286, 42414–42425. [Google Scholar] [CrossRef] [PubMed]
- Von Schimmelmann, M.; Feinberg, P.A.; Sullivan, J.M.; Ku, S.M.; Badimon, A.; Duff, M.K.; Wang, Z.; Lachmann, A.; Dewell, S.; Ma’ayan, A.; et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci. 2016, 19, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- He, X.-B.; Kim, M.; Kim, S.-Y.; Yi, S.-H.; Rhee, Y.-H.; Kim, T.; Lee, E.-H.; Park, C.-H.; Dixit, S.; Harrison, F.E.; et al. Vitamin C facilitates dopamine neuron differentiation in fetal midbrain through TET1- and JMJD3-dependent epigenetic control manner. Stem Cells 2015, 33, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Palomer, E.; Carretero, J.; Benvegnù, S.; Dotti, C.G.; Martin, M.G. Neuronal activity controls Bdnf expression via Polycomb de-repression and CREB/CBP/JMJD3 activation in mature neurons. Nat. Commun. 2016, 7, 11081. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hart, R.P.; Mallimo, E.M.; Swerdel, M.R.; Kusnecov, A.W.; Herrup, K. EZH2-mediated H3K27 trimethylation mediates neurodegeneration in ataxia-telangiectasia. Nat. Neurosci. 2013, 16, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Södersten, E.; Feyder, M.; Lerdrup, M.; Gomes, A.-L.; Kryh, H.; Spigolon, G.; Caboche, J.; Fisone, G.; Hansen, K. Dopamine signaling leads to loss of Polycomb repression and aberrant gene activation in experimental parkinsonism. PLoS Genet. 2014, 10, e1004574. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Tsuji, J.; Labadorf, A.; Roussos, P.; Chen, J.-F.; Myers, R.H.; Akbarian, S.; Weng, Z. The Role of H3K4me3 in Transcriptional Regulation Is Altered in Huntington’s Disease. PLoS ONE 2015, 10, e0144398. [Google Scholar] [CrossRef] [PubMed]
- Seong, I.S.; Woda, J.M.; Song, J.-J.; Lloret, A.; Abeyrathne, P.D.; Woo, C.J.; Gregory, G.; Lee, J.-M.; Wheeler, V.C.; Walz, T.; et al. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 2010, 19, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Biagioli, M.; Ferrari, F.; Mendenhall, E.M.; Zhang, Y.; Erdin, S.; Vijayvargia, R.; Vallabh, S.M.; Solomos, N.; Manavalan, P.; Ragavendran, A.; et al. Htt CAG repeat expansion confers pleiotropic gains of mutant huntingtin function in chromatin regulation. Hum. Mol. Genet. 2015, 24, 2442–2457. [Google Scholar] [CrossRef] [PubMed]
- Hoss, A.G.; Kartha, V.K.; Dong, X.; Latourelle, J.C.; Dumitriu, A.; Hadzi, T.C.; Macdonald, M.E.; Gusella, J.F.; Akbarian, S.; Chen, J.-F.; et al. MicroRNAs located in the Hox gene clusters are implicated in huntington’s disease pathogenesis. PLoS Genet. 2014, 10, e1004188. [Google Scholar] [CrossRef] [PubMed]
- Labadorf, A.; Hoss, A.G.; Lagomarsino, V.; Latourelle, J.C.; Hadzi, T.C.; Bregu, J.; MacDonald, M.E.; Gusella, J.F.; Chen, J.-F.; Akbarian, S.; et al. RNA Sequence Analysis of Human Huntington Disease Brain Reveals an Extensive Increase in Inflammatory and Developmental Gene Expression. PLoS ONE 2015, 10, e0143563. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, N.; Vanderhaeghen, P. Mechanisms of neural specification from embryonic stem cells. Curr. Opin. Neurobiol. 2010, 20, 37–43. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Vertebrate Polycomb Group (PcG) complexes. The Polycomb repressive complex 2 (PRC2) and different forms of Polycomb repressive complex 1 (PRC1) are shown. Core subunits are shown in blue for PRC2 and in green for PRC1. Alternate subunits derived from multiple genes are shown in grey and accessory proteins in apricot. Histone modifications are: orange hexagon; histone H3 trimethylation of lysine 27 (H3K27me3) and violet oval; histone H2A monoubiquitination of lysine 119 (H2AK119ub1). (A) PRC2 consists of four core subunits: EZH1 or 2 (enhancer of zeste homologue 1 or 2), EED (embryonic ectoderm), SUZ12 (suppressor of zeste), and RBBP4/7 (or RbAp46/48) and three accessory proteins, PCL (Polycomb-like homolog), JARID2 (Jumonji, AT-Rich Interactive Domain 2 protein), and AEBP2 (Adipocyte Enhancer-Binding Protein 2). PRC2 di- and tri-methylates histone 3 on lysine 27 (H3K27me2 or 3, respectively) through the SET domain of EZH1 or 2. In addition, PRC2 can bind H3K27me3 via EED; (B) Canonical PRC1 consists of four subunits: RING1A or B, CBX (chromobox), PCGF (PcG ring-finger domain protein), and PHC (Polyhomeotic homologous protein). PRC1 catalyzes H2AK1119ub through RING1A or B. Canonical PRC1 recognizes H3K27me3 via the chromodomain of CBX2 or 7; (C) One class of vertebrate non-canonical PRC1s consists of three core subunits: RING1A or B, PCGF, and RYBP (zinc-finger domain and YY-1 binding protein) or YAF2 (YY1-associated factor 2) and different accessory proteins. The complexes are characterized by different PCGF subunits. For example, PRC1.1 contains PCGF1 in conjunction with the accessory protein KDM2B (H3K36-specific histone demethylase).
Figure 1.
Vertebrate Polycomb Group (PcG) complexes. The Polycomb repressive complex 2 (PRC2) and different forms of Polycomb repressive complex 1 (PRC1) are shown. Core subunits are shown in blue for PRC2 and in green for PRC1. Alternate subunits derived from multiple genes are shown in grey and accessory proteins in apricot. Histone modifications are: orange hexagon; histone H3 trimethylation of lysine 27 (H3K27me3) and violet oval; histone H2A monoubiquitination of lysine 119 (H2AK119ub1). (A) PRC2 consists of four core subunits: EZH1 or 2 (enhancer of zeste homologue 1 or 2), EED (embryonic ectoderm), SUZ12 (suppressor of zeste), and RBBP4/7 (or RbAp46/48) and three accessory proteins, PCL (Polycomb-like homolog), JARID2 (Jumonji, AT-Rich Interactive Domain 2 protein), and AEBP2 (Adipocyte Enhancer-Binding Protein 2). PRC2 di- and tri-methylates histone 3 on lysine 27 (H3K27me2 or 3, respectively) through the SET domain of EZH1 or 2. In addition, PRC2 can bind H3K27me3 via EED; (B) Canonical PRC1 consists of four subunits: RING1A or B, CBX (chromobox), PCGF (PcG ring-finger domain protein), and PHC (Polyhomeotic homologous protein). PRC1 catalyzes H2AK1119ub through RING1A or B. Canonical PRC1 recognizes H3K27me3 via the chromodomain of CBX2 or 7; (C) One class of vertebrate non-canonical PRC1s consists of three core subunits: RING1A or B, PCGF, and RYBP (zinc-finger domain and YY-1 binding protein) or YAF2 (YY1-associated factor 2) and different accessory proteins. The complexes are characterized by different PCGF subunits. For example, PRC1.1 contains PCGF1 in conjunction with the accessory protein KDM2B (H3K36-specific histone demethylase).

Figure 2.
PcG proteins act on different chromatin scales. (A) On the linear chromatin, PcG proteins prevent deposition of active marks (symbolized by green oval). By contrast, PcG proteins place repressive marks (orange or violet oval symbolize H3K27me3 and H2AK119u, respectively) to maintain chromatin compaction; (B) PcG proteins regulate gene expression via recruitment of regulatory DNA elements (green box) to target genes (blue box) through chromatin looping; (C) PcG proteins regulate nuclear chromatin architecture. PcG proteins aggregate via chromatin looping with their targets sites in Polycomb bodies (orange oval). Chromatin is organized into distinct topological associated domains (TADs). In Drosophila, some Polycomb-repressed chromatin domains interact with TADs pointing to a role in global chromatin packaging.
Figure 2.
PcG proteins act on different chromatin scales. (A) On the linear chromatin, PcG proteins prevent deposition of active marks (symbolized by green oval). By contrast, PcG proteins place repressive marks (orange or violet oval symbolize H3K27me3 and H2AK119u, respectively) to maintain chromatin compaction; (B) PcG proteins regulate gene expression via recruitment of regulatory DNA elements (green box) to target genes (blue box) through chromatin looping; (C) PcG proteins regulate nuclear chromatin architecture. PcG proteins aggregate via chromatin looping with their targets sites in Polycomb bodies (orange oval). Chromatin is organized into distinct topological associated domains (TADs). In Drosophila, some Polycomb-repressed chromatin domains interact with TADs pointing to a role in global chromatin packaging.

Figure 3.
Polycomb repressive complex 1 (PRC1) regulates mammalian chromatin in three-dimensional (3D) space during neural induction. Examples of enhancers and associated promoters are numbered 1 to 3. (A) PRC1 assembles target genes in nuclear bodies (pink oval) that represent sites of Polycomb mediated gene silencing in undifferentiated mouse embryonic stem cells (ESCs). PRC1 dependent contacts maintain the promoters of target genes in a poised, albeit repressed state (deep orange box). Polycomb proteins also block the activational effects from contacts between poised enhancers (light orange box) and the associated promoters on transcription of target genes (examples 1 and 2). By contrast, poised enhancers (light green box) strongly activate associated promoters (dark green box) outside PcG bodies and confer potent transcription to a target gene (example 3); (B) The PRC1 promoter interactome disintegrates in mouse ESCs deficient in RING1A and 1B (RING1A/B). This enables promoters and associated enhancers to transit from a poised to an active state (examples 1 and 2); (C) Model for the activation of PcG target genes during neural induction of mESCs. Selected genes are thought to be let off from the PRC1 interactome followed by transcriptional activation (example 2). Conversely, genes with a role in pluripotency may be incorporated into the PRC1 interactome upon transcriptional deactivation (example 3). Lastly, some genes (example 1) reside already in PcG bodies and stay unaffected by neural induction.
Figure 3.
Polycomb repressive complex 1 (PRC1) regulates mammalian chromatin in three-dimensional (3D) space during neural induction. Examples of enhancers and associated promoters are numbered 1 to 3. (A) PRC1 assembles target genes in nuclear bodies (pink oval) that represent sites of Polycomb mediated gene silencing in undifferentiated mouse embryonic stem cells (ESCs). PRC1 dependent contacts maintain the promoters of target genes in a poised, albeit repressed state (deep orange box). Polycomb proteins also block the activational effects from contacts between poised enhancers (light orange box) and the associated promoters on transcription of target genes (examples 1 and 2). By contrast, poised enhancers (light green box) strongly activate associated promoters (dark green box) outside PcG bodies and confer potent transcription to a target gene (example 3); (B) The PRC1 promoter interactome disintegrates in mouse ESCs deficient in RING1A and 1B (RING1A/B). This enables promoters and associated enhancers to transit from a poised to an active state (examples 1 and 2); (C) Model for the activation of PcG target genes during neural induction of mESCs. Selected genes are thought to be let off from the PRC1 interactome followed by transcriptional activation (example 2). Conversely, genes with a role in pluripotency may be incorporated into the PRC1 interactome upon transcriptional deactivation (example 3). Lastly, some genes (example 1) reside already in PcG bodies and stay unaffected by neural induction.

Figure 4.
Poised enhancers preset gene activation during anterior neural differentiation. Histone marks are: trimethylation of histone H3 on lysine 27 (H3K27me3; orange hexagon) and acetylation of histone H3 on lysine 27 (H3K27ac; olive dot). (A) Polycomb repressive complex 2 (PRC2) mediates contacts with poised enhancers (light orange box) and their associated gene promoters (grey box) in undifferentiated embryonic stem cells (ESCs). Following neural differentiation, PRC2 disengages and poised enhancers acquire an active state (light green box). This transition stimulates promoter activity (dark green box) of anterior neural genes and RNA polymerase II (Pol II) dependent transcription; (B) Poised enhancers lack H3K27me3 in the absence of the PRC2 core components Suz12 and Eed and do not contact their target promoters in undifferentiated ESCs. Following neural differentiation, poised enhancers do not acquire H3K27ac, and fail in activation and transcription of their associated anterior neural genes.
Figure 4.
Poised enhancers preset gene activation during anterior neural differentiation. Histone marks are: trimethylation of histone H3 on lysine 27 (H3K27me3; orange hexagon) and acetylation of histone H3 on lysine 27 (H3K27ac; olive dot). (A) Polycomb repressive complex 2 (PRC2) mediates contacts with poised enhancers (light orange box) and their associated gene promoters (grey box) in undifferentiated embryonic stem cells (ESCs). Following neural differentiation, PRC2 disengages and poised enhancers acquire an active state (light green box). This transition stimulates promoter activity (dark green box) of anterior neural genes and RNA polymerase II (Pol II) dependent transcription; (B) Poised enhancers lack H3K27me3 in the absence of the PRC2 core components Suz12 and Eed and do not contact their target promoters in undifferentiated ESCs. Following neural differentiation, poised enhancers do not acquire H3K27ac, and fail in activation and transcription of their associated anterior neural genes.

Figure 5.
Ring1B mediates Meis2 expression states during neurodevelopment. (A) In undifferentiated mouse embryonic stem cells (ESCs), Ring1B mediates an interaction with the promoter (grey box) and a downstream silencer of the Meis2 gene (red box). This interaction maintains Meis2 in a repressed, though preactivated, state; (B) During early embryonic midbrain development, a tissue-specific Meis2 enhancer (light green box) is recruited to the promoter-silencer complex to establish a tripartite repressive interaction. Ring1B and/or associated PcG proteins may directly assemble the tissue-specific Meis2 enhancer or serve as a platform for unknown bridging factors; (C) At later stages of embryonic midbrain development, the tissue-specific silencer disengages together with Ring1B from this tripartite complex and leaves back a transcriptionally active promoter-enhancer complex (light and dark green box, respectively); (D) A Ring1B knockout (KO) prevents formation of the promoter-silencer-Ring1B protein complex in mouse ESCs. Later on, the tissue-specific enhancer is not recruited (crossed out arrow) to the Meis2 promoter during embryonic midbrain development and Meis2 is not expressed.
Figure 5.
Ring1B mediates Meis2 expression states during neurodevelopment. (A) In undifferentiated mouse embryonic stem cells (ESCs), Ring1B mediates an interaction with the promoter (grey box) and a downstream silencer of the Meis2 gene (red box). This interaction maintains Meis2 in a repressed, though preactivated, state; (B) During early embryonic midbrain development, a tissue-specific Meis2 enhancer (light green box) is recruited to the promoter-silencer complex to establish a tripartite repressive interaction. Ring1B and/or associated PcG proteins may directly assemble the tissue-specific Meis2 enhancer or serve as a platform for unknown bridging factors; (C) At later stages of embryonic midbrain development, the tissue-specific silencer disengages together with Ring1B from this tripartite complex and leaves back a transcriptionally active promoter-enhancer complex (light and dark green box, respectively); (D) A Ring1B knockout (KO) prevents formation of the promoter-silencer-Ring1B protein complex in mouse ESCs. Later on, the tissue-specific enhancer is not recruited (crossed out arrow) to the Meis2 promoter during embryonic midbrain development and Meis2 is not expressed.

Figure 6.
PCR2 safeguards bivalent domains in mature neurons. Histone marks are: trimethylation of histone 3 on lysine 27 (H3K27me3; orange hexagon) and trimethylation of histone H3 on lysine 4 (H3K4me3; green dot). (A) In medium spiny neurons (MSNs) PRC2’s catalytic subunits Ezh1 and 2 (enhancer of zeste homologue 1 and 2) jointly establish and maintain H3K27me3. Bivalent domains feature silencing H3K27me3 and activating H3K4me3 chromatin marks. Chromatin bivalency is enriched in genes encoding transcription factors (TFs) expressed during development, or in non-neuronal or non-MSN cell types in adults, but also in genes controlling cell death. By contrast, MSN specific genes feature only activating H3K4me3; (B) Combined loss of the catalytic subunits Ezh1 and 2 in adult MSNs abolishes chromatin bivalency. In turn, auto- and coregulatory transcriptional networks are switched on and repress MSN specific genes. This progressive process disrupts postmitotic neuronal function and viability and manifests molecular, cellular, and organismal pathologies closely resembling the ones from mouse models of Huntington disease.
Figure 6.
PCR2 safeguards bivalent domains in mature neurons. Histone marks are: trimethylation of histone 3 on lysine 27 (H3K27me3; orange hexagon) and trimethylation of histone H3 on lysine 4 (H3K4me3; green dot). (A) In medium spiny neurons (MSNs) PRC2’s catalytic subunits Ezh1 and 2 (enhancer of zeste homologue 1 and 2) jointly establish and maintain H3K27me3. Bivalent domains feature silencing H3K27me3 and activating H3K4me3 chromatin marks. Chromatin bivalency is enriched in genes encoding transcription factors (TFs) expressed during development, or in non-neuronal or non-MSN cell types in adults, but also in genes controlling cell death. By contrast, MSN specific genes feature only activating H3K4me3; (B) Combined loss of the catalytic subunits Ezh1 and 2 in adult MSNs abolishes chromatin bivalency. In turn, auto- and coregulatory transcriptional networks are switched on and repress MSN specific genes. This progressive process disrupts postmitotic neuronal function and viability and manifests molecular, cellular, and organismal pathologies closely resembling the ones from mouse models of Huntington disease.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hoffmann, A.; Sportelli, V.; Ziller, M.; Spengler, D. Switch-Like Roles for Polycomb Proteins from Neurodevelopment to Neurodegeneration. Epigenomes 2017, 1, 21. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes1030021
AMA Style
Hoffmann A, Sportelli V, Ziller M, Spengler D. Switch-Like Roles for Polycomb Proteins from Neurodevelopment to Neurodegeneration. Epigenomes. 2017; 1(3):21. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes1030021
Chicago/Turabian StyleHoffmann, Anke, Vincenza Sportelli, Michael Ziller, and Dietmar Spengler. 2017. "Switch-Like Roles for Polycomb Proteins from Neurodevelopment to Neurodegeneration" Epigenomes 1, no. 3: 21. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes1030021