Dynamics of the Methylome and Transcriptome during the Regeneration of Rice


Abstract
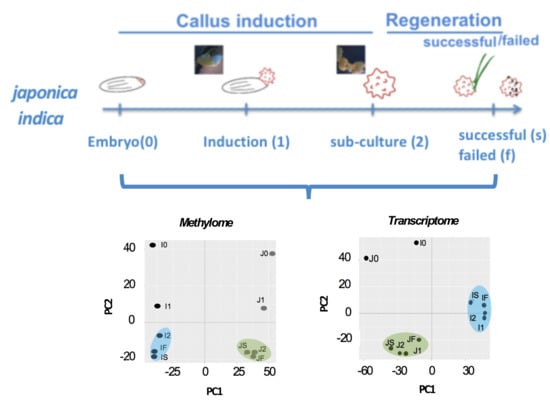
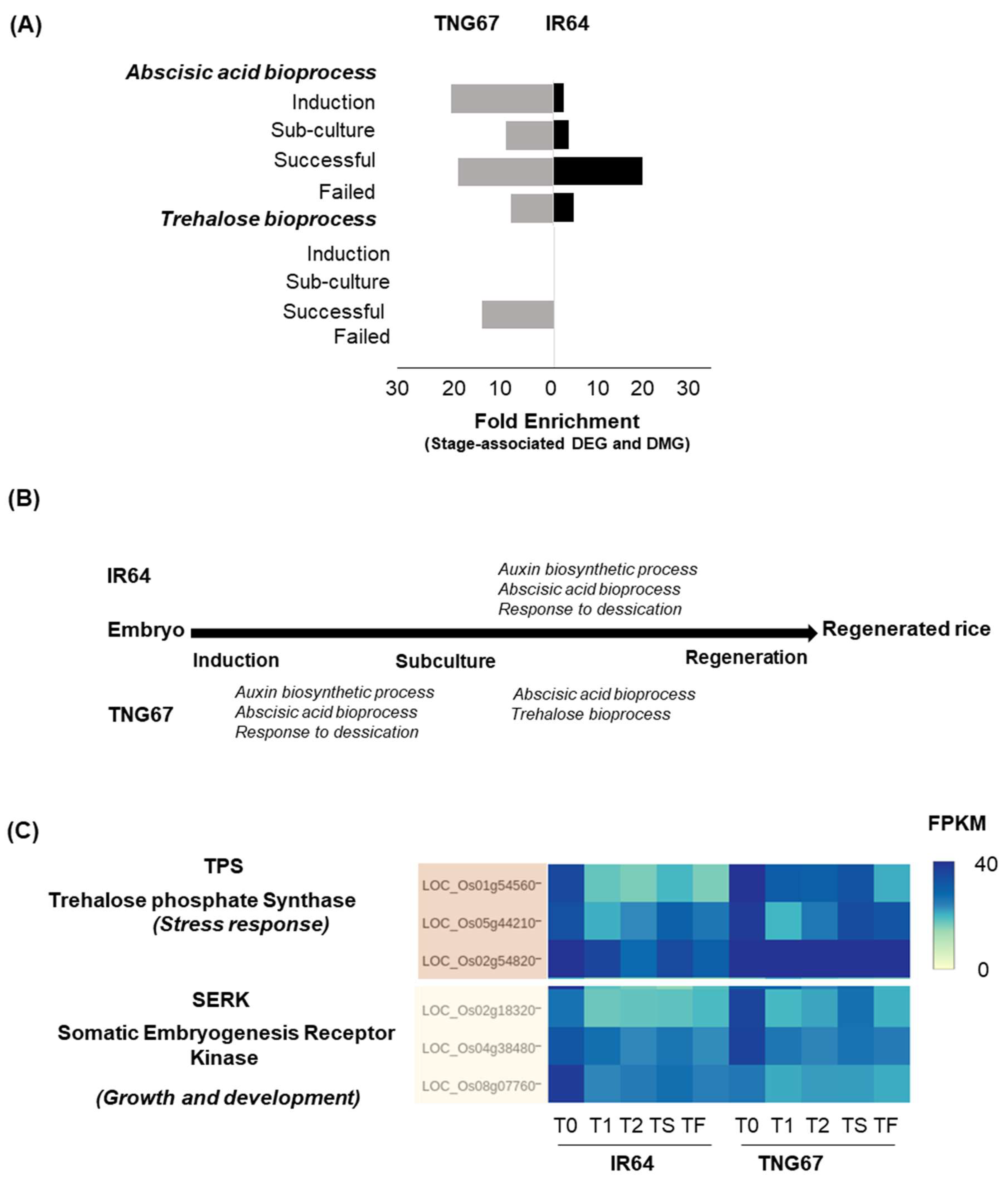
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
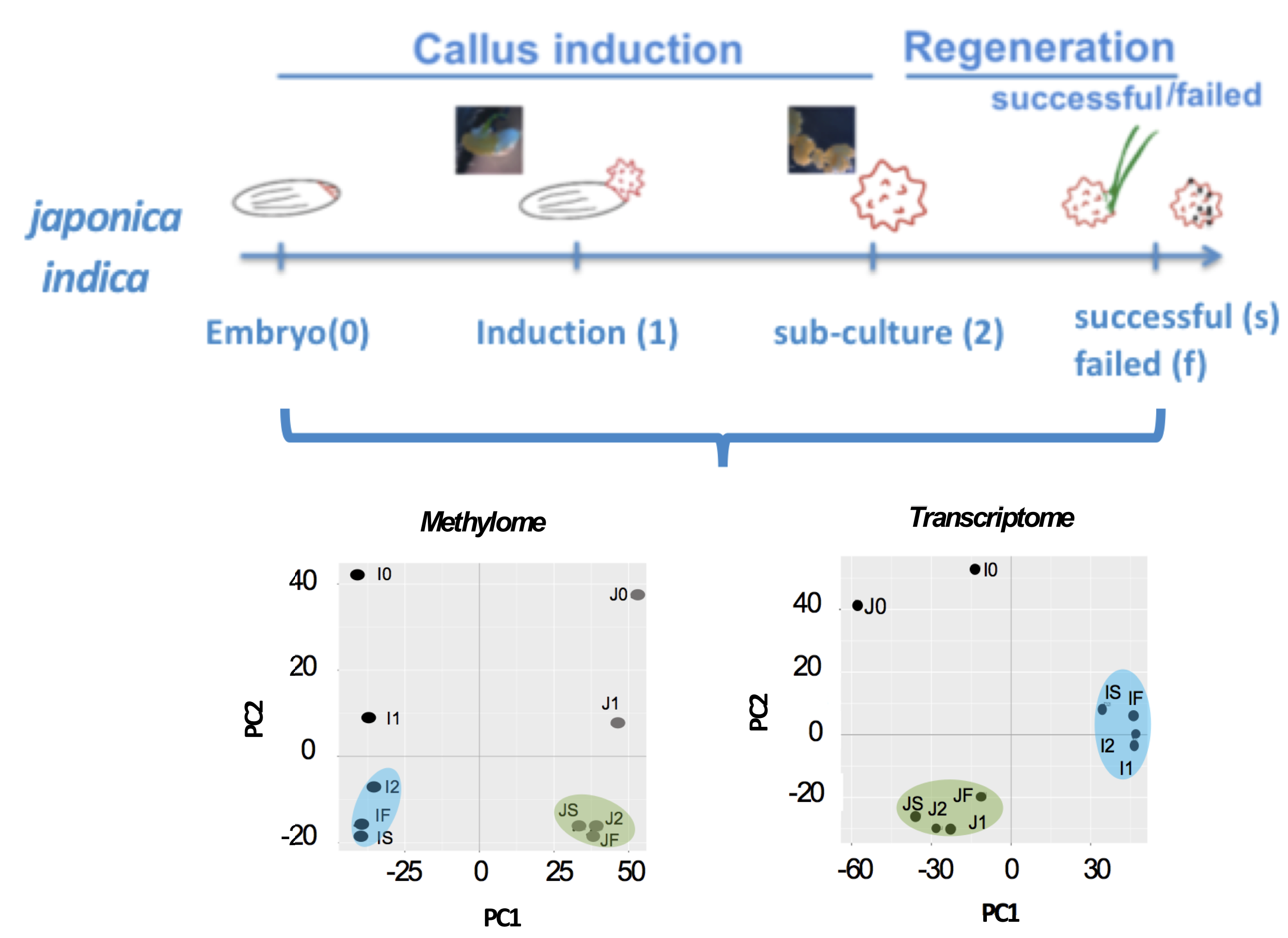
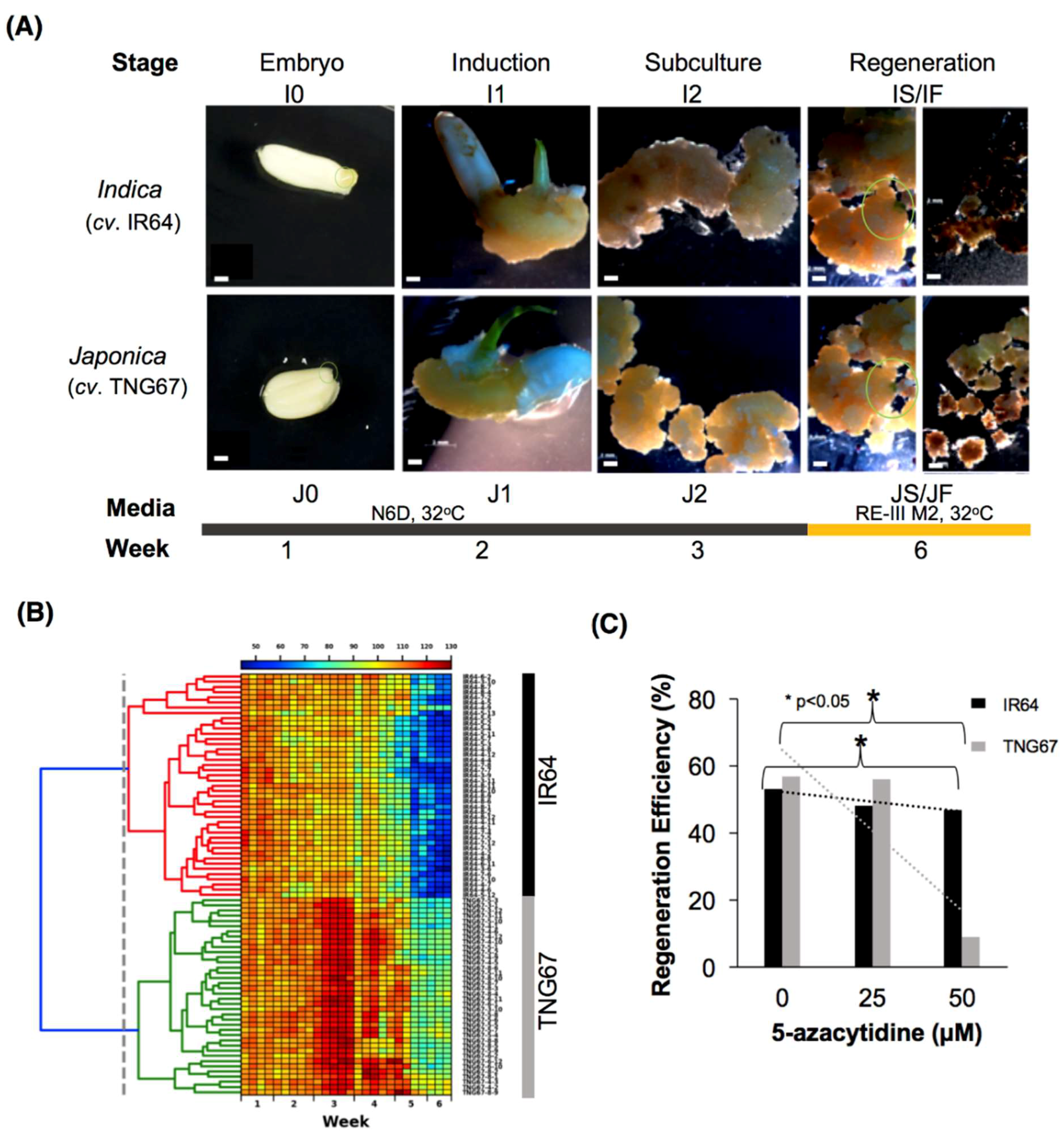
2.1. Morphological Evidence of the Differential Impact of Tissue Culture
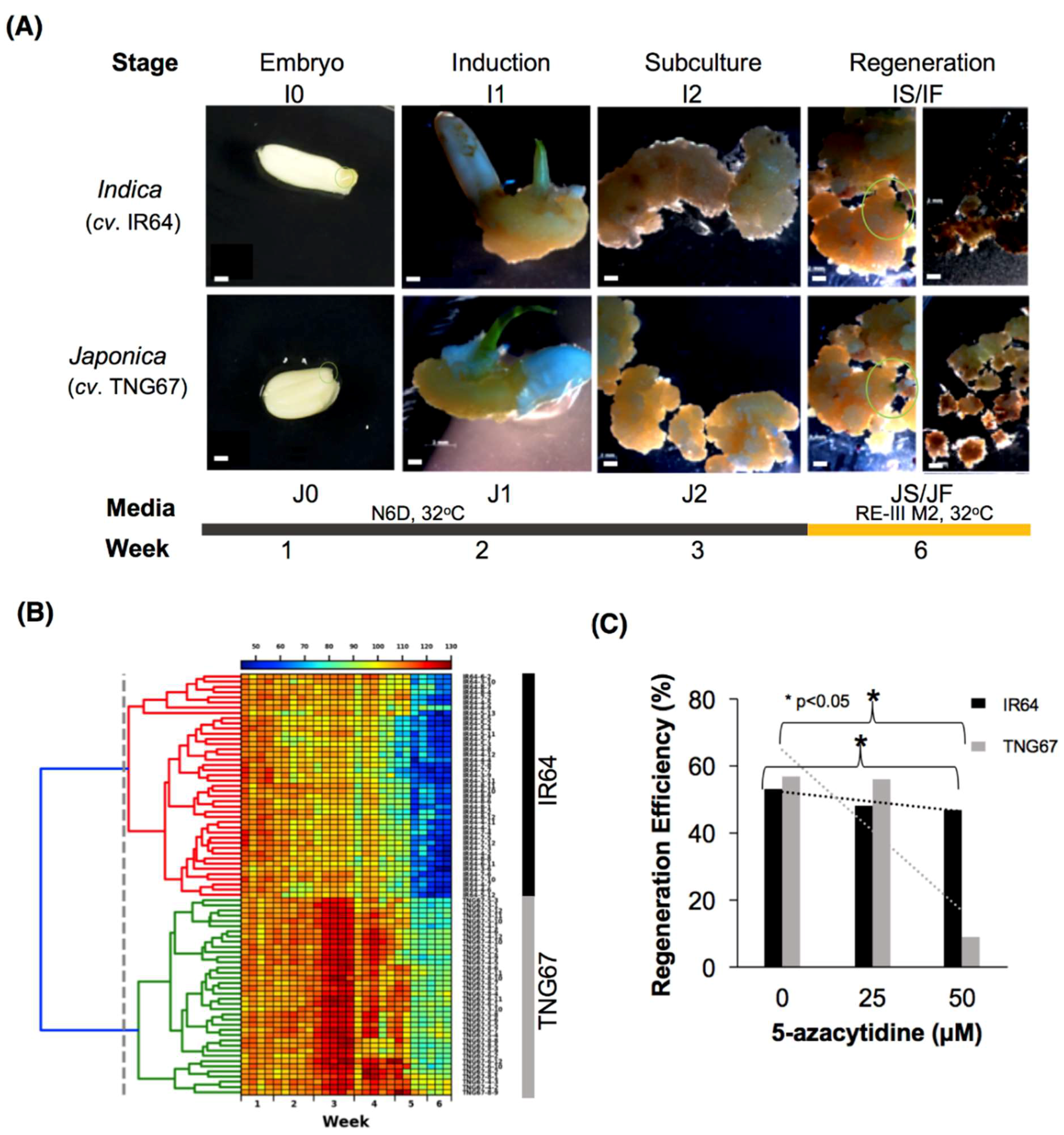
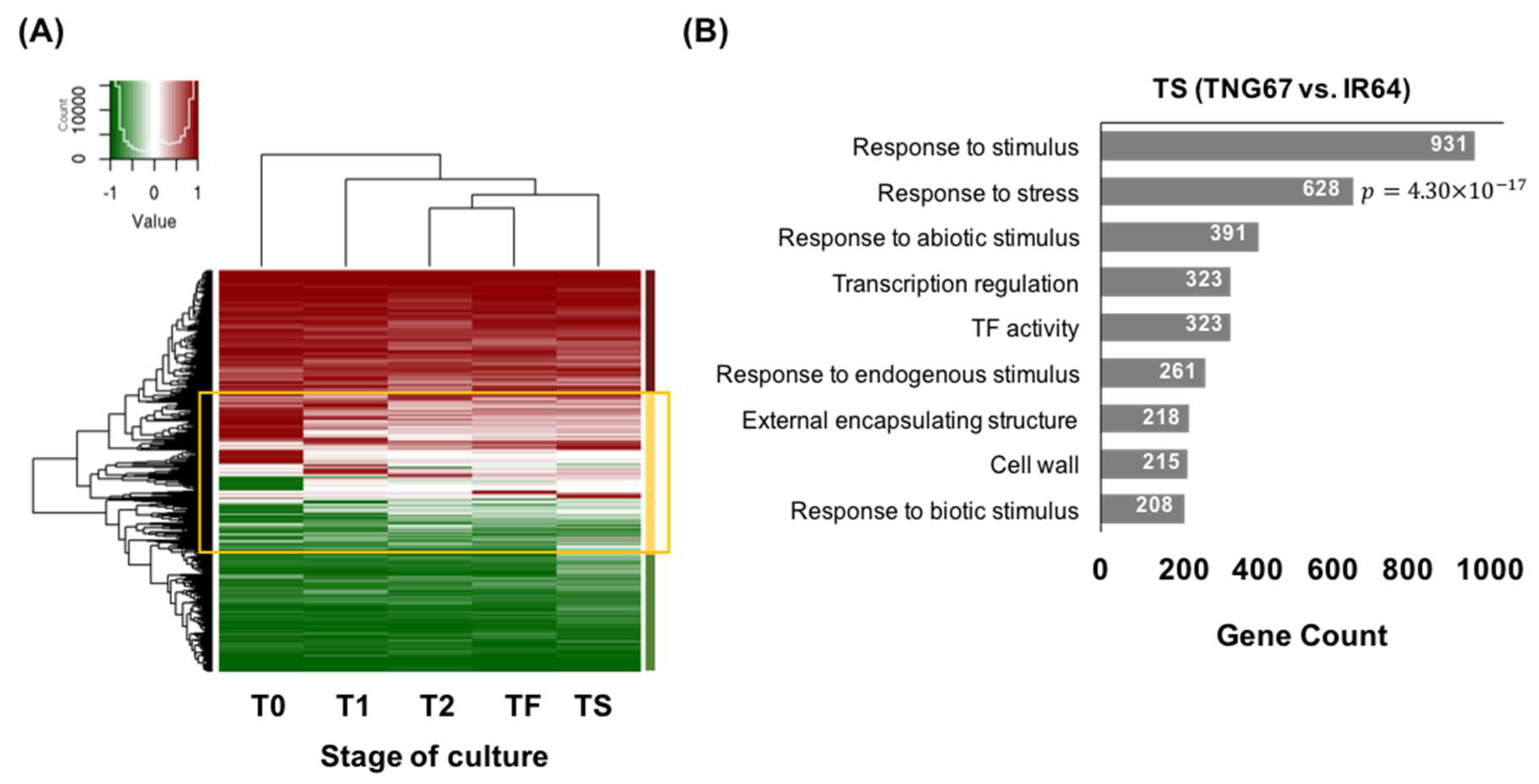
2.2. Stage-Associated Impact of Tissue Culture on IR64 and TNG67
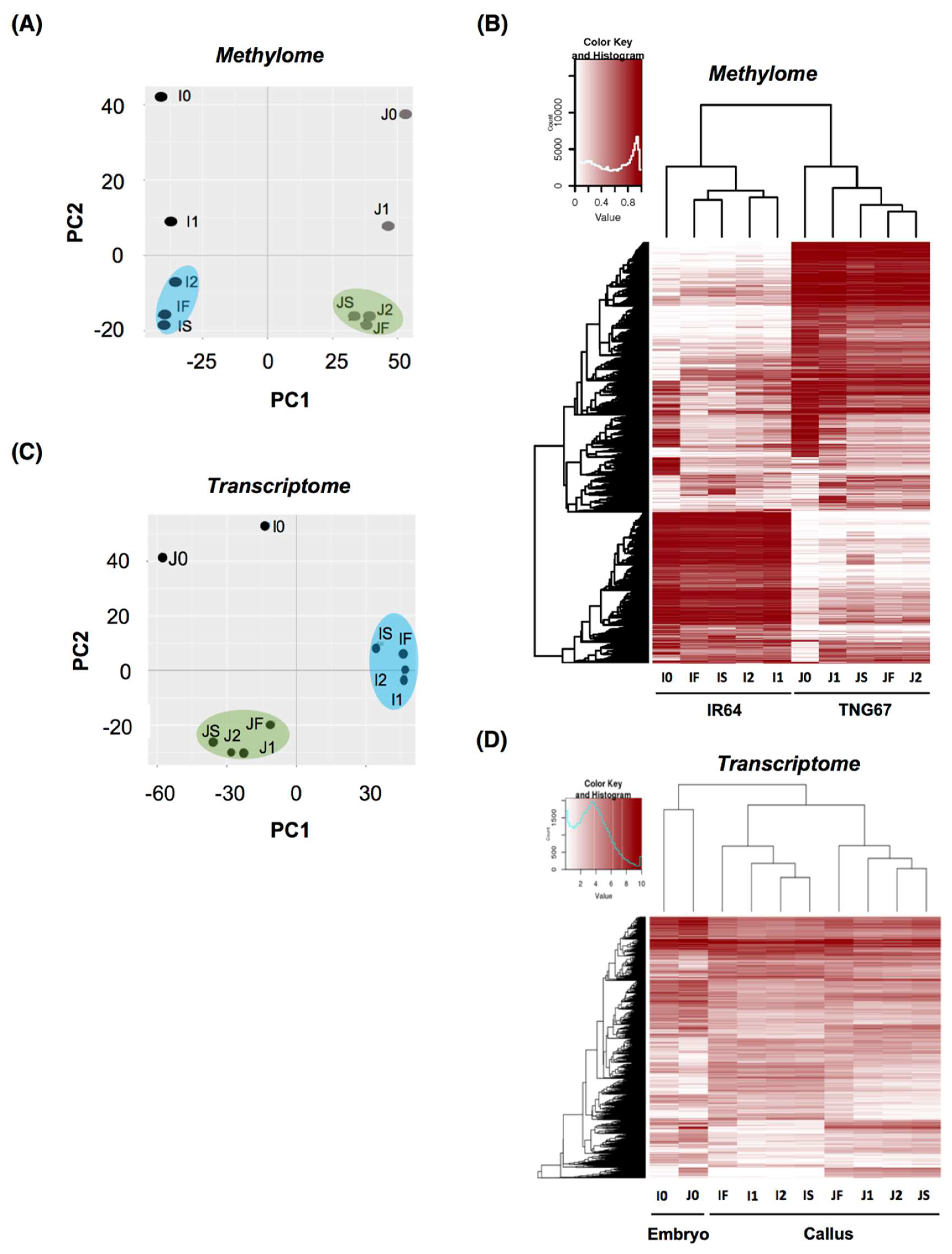
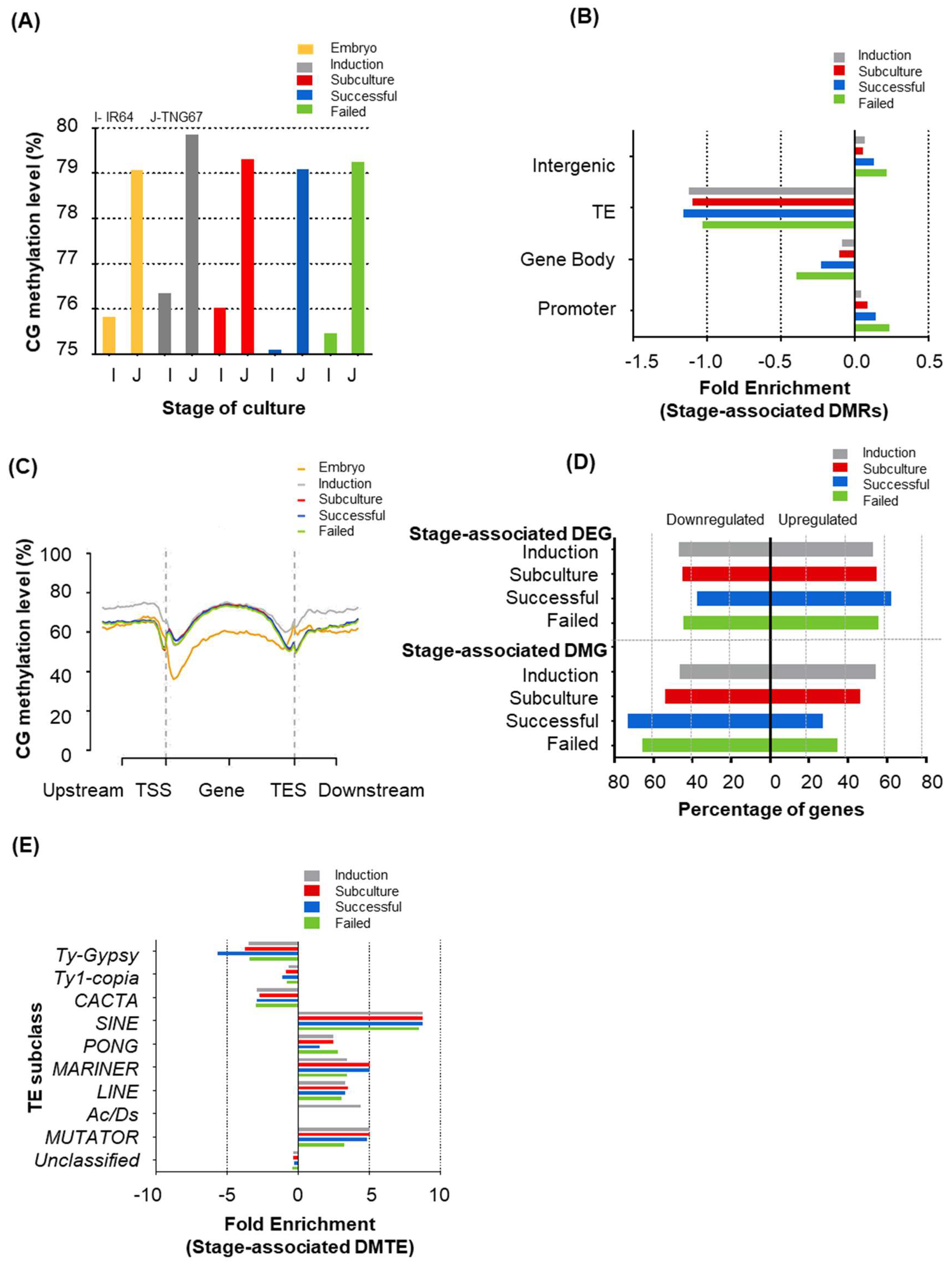
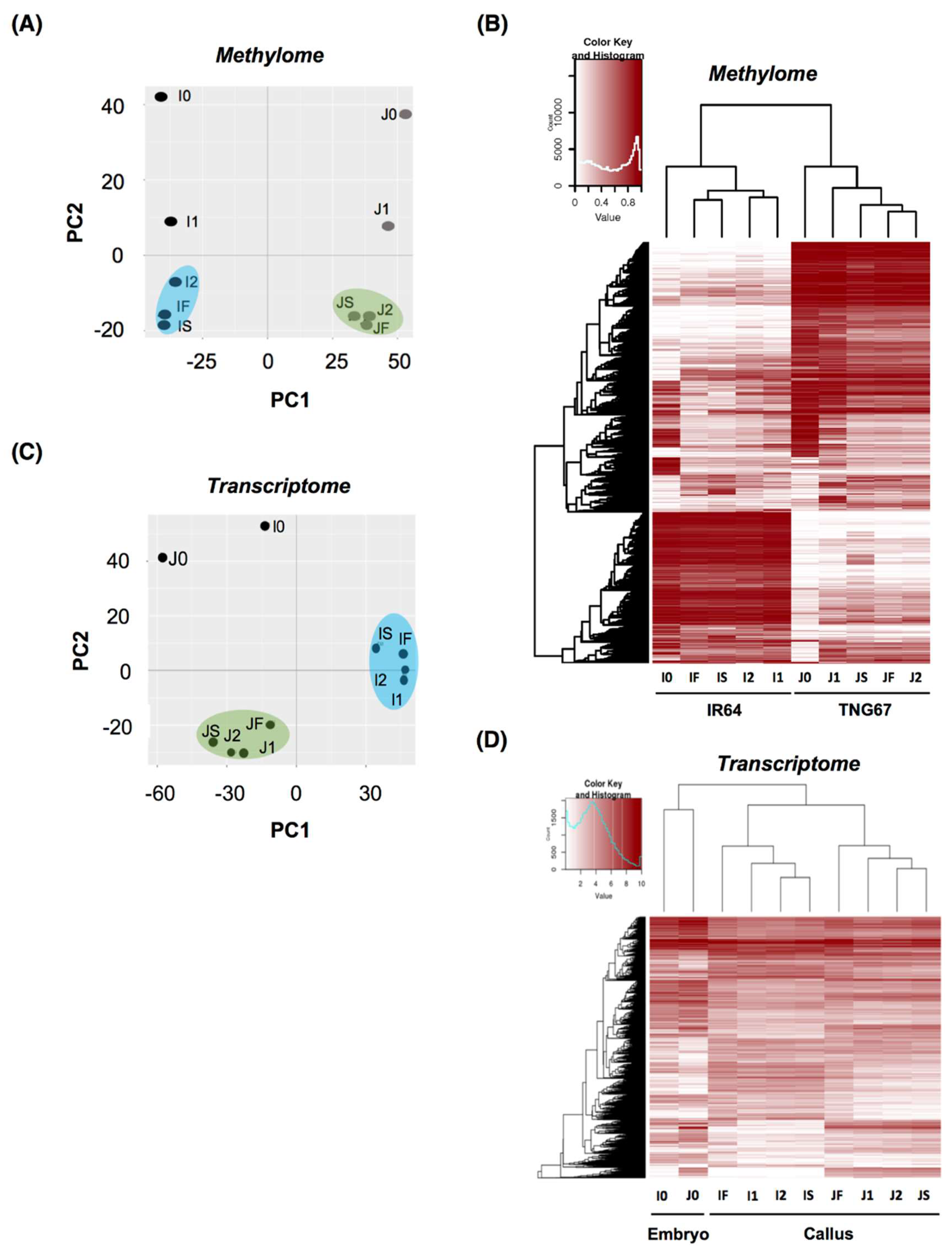
2.3. Stage of Culture Modulates Level and Distribution of DNA Methylation
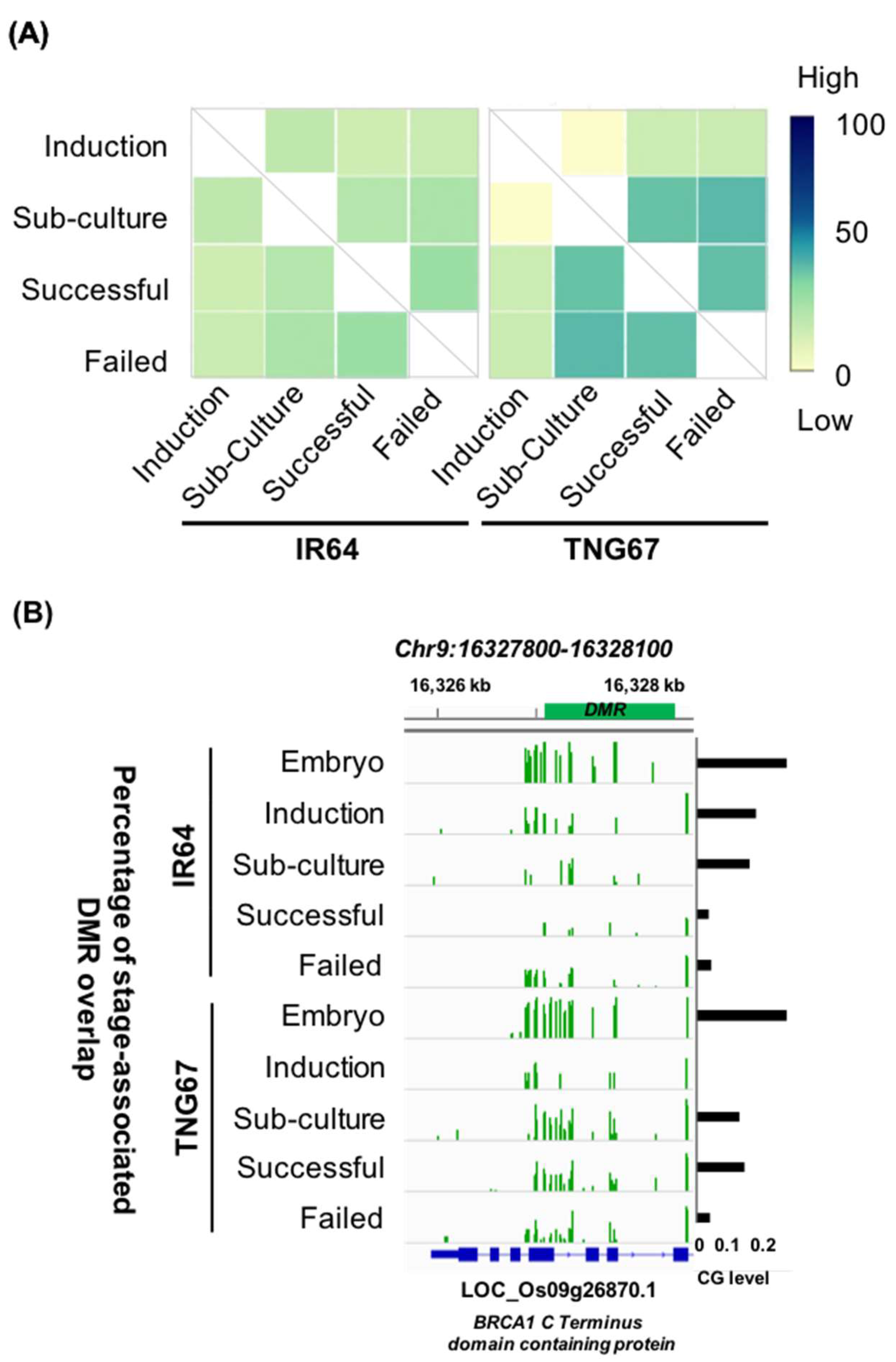
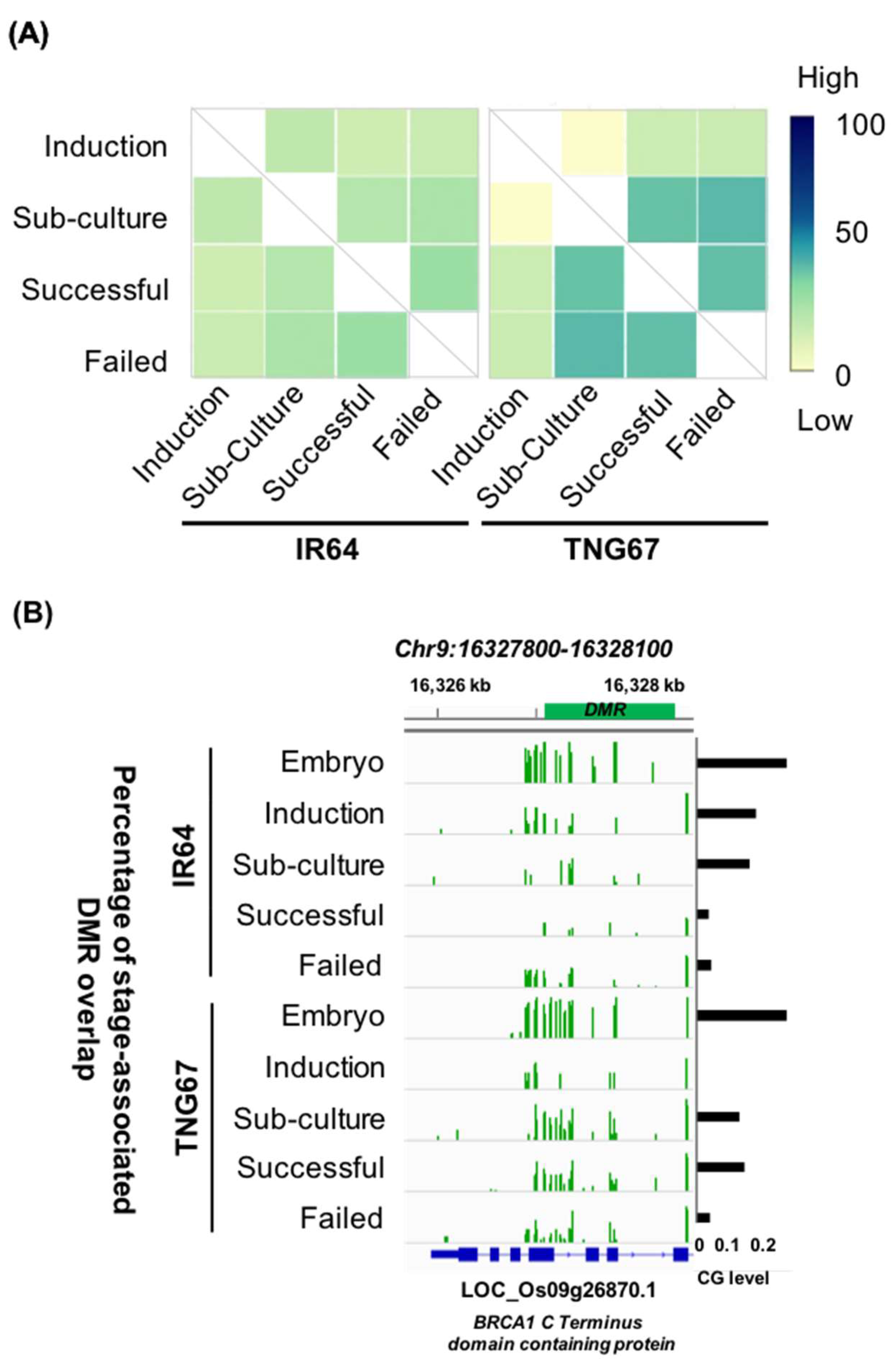
2.4. Stage-Associated Hotspots for Methylation Changes
2.5. Stage Differently Impacts TE Methylation in IR64 and TNG67
2.6. Cultivar-Associated Impact during Culture
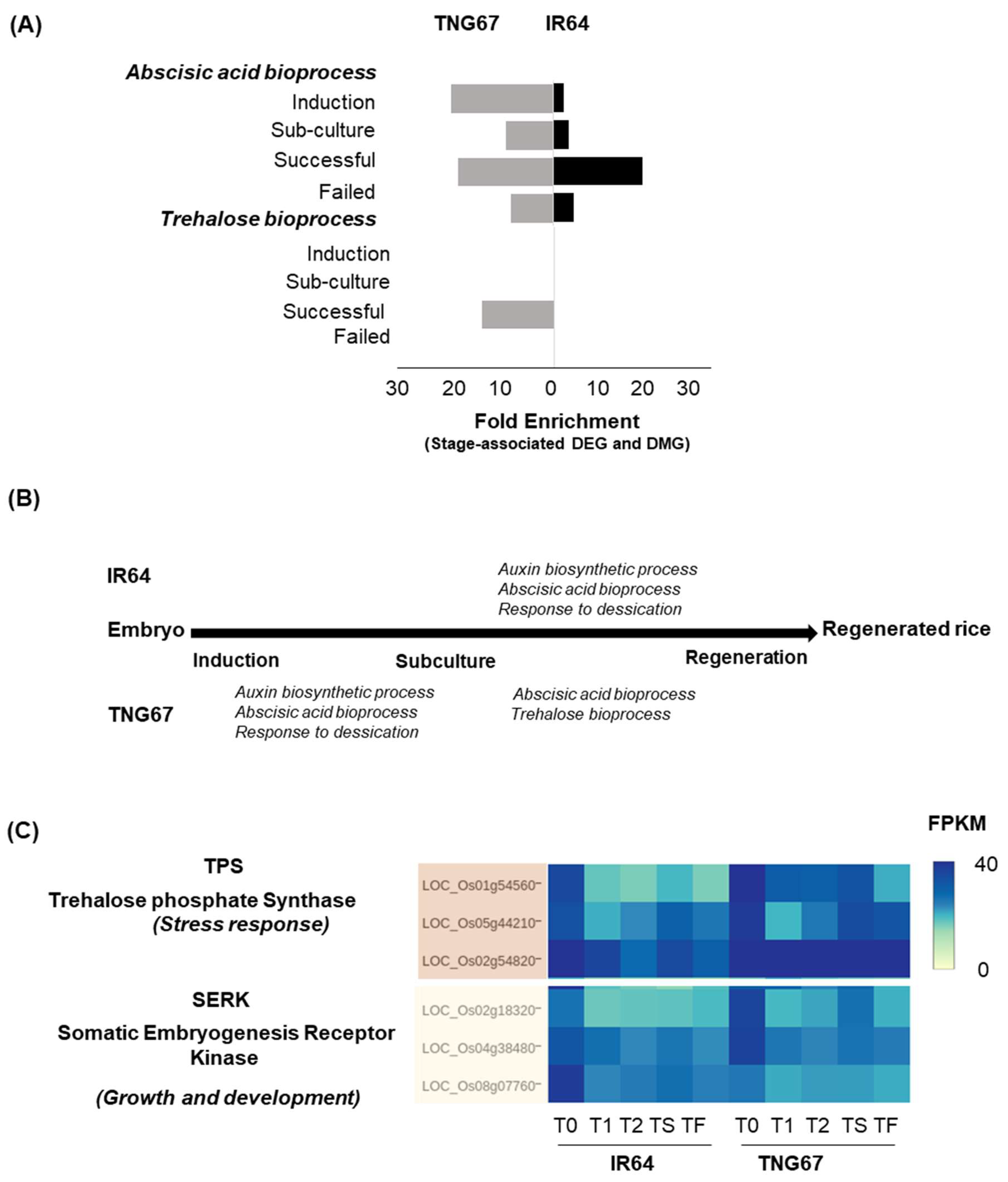
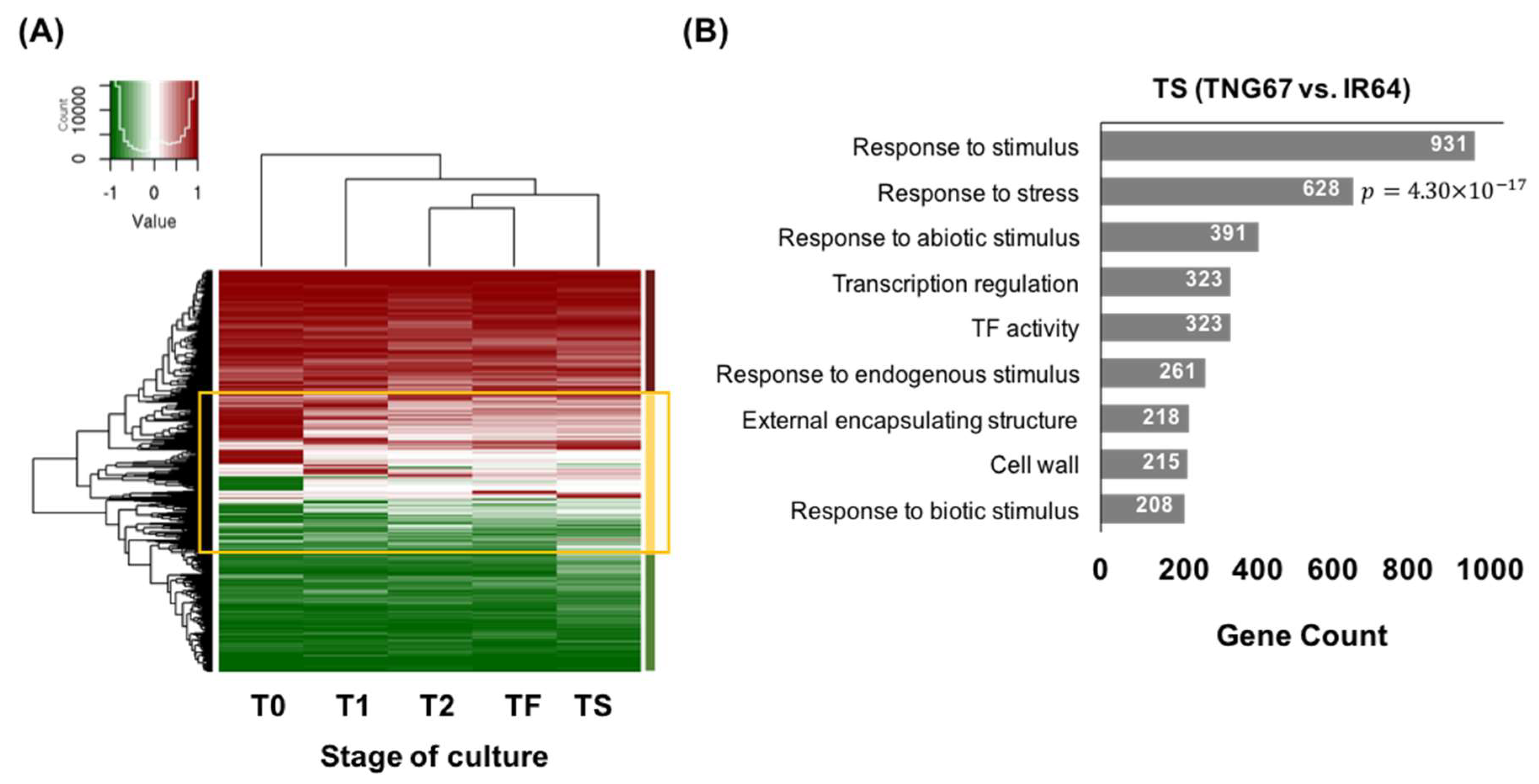
2.7. Regulation of Stress-Responsive Genes by DNA Methylation May Impart Resilience to TNG67
3. Discussion
3.1. Modulation of DNA Methylation in Culture Is Crucial to Regeneration Outcome
3.2. Differential Adaptive Response of Cultivars in Culture
3.3. Integration of Stage- and Cultivar-Associated Changes Determines Regeneration Outcome
4. Materials and Methods
4.1. Callus Induction from Rice Seeds
4.2. Regeneration of Embryogenic Callus
4.3. Morphological Evaluation of Callus Culture
4.4. 5-Azacytidine Demethylation Assay
4.5. RNA-Seq Library Construction and Data Processing
4.6. Library Preparation for Whole-Genome Bisulfite Sequencing (WGBS)
4.7. Identifying Differentially Methylated Regions and Associated Genes
4.8. Estimating False Discovery Rate (FDR)
4.9. Estimation of Bisulfite Conversion Efficiency
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Materials
Abbreviations
References
- Krikorian, A.D.; Berquam, D.L. Plant cell and tissue cultures: The role of Haberlandt. In Plant Tissue Culture; Springer: Vienna, Austria, 2003. [Google Scholar]
- Bhat, S.R.; Srinivasan, S. Molecular and genetic analyses of transgenic plants: Considerations and approaches. Plant Sci. 2002, 163, 673–681. [Google Scholar] [CrossRef]
- Sun, Y.; Li, J.; Xia, L. Precise Genome Modification via Sequence-Specific Nucleases-Mediated Gene Targeting for Crop Improvement. Front. Plant Sci. 2016, 7, 1928. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liang, Z.; Zong, Y.; Wang, Y.; Liu, J.; Chen, K.; Qiu, J.L.; Gao, C. Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA. Nat. Commun. 2016, 7, 12617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulter, J. Regeneration in Plants. Biblic. World 1914, 43, 377–381. [Google Scholar] [CrossRef]
- Larkin, P.J.; Scowcroft, W.R. Somaclonal variation—A novel source of variability from cell cultures for plant improvement. Theor. Appl. Genet. 1981, 60, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Litz, R.E.; Gray, D.J. Somatic embryogenesis for agricultural improvement. World J. Microbiol. Biotechnol. 1995, 11, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.M.; Brar, D.S.; Ahloowalia, B.S. Somaclonal Variations and Induced Mutations in Crop Improvement; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1998. [Google Scholar]
- Nakajima, K. Biotechnology for crop improvement and production in Japan. In Proceedings of the Regional Expert Consultation on the Role of Biotechnology in Crop Production, Bangkok, Thailand, 18–21 June 1991. [Google Scholar]
- Ong-Abdullah, M.; Ordway, J.M.; Jiang, N.; Ooi, S.E.; Kok, S.Y.; Sarpan, N.; Azimi, N.; Hashim, A.T.; Ishak, Z.; Rosli, S.K.; et al. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature 2015, 525, 533–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, H.; Alizadeh, M.; Singh, D.; Singh, U.; Chauhan, N.; Eftekhari, M.; Sadh, R.K. Somaclonal variations and their applications in horticultural crops improvement. 3 Biotech 2016, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Z.; Wang, N.; Gao, Y.; Liu, Y.; Wu, Y.; Bai, Y.; Zhang, Z.; Lin, X.; Dong, Y.; et al. Tissue culture-induced heritable genomic variation in rice, and their phenotypic implications. PLoS ONE 2014, 9, e96879. [Google Scholar] [CrossRef] [PubMed]
- Neelakandan, A.K.; Wang, K. Recent progress in the understanding of tissue culture-induced genome level changes in plants and potential applications. Plant Cell Rep. 2012, 31, 597–620. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Futsuhara, Y. Genotypic variability for callus formation and plant regeneration in rice (Oryza sativa L.). Theor. Appl. Genet. 1986, 72, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Futsuhara, Y. Selection of higher regenerative callus and change in isozyme pattern in rice (Oryza sativa L.). Theor. Appl. Genet. 1989, 78, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, R.; Lin, X.; Bai, Y.; Song, C.; Yu, X.; Xu, C.; Zhao, N.; Dong, Y.; Liu, B. Tissue culture-induced genetic and epigenetic alterations in rice pure-lines, F1 hybrids and polyploids. BMC Plant Biol. 2013, 13, 77. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.J.; Kuang, L.Y.; Oung, H.M.; Cheng, S.Y.; Wu, H.P.; Huang, L.T.; Tseng, Y.T.; Chiou, W.Y.; Hsieh-Feng, V.; Chung, C.H.; et al. Somaclonal variation does not preclude the use of rice transformants for genetic screening. Plant J. 2016, 85, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Stelpflug, S.C.; Eichten, S.R.; Hermanson, P.J.; Springer, N.M.; Kaeppler, S.M. Consistent and heritable alterations of DNA methylation are induced by tissue culture in maize. Genetics 2014, 198, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Kaeppler, S.M.; Kaeppler, H.F.; Rhee, Y. Epigenetic aspects of somaclonal variation in plants. Plant Mol. Biol. 2000, 43, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Kaeppler, S.M.; Phillips, R.L. Tissue culture-induced DNA methylation variation in maize. Proc. Natl. Acad. Sci. USA 1993, 90, 8773–8776. [Google Scholar] [CrossRef] [PubMed]
- Aydin, M.; Arslan, E.; Taspinar, M.S.; Karadayi, G.; Agar, G. Analyses of somaclonal variation in endosperm-supported mature embryo culture of rye (Secale cereale L.). Biotechnol. Biotechnol. Equip. 2016, 30, 1082–1089. [Google Scholar]
- Machczynska, J.; Zimny, J.; Bednarek, P.T. Tissue culture-induced genetic and epigenetic variation in triticale (x Triticosecale spp. Wittmack ex A. Camus 1927) regenerants. Plant Mol. Biol. 2015, 89, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, M.D.; Yanez-Santos, A.M.; Paz, R.C.; Quiroga, M.P.; Marfil, C.F.; Conci, V.C.; Garcia-Lampasona, S.C. Assessment of genetic and epigenetic changes in virus-free garlic (Allium sativum L.) plants obtained by meristem culture followed by in vitro propagation. Plant Cell Rep. 2016, 35, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhong, J.; Li, S.; Wang, X. Tissue culture-induced somaclonal variation of decreased pollen viability in torenia (Torenia fournieri Lind.). Bot. Stud. 2013, 54, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, H.; Youssefian, S. A novel ras-related rgp1 gene encoding a GTP-binding protein has reduced expression in 5-azacytidine-induced dwarf rice. Mol. Gen. Genet. 1991, 228, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Kumpatla, S.P.; Hall, T.C.; Teng, W.; Buchholz, W.G.; Hall, T.C. Epigenetic transcriptional silencing and 5-azacytidine-mediated reactivation of a complex transgene in rice. Plant Physiol. 1997, 115, 361–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumpatla, S.P.; Hall, T.C. Longevity of 5-azacytidine-mediated gene expression and re-establishment of silencing in transgenic rice. Plant Mol. Biol. 1998, 38, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Munsamy, A.; Rutherford, R.S.; Snyman, S.J.; Watt, M.P. 5-Azacytidine as a tool to induce somaclonal variants with useful traits in sugarcane (Saccharum spp.). Plant Biotechnol. Rep. 2013, 7, 489–502. [Google Scholar] [CrossRef]
- Lee, Y.H.; Lee, J.S.; Kim, S.Y.; Sohn, S.H.; Kim, D.Y.; Yoon, I.S. Effects of 5-azacytidine, a DNA methylation inhibitor, on embryogenic callus formation and shoot regeneration from rice mature seeds. J. Plant Biotechnol. 2008, 35, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Sano, Y. The genic nature of gamete eliminator in rice. Genetics 1990, 125, 183–191. [Google Scholar] [PubMed]
- Muller, E.; Brown, P.T.; Hartke, S.; Lorz, H. DNA variation in tissue-culture-derived rice plants. Theor. Appl. Genet. 1990, 80, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Fieldes, M.A. Heritable effects of 5-azacytidine treatments on the growth and development of flax (Linum usitatissimum) genotrophs and genotypes. Genome 1994, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Janousek, B.; Siroky, J.; Vyskot, B. Epigenetic control of sexual phenotype in a dioecious plant, Melandrium album. Mol. Gen. Genet. 1996, 250, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Orlowska, R.; Machczynska, J.; Oleszczuk, S.; Zimny, J.; Bednarek, P.T. DNA methylation changes and TE activity induced in tissue cultures of barley (Hordeum vulgare L.). J. Biol. Res. (Thessalon) 2016, 23, 19. [Google Scholar] [CrossRef] [PubMed]
- Amado, L.; Abranches, R.; Neves, N.; Viegas, W. Development-dependent inheritance of 5-azacytidine-induced epimutations in triticale: Analysis of rDNA expression patterns. Chromosome Res. 1997, 5, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Soppe, W.J.; Jacobsen, S.E.; Alonso-Blanco, C.; Jackson, J.P.; Kakutani, T.; Koornneef, M.; Peeters, A.J. The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol. Cell 2000, 6, 791–802. [Google Scholar] [CrossRef]
- Manning, K.; Tor, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Agetsuma, M.; Kitano, H.; Yoshimura, A.; Matsuoka, M.; Jacobsen, S.E.; Ashikari, M. A metastable DWARF1 epigenetic mutant affecting plant stature in rice. Proc. Natl. Acad. Sci. USA 2009, 106, 11218–11223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroud, H.; Ding, B.; Simon, S.A.; Feng, S.; Bellizzi, M.; Pellegrini, M.; Wang, G.L.; Meyers, B.C.; Jacobsen, S.E. Plants regenerated from tissue culture contain stable epigenome changes in rice. Elife 2013, 2, e00354. [Google Scholar] [CrossRef] [PubMed]
- Vining, K.; Pomraning, K.R.; Wilhelm, L.J.; Ma, C.; Pellegrini, M.; Di, Y.; Mockler, T.C.; Freitag, M.; Strauss, S.H. Methylome reorganization during in vitro dedifferentiation and regeneration of Populus trichocarpa. BMC Plant Biol. 2013, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Ran, L.; Li, M.; Fan, H.X.; Jiang, J.J.; Wang, Y.P.; Sokolov, V. Epigenetic variation in the callus of Brassica napus under different inducement conditions. Russ. J. Genet. 2016, 52, 802–809. [Google Scholar] [CrossRef]
- Grandbastien, M.A. Activation of plant retrotransposons under stress conditions. Trends Plant Sci. 1998, 3, 181–187. [Google Scholar] [CrossRef]
- Garg, A.K.; Kim, J.K.; Owens, T.G.; Ranwala, A.P.; Choi, Y.D.; Kochian, L.V.; Wu, R.J. Trehalose accumulation in rice plants confers high tolerance levels to different abiotic stresses. Proc. Natl. Acad. Sci. USA 2002, 99, 15898–15903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nic-Can, G.I.; López-Torres, A.; Barredo-Pool, F.; Wrobel, K.; Loyola-Vargas, V.M.; Rojas-Herrera, R.; De-la-Peña, C. New insights into somatic embryogenesis: LEAFY COTYLEDON1, BABY BOOM1 and WUSCHEL-RELATED HOMEOBOX4 are epigenetically regulated in Coffea canephora. PLoS ONE 2013, 8, e72160. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, A.; Zebrowski, J.; Oklejewicz, B.; Czarnik, J.; Halibart-Puzio, J.; Wnuk, M. The age-dependent epigenetic and physiological changes in an Arabidopsis T87 cell suspension culture during long-term cultivation. Biochem. Biophys. Res. Commun. 2014, 447, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ma, L.; Yang, X.; Zhang, L.; Zeng, X.; Xie, S.; Peng, H.; Gao, S.; Lin, H.; Pan, G.; et al. Integrative analysis of DNA methylation, mRNAs, and small RNAs during maize embryo dedifferentiation. BMC Plant Biol. 2017, 17, 105. [Google Scholar] [CrossRef] [PubMed]
- Sena, G.; Birnbaum, K.D. Built to rebuild: In search of organizing principles in plant regeneration. Curr. Opin. Genet. Dev. 2010, 20, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Leibfried, A.; To, J.P.; Busch, W.; Stehling, S.; Kehle, A.; Demar, M.; Kieber, J.J.; Lohmann, J.U. WUSCHEL controls meristem function by direct regulation of cytokinin-inducible response regulators. Nature 2005, 438, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.P.; Heisler, M.G.; Reddy, G.V.; Ohno, C.; Das, P.; Meyerowitz, E.M. Pattern formation during de novo assembly of the Arabidopsis shoot meristem. Development 2007, 134, 3539–3548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeuchi, M.; Sugimoto, K.; Iwase, A. Plant callus: Mechanisms of induction and repression. Plant Cell 2013, 25, 3159–3173. [Google Scholar] [CrossRef] [PubMed]
- Skoog, F.; Miller, C.O. Chemical regulation of growth and organ formation in plant tissues cultured in vitro. Symp. Soc. Exp. Biol. 1957, 11, 118–130. [Google Scholar] [PubMed]
- De Klerk, G.J.; Arnholdt-Schmitt, B.; Lieberei, R.; Neumann, K.H. Human genetics in the Crescent City. Nat. Genet. 2006, 38, 1221. [Google Scholar] [CrossRef]
- Arnholdt-Schmitt, B.; Herterich, S.; Neumann, K.H. Physiological aspects of genome variability in tissue culture. I. Growth phase-dependent differential DNA methylation of the carrot genome (Daucus carota L.) during primary culture. Theor. Appl. Genet. 1995, 91, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, K.D.; Roudier, F. Epigenetic memory and cell fate reprogramming in plants. Regeneration 2017, 4, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Morozova, N.; Williams, L.; Libs, L.; Avivi, Y.; Grafi, G. Two phases of chromatin decondensation during dedifferentiation of plant cells: Distinction between competence for cell fate switch and a commitment for S phase. J. Biol. Chem. 2001, 276, 22772–22778. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Su, Y.H.; Cheng, Z.J.; Zhang, X.S. Cell fate switch during in vitro plant organogenesis. J. Integr. Plant Biol. 2008, 50, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Meshorer, E.; Yellajoshula, D.; George, E.; Scambler, P.J.; Brown, D.T.; Misteli, T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell 2006, 10, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; Jeddeloh, J.A.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 2003, 163, 1109–1122. [Google Scholar] [PubMed]
- Li, W.; Liu, H.; Cheng, Z.J.; Su, Y.H.; Han, H.N.; Zhang, Y.; Zhang, X.S. DNA methylation and histone modifications regulate de novo shoot regeneration in arabidopsis by modulating wuschel expression and auxin signaling. PLoS Genet. 2011, 7, e1002243. [Google Scholar] [CrossRef] [PubMed]
- González, A.I.; Saiz, A.; Acedo, A.; Ruiz, M.L.; Polanco, C. Analysis of genomic DNA methylation patterns in regenerated and control plants of rye (Secale cereale L.). Plant Growth Regul. 2013, 70, 227–236. [Google Scholar] [CrossRef]
- Uthup, T.K.; Ravindran, M.; Bini, K.; Thakurdas, S. Divergent DNA methylation patterns associated with abiotic stress in Hevea brasiliensis. Mol. Plant 2011, 4, 996–1013. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Narayana Chevala, V.; Shankar, R.; Jain, M. Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci. Rep. 2015, 5, 14922. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, D.; Wang, Z.; Xun, H.; Ma, J.; Wang, H.; Huang, W.; Liu, Y.; Lin, X.; Li, N.; et al. Mutation of the RDR1 gene caused genome-wide changes in gene expression, regional variation in small RNA clusters and localized alteration in DNA methylation in rice. BMC Plant Biol. 2014, 14, 177. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.S.; Sano, H. Abiotic-stress induces demethylation and transcriptional activation of a gene encoding a glycerophosphodiesterase-like protein in tobacco plants. Mol. Genet. Genom. 2007, 277, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Guerra, M.P.; Steiner, N.; Farias-Soares, F.L.; Vieira Ldo, N.; Fraga, H.P.; Rogge-Renner, G.D.; Maldonado, S.B. Somatic Embryogenesis in Araucaria angustifolia (Bertol.) Kuntze (Araucariaceae). Methods Mol. Biol. 2016, 1359, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Editorial. Between genotype and phenotype. Nat. Genet. 2006, 38, 1355. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 17 July 2018).
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2go: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Jung, K.H.; Choi, D.; Hwang, D.; Zhu, J.; Ronald, P.C. The Rice Oligonucleotide Array Database: An atlas of rice gene expression. Rice 2012, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Fiziev, P.; Yan, W.; Cokus, S.; Sun, X.; Zhang, M.Q.; Chen, P.Y.; Pellegrini, M. BS-Seeker2: A versatile aligning pipeline for bisulfite sequencing data. BMC Genom. 2013, 14, 774. [Google Scholar] [CrossRef] [PubMed]
- Baranek, M.; Cechova, J.; Kovacs, T.; Eichmeier, A.; Wang, S.; Raddova, J.; Necas, T.; Ye, X. Use of Combined MSAP and NGS Techniques to Identify Differentially Methylated Regions in Somaclones: A Case Study of Two Stable Somatic Wheat Mutants. PLoS ONE 2016, 11, e0165749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Kitimu, S.R.; Taylor, J.; March, T.J.; Tairo, F.; Wilkinson, M.J.; Rodriguez Lopez, C.M. Meristem micropropagation of cassava (Manihot esculenta) evokes genome-wide changes in DNA methylation. Front. Plant Sci. 2015, 6, 590. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, F.-M.; Gohain, M.; Allishe, A.; Huang, Y.-J.; Liao, J.-L.; Kuang, L.-Y.; Chen, P.-Y. Dynamics of the Methylome and Transcriptome during the Regeneration of Rice. Epigenomes 2018, 2, 14. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes2030014
Hsu F-M, Gohain M, Allishe A, Huang Y-J, Liao J-L, Kuang L-Y, Chen P-Y. Dynamics of the Methylome and Transcriptome during the Regeneration of Rice. Epigenomes. 2018; 2(3):14. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes2030014
Chicago/Turabian StyleHsu, Fei-Man, Moloya Gohain, Archana Allishe, Yan-Jiun Huang, Jo-Ling Liao, Lin-Yun Kuang, and Pao-Yang Chen. 2018. "Dynamics of the Methylome and Transcriptome during the Regeneration of Rice" Epigenomes 2, no. 3: 14. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes2030014