Nuclear Envelope Regulation of Oncogenic Processes: Roles in Pancreatic Cancer


1
Genetics and Genomics, Sanford Research, Sioux Falls, SD 57104, USA
2
Department of Pediatrics, Sanford School of Medicine of the University of South Dakota, Sioux Falls, SD 57105, USA
*
Author to whom correspondence should be addressed.
Epigenomes 2018, 2(3), 15; https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes2030015
Submission received: 10 August 2018
/
Revised: 30 August 2018
/
Accepted: 31 August 2018
/
Published: 2 September 2018
(This article belongs to the Special Issue Epigenetics of Pancreatic Cancer)
Abstract
:Pancreatic cancer is an aggressive and intractable malignancy with high mortality. This is due in part to a high resistance to chemotherapeutics and radiation treatment conferred by diverse regulatory mechanisms. Among these, constituents of the nuclear envelope play a significant role in regulating oncogenesis and pancreatic tumor biology, and this review focuses on three specific components and their roles in cancer. The LINC complex is a nuclear envelope component formed by proteins with SUN and KASH domains that interact in the periplasmic space of the nuclear envelope. These interactions functionally and structurally couple the cytoskeleton to chromatin and facilitates gene regulation informed by cytoplasmic activity. Furthermore, cancer cell invasiveness is impacted by LINC complex biology. The nuclear lamina is adjacent to the inner nuclear membrane of the nuclear envelope and can actively regulate chromatin in addition to providing structural integrity to the nucleus. A disrupted lamina can impart biophysical compromise to nuclear structure and function, as well as form dysfunctional micronuclei that may lead to genomic instability and chromothripsis. In close relationship to the nuclear lamina is the nuclear pore complex, a large megadalton structure that spans both outer and inner membranes of the nuclear envelope. The nuclear pore complex mediates bidirectional nucleocytoplasmic transport and is comprised of specialized proteins called nucleoporins that are overexpressed in many cancers and are diagnostic markers for oncogenesis. Furthermore, recent demonstration of gene regulatory functions for discrete nucleoporins independent of their nuclear trafficking function suggests that these proteins may contribute more to malignant phenotypes beyond serving as biomarkers. The nuclear envelope is thus a complex, intricate regulator of cell signaling, with roles in pancreatic tumorigenesis and general oncogenic transformation.
1. Introduction
Pancreatic cancer is among the most aggressive forms of cancer in the world with a higher occurrence observed in developed countries [1,2]. In the United States, pancreatic cancer is the third most deadliest form of cancer, with a 5-year survival rate of only 8.5% and an incidence that has slowly but steadily risen in the past 15 years [3,4]. Exocrine pancreatic cancers, often located at the head of the pancreas, account for ≈95% of all diagnosed cases with the most common type being pancreatic ductal adenocarcinomas (PDAC). Other less common tumors, i.e., pancreatic neuroendocrine tumors, account for less than 5% of cases and have a more favorable 5-year survival rate at (50–65%) [2,5]. Still, PDAC is often synonymous for pancreatic cancer since it influences the vast majority of the epidemiological data of this disease.
PDAC is characterized by a large, firm mass with poorly defined margins and protrusions that extend inside the pancreas [6]. This aggressive tumor produces a strong desmoplastic reaction that contributes to its pervasive chemoresistance [7,8]. Gross pathology of PDAC biopsies typically present as whitish masses, that in conjunction with other distinctive histological features (i.e., presence of infiltrative but well-differentiated neoplastic glands, luminal necrosis, perineural, and vascular invasion), support the diagnosis of pancreatic adenocarcinoma [6]. PDAC elicits vague symptoms during early stages, with the most common being anorexia, fatigue, and weight loss; moreover, an increase in the interval from symptom onset to diagnosis has been observed in late stage exocrine pancreatic cancer [9,10,11].
There have been many established risk factors implicated in the risk of pancreatic cancer, including chronic diabetes mellitus, non-hereditary chronic pancreatitis, and some environmental risk factors, i.e., meat and fat high diets, obesity, alcohol, and tobacco use [12]. The genetic risk factors associated with pancreatic adenocarcinomas is a field that is in continuous growth and exploration [13,14,15]. Detailed in Table 1 are the most common somatic and germline mutations that have so far been detected in patients with PDAC. Dissecting the functional gene ontology of these genes implicates possible alternative pathways associated with pancreatic cancer that deserve exploration. The functional enrichment analysis of these common somatic and germline mutation genes contain functional terms common to both groups, being nucleoplasmic function and DNA regulation (Table 2 and Table 3).
The development and progression of cancer has been attributed to: mitotic aberrations leading to aneuploidy; chromosomal abnormalities such as translocations, deletions, inversions, and duplications; and genome-wide chromothripsis [16]. Dysregulation of expression or regulation of tumor suppressors, oncogenes, and cell cycling controls can also initiate pathological signaling leading to oncogenesis [17,18,19,20]. Furthermore, epigenetic reader, writer, and eraser malfunction, with concomitant changes in chromatin structure, alters accessibility and disrupts proper programming [21]. Other nuclear regulatory modalities may play a role in cancer development given the emerging role of nuclear associated genes in pancreatic adenocarcinoma (Table 2 and Table 3). Given the increased functional repertoire of the nuclear envelope in a variety of biological processes, nuclear envelope-related mechanisms of oncogenesis and their relevance to cancer biology are of critical importance and consideration.
2. Chromatin Organization and Dynamics
In general, chromatin access is highly regulated, underscored by dynamic rates of heterochromatin maintenance, and relaxation that control cellular processes in regular development and cancer [22]. Mechanisms of chromatin organization and accessibility include soluble nucleoplasmic machinery and nuclear envelope-bound components, with discrete temporospatial regulatory events that can occur within specific subnuclear domains [23,24,25,26]. For example, chromosome territories are the dynamic three-dimensional volumes occupied by each chromosome within the nucleus [27,28]. These non-static volumes are subject to spatial shifting in response to mitotic signaling [29,30], cellular aging cues [31], and DNA damage [32,33]. Chromosome territory dynamics facilitate gene positioning for transcriptional control [34], as well as gene triage for DNA repair [35]. Gene regulation also occurs within the spaces separating chromosome territories, known as interchromatin domains or compartments [36,37,38]. Within these spaces, post-transcriptional processing centers, such as nuclear speckles, can assemble to regulate transcriptional splicing [39,40]. This can occur throughout the nucleus, coordinating discrete chromosome territories to facilitate cis- and trans-regulation [40]. Heterochromatic compaction is subject to mechanisms that control chromosome dynamics. Cycling between heterochromatic relaxation and maintenance insures efficient ad hoc access to specific gene programs while simultaneously restricting access to inactive genes [41,42,43]. In concert with nucleosomal sliding and eviction, chromatin structural remodeling emerges as a highly dynamic system of global gene regulation and expression control. A key function of these chromatin dynamics is to bring super enhancers in contact with target genes, facilitating distal gene regulation [44,45,46]. This can be disrupted in cancer, where dysregulation of enhancer accessibility can promote malignancy [47,48].
Normal cellular function is dependent on proper maintenance of these various mechanisms of chromatin control, and their disruption has significant consequences for development and oncogenesis. The nuclear envelope interacts with this machinery and contributes to the regulation of chromatin structure and dynamics in addition to its function as the specialized intermediary between extranuclear and intranuclear compartments [49,50]. In this regard, the nuclear lamina, the LINC complex, and the nuclear pore complex emerge as key regulators of chromatin accessibility and cellular function, with relevant implications for cancer biology [51,52].
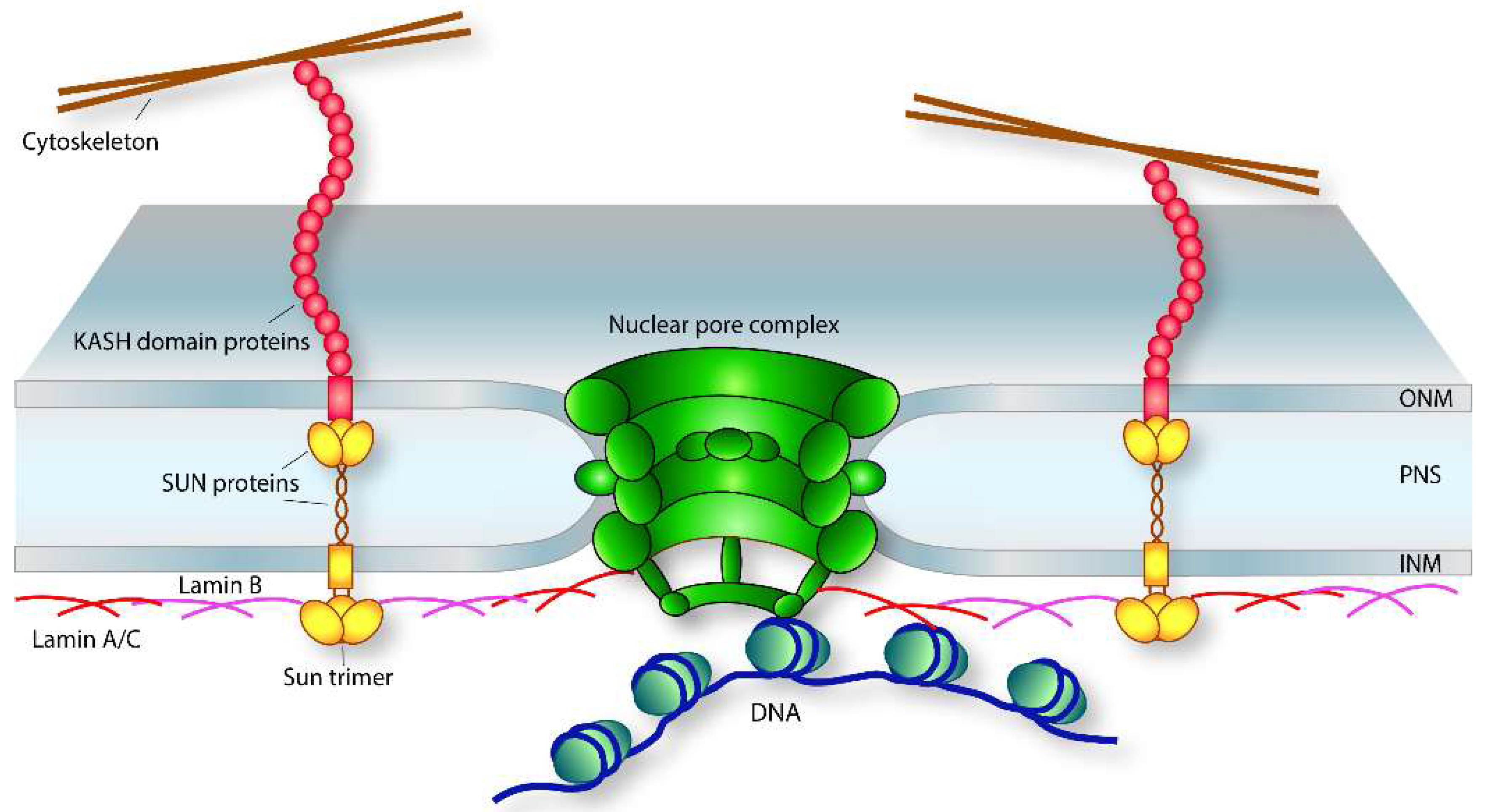
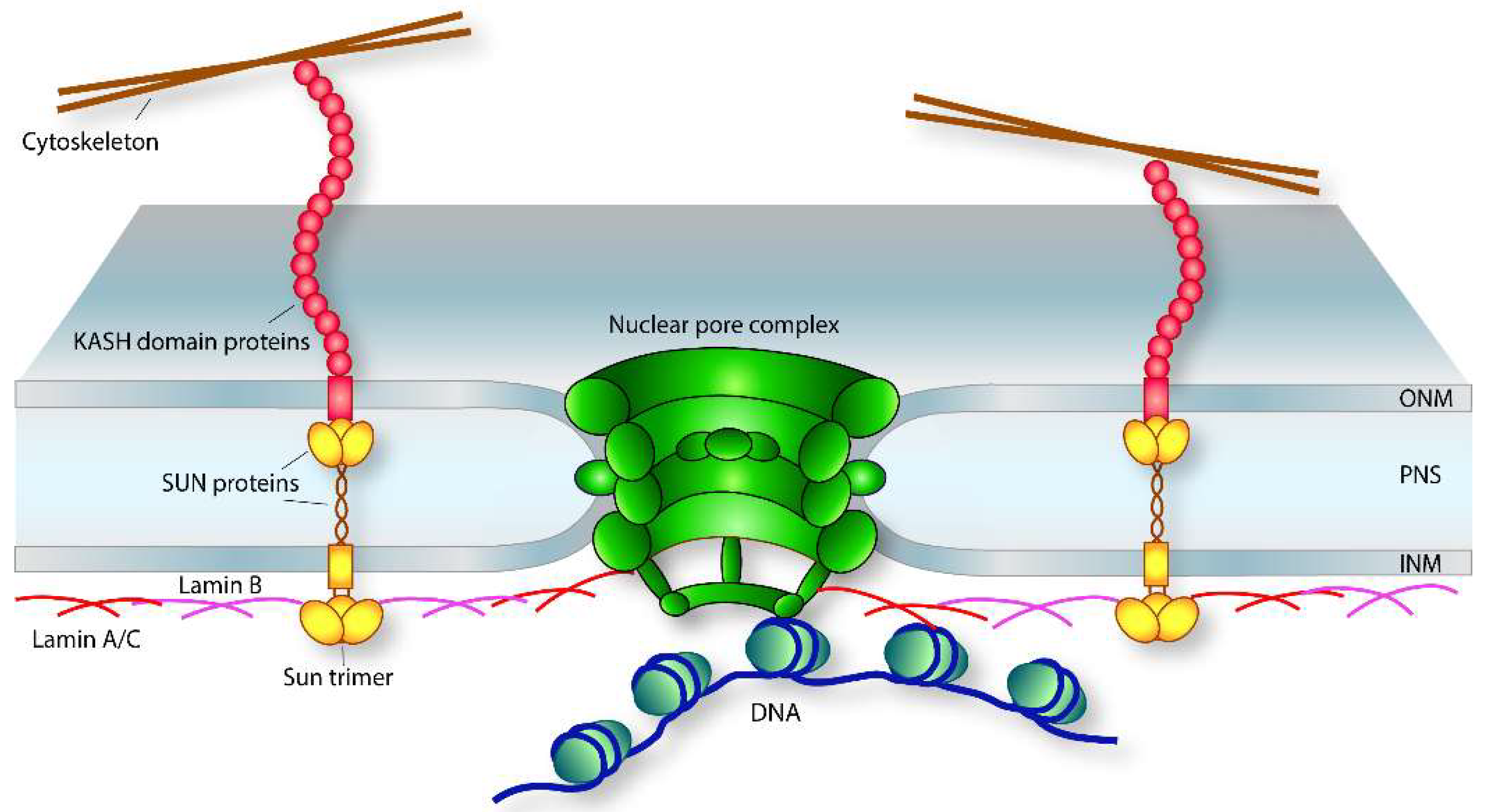
As the barrier that physically and functionally sequesters chromatin from the cytoplasm, the nuclear envelope is the first regulatory entity that coordinates extranuclear stimulus with the nuclear response. It is a complex structure composed of two lipid bilayer systems, the inner nuclear membrane (INM) that immediately surrounds the chromatin, and the outer nuclear membrane (ONM) contiguous with the endoplasmic reticulum network (Figure 1). Interposed between the INM and ONM is the perinuclear space (PNS), which is functionally and topologically connected to the endoplasmic reticulum lumen [53].
3. The Nuclear Envelope
3.1. LINC Complexes
The linker of nucleoskeleton to cytoskeleton (LINC) complex mediates cytoskeletal communication with the nuclear interior [54]. The core of the LINC complex structure is the interaction between SUN (Sad1, UNC-84) and KASH (Klarsicht, ANC-1, Syne homology) domain proteins localized to the inner (INM) and outer (ONM) nuclear membranes of the nuclear envelope, respectively (Figure 1) [55]. The C-termini of both are located in the periplasmic space where they interact to transmit biomechanical stimuli from the cytoplasmic milieu into the nuclear interior to adjust biophysical properties of the nucleus. This interaction is critical to regulate nuclear positioning and deformability required for cellular migration [54]. In addition, the interactions between the C-termini of the SUN and KASH domain proteins maintain uniformity of the periplasmic space [56]. As a gene expression regulator, SUN domain transmembrane proteins in the INM interact with the nuclear lamina via their N-terminuses [57], which in turn interact with nuclear pore complexes and chromatin [58,59].
The KASH domain proteins consist primarily of nesprins that are found in the ONM, whereas SUN domain proteins are transported to the INM by diffusing from the ONM through the nuclear pore complex (NPC) to finally reside in the INM [60]. Upon proper localization, the interactions between the KASH and SUN domain proteins couple cytosolic microtubule, actin, and plectin networks to the nuclear interior [61,62,63], and are determinants of nuclear envelope thickness [64]. This distance ranges from 30–50 nm and is possible because of the varying C-terminal lengths of the different SUN protein isoforms [64]. Binding of SUN and KASH domain proteins can occur in varying ratios, with as many as three KASH proteins able to simultaneously bind one SUN trimer [65].
Recent work suggests the intriguing possibility that biomechanical forces transduced by the LINC complex are capable of directly regulating the nuclear transport function of the nuclear pore complex [66,67,68]. Interactions of SUN1 and SUN2 with nuclear export factor 1 (NXF1) as well as with components of the pre-export messenger ribonucleoprotein particle (mRNP) were disrupted following the SUN domain protein knockdown [68]. This caused mRNP accumulation within the nucleus, confirming the functional role of SUN domain proteins on nuclear export [68]. This wasfurther supported by evidence of SUN1 interactions with the nuclear basket nucleoporin NUP153, a critical component of the nuclear transport pathway [68].
With respect to oncogenesis, it remains to be seen if the biophysical functions of the LINC complex contribute to oncogenic gene expression dysregulation per se, though reports suggest that cancer cell migration requires intact LINC complex structure and function [69,70,71]. Recent work by Infante et al. demonstrated that interactions between nesprin-2 and the dynein adaptor Lis1 coordinate with an extracellular matrix “digest-on-demand” mechanism to regulate cancer cell invasion. In this manner, while not a mechanism for initiating genomic catastrophe that precedes oncogenic transformation, LINC complex biology contributes to metastasis and persistence of malignancy.
3.2. Nuclear Lamina
The nuclear lamina is located adjacent to the inner nuclear membrane of the nuclear envelope (Figure 1) and regulates cell signaling by coordinating the assembly of multiple protein complexes [72,73,74]. It participates in chromatin organization, repressive transcriptional control, and DNA replication [75,76,77]. In addition, the nuclear lamina provides structural support to the nucleus [78]. The meshwork pattern observed for the lamina is composed of either A/C or B-type lamins that impart different physical properties to the lamin network [79]. Several integral INM proteins are associated with the nuclear lamina. In addition to SUN-domain proteins described above, LEM domain proteins (LAP2B, emerin, and MAN1), and lamin-b receptor (LBR) are associated with the nuclear lamina and contribute to gene expression regulation and nuclear form and function [80].
Nuclear deformability reflects lamin composition, with lamin A/C as the main lamin isoform that provides biomechanical stiffness to the nucleus [81,82]. In studies using cells lacking the A/C isoform, nuclei were fragile, less resistant to strain, and were frequently misshapen. In contrast, lamin B1 deficiency exhibited normal nuclear mechanics and biophysical properties despite increased nuclear blebbing. This led to the conclusion that lamin B1 was critical for nuclear integrity, but not stiffness. In support of this, micromanipulation of isolated nuclei to simulate physiological distension revealed that lamin A/C behaves as a polymeric shell with strain-stiffening properties that provide resistance to deformation [83]. A tunable auxetic property appears to be a dynamic function of the nuclear lamina [84,85]. At the extreme opposite end of this dynamism, cells can assemble a transient perinuclear actin network to mechanically rupture the lamina without damaging the nuclear envelope [86]. This facilitates cell deformability without cell death, making cells resilient as well as competent for migration.
In oncogenic models, a dysfunctional lamina is associated with pathological disruption of the nuclear envelope, which can lead to micronuclei formation that precedes genomic instability and aneuploidy [87,88,89]. Micronuclei possess transcriptional competency and may initiate chromothripsis, potentiating the development of cancer [90,91]. Indeed, morphological changes to the nuclear envelope, as well as micronuclei formation, have been used as criteria to diagnose or predict cancer [92]. For example, micronuclei detection in peripheral blood lymphocytes have been proposed to serves as predictive biomarkers for pancreatic cancer [93].
Nuclear scaling and size regulation is also impacted by lamins [94]. In general, an inverse relationship exists between lamin expression and nuclear size [95]. For example, depletion of lamin B1 increases the size of the lamina mesh and overall nuclear size in HeLa cells [96]. Abnormal lamin expression and/or localization that leads to enlarged nuclei is used as a cytopathological marker of cancer development and progression [52]. Thus, the nuclear lamina demonstrates roles in diagnosis and functional impacts on malignant growth, a characteristic exhibited by other NE components.
3.3. Nuclear Pore Complex
The NPC is a large multiproteinaceous structure that spans both membranes of the nuclear envelope (Figure 1) and regulates nucleocytoplasmic trafficking of molecules between nuclear and cytoplasmic compartments. The NPC is separated into three domains referred to as the cytoplasmic, central channel, and nucleoplasmic regions [97], and is composed of repetitive copies of ≈30 nuclear pore proteins, collectively termed nucleoporins. These are arranged in specific subcomplexes to form distinctive pore domains within each of the three regions [98,99]. These domains assemble to form a channel with a semipermeable hydrogel composed primarily of phenylalanine-glycine (FG) repeats [100,101]. A RanGTP/GDP cycle powers transport and acts in concert with soluble karyopherins to regulate molecular cargo transit [102].
NPC-mediated nuclear transport can drive cancer via mislocalization of cancer-related proteins [103] or through dysfunctional transport pathways. In general, a variety of import and export karyopherins, respectively termed importins and exportins, are overexpressed in a multitude of carcinomas [104]. This is advantageous as it offers candidates for therapeutic targeting [105,106,107], though rigorous characterization and testing are imperative to identify molecules with low to no adverse effects. Leptomycin B and its analog, selinexor, are inhibitors of exportin-1 (XPO1) that have been tested for their efficacy in treating pancreatic cancer [108,109]. Treatment of pancreatic cancer cell lines with these pharmacological inhibitors demonstrated effectiveness by deactivating NF-κB signaling and restoring tumor suppressive miR-145 [109,110]. In combination with gemcitabine, selinexor exhibited enhanced antitumor activity [111]. Recent identification of a novel karyopherin family member, KPNA7, is another potential target with pancreatic specificity. Initial reports identified overexpressed KPNA7 in multiple pancreatic cancer cell lines that implicated overactive transport of KPNA7 cargoes [112,113]. In an elegant series of experiments, Laurila et al. discovered that KPNA7 controlled pancreatic cancer cell proliferation by regulating p21 induction. This was supported by later work in which Vuorinen et al. demonstrated increased mitotic defects and nuclear deformation due to remodeling of the nuclear lamina and a shift in favor of lamin B1 [114].
Beyond anticipated roles in dysfunctional nuclear trafficking, a role for individual nucleoporins in cancer was discovered by the identification of nucleoporin fusion proteins in hematologic malignancies [115,116]. This early work identified fusions of NUP98 and NUP214 that led to a variety of de novo hematologic malignancies [117]. Further work identified other nups that promoted, as well as served as diagnostic markers, for a variety of other cancers [118,119,120]. To date, the individual nups associated with malignancy include NUP37, NUP88, NUP98, NUP160, NUP214, NUP358, and TPR [121,122,123].
NUP37 overexpression in a model of hepatocellular carcinoma was associated with enhanced metastasis and invasion that decreased upon NUP37 knockdown [123]. NUP88 was first identified as a marker for a variety of cancers, where its overexpression was diagnostic for tumorigenesis [119,120,124]. Recent identification of a phosphoregulatory function for NUP88 has been reported, where excess NUP88 inhibited dephosphorylation of vimentin in HeLa cells [118]. Dysregulation of the vimentin filament network can have consequences on pancreatic chemosensitivity, as vimentin is an active factor in the epithelial-to-mesenchymal transition that grants pancreatic tumors high chemoresistance [125,126]. NUP98, NUP160, and NUP214 have both been reported as fusion proteins with a variety of partners that lead to hematologic malignancies and angiosarcoma [117,122]. NUP358/RANBP2 also behaves as an oncogenic fusion protein [127], in addition to its transport associated mechanisms [128,129]. Novel functions for NUP358 were described in a recent report by Vecchione et al., where the authors discussed how NUP358 interacts with microtubules in a subset of colon cancer cells to promote their survival [130]. Similar to oncogenic mechanisms identified for the nups described above, TPR was identified in a variety of malignant fusion proteins that possessed constitutive tyrosine receptor kinase activity [131], whereas its overexpression promoted cancer cell survival through sustained mitosis. These examples demonstrate that a wide variety of mechanisms mediated by, or associated with nups can give rise to oncogenesis. Current work characterizing the expanding number and functional repertoire of nups that contribute to development may thus uncover novel mechanisms underlying general as well as tumor biology.
3.4. Potential Regulation of Epigenomic Dynamics by the Nuclear Envelope in the Setting of Pancreatic Cancer
An extant diversity of underlying epigenetic and epigenomic mechanisms drive pancreatic oncogenesis [132]. Among these, differential histone modifications can give rise to chromatin states that lead to the development of different subtypes of PDAC [21]. Methylation and acetylation states of various lysine residues on histone H3, i.e., H3K4, H3K9, and H3K27, as well as their localization within the nuclei, functionally and physically anchor discrete gene networks that establish pancreatic tumor aggression, metastasis, and resistance to therapy. The intimate relationship of histones, nuclear pore complexes, and nuclear lamina suggests a role for the nuclear envelope in regulating histone modification. This occurs in normal developmental processes mediated by lamina associated domains (LADs). Earlier work confirmed constitutive wild type LADs linked to transcriptional repression and histone H3 methylation [133,134]. This mechanism may have implications in chromatin remodeling that occurs in PDAC, as previous work identified lamin B1 overexpression in human pancreatic cancer associated with poor prognosis [135]. In pancreatic cancer cell models, lamin B1 knockdown significantly diminished proliferative aggressiveness and invasiveness [135]. Later independent work demonstrated that H3K27Me3 dispersed in a dramatic fashion from the nuclear periphery to the nuclear interior upon lamin B1 silencing [136], supporting the notion of the nuclear lamina playing a regulatory role in histone dynamics underlying oncogenesis.
Furthermore, the identification of enzymes such as histone deacetylases associated with individual nucleoporins supports the proposed regulatory roles of the nuclear envelope in histone modification and chromatin accessibility [137]. As an additional modality, NUP fusions to histone readers, as in the case of NUP98-PHD finger protein fusions, result in the “reading” of H3K4Me3 that locks associated genomic loci in an active state [138]. This type of mechanism is critical for hematologic transformation and establishes a rationale for its potential role in other cancer types. Indeed, the functional precedence for this system of H3K4Me3 detection and regulation exists for wild type NUP98 [139], and is significant when considering that H3K4Me3 is critical for anti-apoptotic gene activation in PDAC [140].
Epigenetic/epigenomic modifications and their association with the nuclear envelope in the setting of pancreatic oncogenesis is underscored in the “Triple Code Hypothesis,” where crosstalk among genetics, epigenetics, and nuclear structure plays an important role in the evolution of PDAC [141]. In support of this, the nuclear envelope’s regulatory contributions are mediated by the nuclear lamina, nucleoporins of the nuclear pore complex, and by the LINC complex. Evidence for the latter suggests that the LINC complexplays a larger role in metastatic dissemination and cancer metabolism over direct epigenomic regulation [70,142,143]. However, these biophysical and biochemical functions downstream of epigenomic remodeling furthermore do not preclude its potential primary contribution to oncogenic chromatin dynamics. This was recently demonstrated in work by Aymard et al., where the authors identify an active role for the LINC complex in clustering double-stranded DNA breaks induced in active genes [144]. This is particularly relevant as these gene lesions are critical for initiating and promoting tumorigenesis, and account for a major subtype of PDAC [145]. In addition, independent work showed that the expression of spectrin repeat containing nuclear envelope protein 2 (SYNE2/Nesprin-2), a component of the LINC complex, is dysregulated in PDAC by small nucleolar noncoding RNA SNORA23, and is associated with increased tumor invasion and metastasis in mice [146]. Given these initial findings, future studies are critical to define the role of the LINC complex in the context of tumorigenesis and PDAC.
4. Summary and Future Directions
While individual components of the nuclear envelope described above possess the capacity to regulate nuclear function in discrete ways, functional crosstalk among these components is critical to maintain a normal phenotype. The nuclear envelope thus emerges as an intricate regulatory body that integrates an array of biomechanical and biochemical signaling pathways to modulate nuclear structure and function. For example, developmental disruptions in any of the LINC complex, nuclear lamina, or NPC proteins are related to a class of diseases termed laminopathies that can affect multiple physiological systems [147]. This observation, in line with the enriched functional bias of known pancreatic cancer genes towards nucleoplasmic function and DNA regulation described earlier, supports the notion that the nucleus may have more roles in pancreatic cancer biology that are unexplored.
The high mortality rate associated with pancreatic cancer is a conflation of strong chemoresistance and high metastasis [148], and this underscores the priority to identify and develop specific and robust treatments with improved therapeutic potential. Exploiting the unique regulatory biology of the nuclear envelope may offer unique strategies for the design of novel therapeutic approaches to cancer. When considered in combination with extant treatment paradigms, such as cell cycle checkpoint inhibitors, oncolytic viruses, and pharmacological MEK antagonists [149], nuclear envelope interventions may contribute synergistic advantages to personalized cancer treatment regimens. Currently, immunotherapeutic approaches to pancreatic cancer are the most advanced paradigm being developed to address pancreatic malignancy [150] and involve the recruitment of activated T-cells. Adoptive T-cell therapy, or CAR-T cell therapy, has been promising in addressing hematologic malignancies [151,152] and has promise for treating pancreatic cancer. In this approach, chimeric antigen receptor (CAR) T-cells are autologously generated from extracted patient cells, then infused back into original donors. Despite the demonstrated success in a liquid tumor, challenges remain for its application to solid tumors. Progress in this field is ongoing, and it is anticipated CAR-T cell therapy has the potential to efficiently overcome the low immunogenicity of the pancreatic tumor microenvironment and facilitate its clearance and regression [149,153].
The functional priority of the nucleus associated with somatic mutations driving pancreatic malignancy provides the rationale to focus on nuclear elements that contribute to the pathology of pancreatic cancer. This is critical to developing novel strategies that synergize with current approaches to treat pancreatic malignancy. Thus, characterization of the nuclear envelope and its role with respect to oncogenesis will supplement the growing therapeutic armamentarium to address carcinomas and offers a body of knowledge that may be leveraged to overcome the innate resistance of pancreatic tumors.
Funding
This study was supported by Sanford Research, and an NIH CoBRE grant (P20GM103620).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- McGuire, S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv. Nutr. 2016, 7, 418–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronin, K.A.; Lake, A.J.; Scott, S.; Sherman, R.L.; Noone, A.M.; Howlader, N.; Henley, S.J.; Anderson, R.N.; Firth, A.U.; Ma, J.; et al. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer 2018, 124, 2785–2800. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Sharma, P.; Zakalik, D. Comparison of Demographics, Tumor Characteristics, and Survival Between Pancreatic Adenocarcinomas and Pancreatic Neuroendocrine Tumors: A Population-based Study. Am. J. Clin. Oncol. 2018, 41, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Klimstra, D.S. Adenocarcinoma of the pancreas. Semin. Diagn. Pathol. 2014, 31, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, A.; Khazaei, M.; Bagherieh, F.; Ghayour-Mobarhan, M.; Maftouh, M.; Hassanian, S.M.; Avan, A. Targeting stroma in pancreatic cancer: Promises and failures of targeted therapies. J. Cell. Physiol. 2017, 232, 2931–2937. [Google Scholar] [CrossRef] [PubMed]
- Bakkevold, K.E.; Arnesjo, B.; Kambestad, B. Carcinoma of the pancreas and papilla of Vater: Presenting symptoms, signs, and diagnosis related to stage and tumour site. A prospective multicentre trial in 472 patients. Norwegian Pancreatic Cancer Trial. Scand. J. Gastroenterol. 1992, 27, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Porta, M.; Fabregat, X.; Malats, N.; Guarner, L.; Carrato, A.; de Miguel, A.; Ruiz, L.; Jariod, M.; Costafreda, S.; Coll, S.; et al. Exocrine pancreatic cancer: Symptoms at presentation and their relation to tumour site and stage. Clin. Transl. Oncol. 2005, 7, 189–197. [Google Scholar] [CrossRef] [PubMed]
- El Guesmi, S.; Ben Nasr, S.; Afrit, M.; Labidi, S.; Boussen, H. Exocrine Pancreatic carcinoma in Tunisia: A retrospective study about 158 cases. Tunis. Med. 2015, 93, 73–75. [Google Scholar] [PubMed]
- Lowenfels, A.B.; Maisonneuve, P. Epidemiology and risk factors for pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 2006, 20, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.P.; Wolpin, B.M.; Risch, H.A.; Stolzenberg-Solomon, R.Z.; Mocci, E.; Zhang, M.; Canzian, F.; Childs, E.J.; Hoskins, J.W.; Jermusyk, A.; et al. Genome-wide meta-analysis identifies five new susceptibility loci for pancreatic cancer. Nat. Commun. 2018, 9, 556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, L.G.; Chan, T.A. Therapeutic targeting of tumor suppressor genes. Cancer 2015, 121, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Muller, W.J. Oncogenes and tumor suppressor genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a003236. [Google Scholar] [CrossRef] [PubMed]
- Inokawa, H.; Katayama, N.; Nakao, M. Evaluation of multidrug cancer chronotherapy based on cell cycle model under influences of circadian clock. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2016, 2016, 1439–1442. [Google Scholar] [PubMed]
- Sotak, M.; Sumova, A.; Pacha, J. Cross-talk between the circadian clock and the cell cycle in cancer. Ann. Med. 2014, 46, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; dal Zuffo, R.; Matti, V.; d’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrimore, J.; Barry, T.M.; Barry, R.M.; York, A.C.; Friedman, B.; Cook, D.M.; Akialis, K.; Tyler, J.; Vasquez, P.; Yeh, E.; et al. Microtubule dynamics drive enhanced chromatin motion and mobilize telomeres in response to DNA damage. Mol. Biol. Cell 2017, 28, 1701–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amendola, M.; van Steensel, B. Mechanisms and dynamics of nuclear lamina-genome interactions. Curr. Opin. Cell Biol. 2014, 28, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, I.A.; Dundr, M. Chromatin loops and causality loops: The influence of RNA upon spatial nuclear architecture. Chromosoma 2017, 126, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, I.A.; Dundr, M. Nuclear bodies: Built to boost. J. Cell Biol. 2016, 213, 509–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serizay, J.; Ahringer, J. Genome organization at different scales: Nature, formation and function. Curr. Opin. Cell Biol. 2018, 52, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Grosveld, F.G.; Papantonis, A. Forces driving the three-dimensional folding of eukaryotic genomes. Mol. Syst. Biol. 2018, 14, e8214. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.D.; Tanizawa, H.; Iwasaki, O.; Noma, K. Transcription factors mediate condensin recruitment and global chromosomal organization in fission yeast. Nat. Genet. 2016, 48, 1242–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, O.; Corcoran, C.J.; Noma, K. Involvement of condensin-directed gene associations in the organization and regulation of chromosome territories during the cell cycle. Nucleic Acids Res. 2016, 44, 3618–3628. [Google Scholar] [CrossRef] [PubMed]
- Miloshev, G.; Staneva, D.; Uzunova, K.; Vasileva, B.; Draganova-Filipova, M.; Zagorchev, P.; Georgieva, M. Linker histones and chromatin remodelling complexes maintain genome stability and control cellular ageing. Mech. Ageing Dev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fatakia, S.N.; Kulashreshtha, M.; Mehta, I.S.; Rao, B.J. Chromosome territory relocation paradigm during DNA damage response: Some insights from molecular biology to physics. Nucleus 2017, 8, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, I.S.; Kulashreshtha, M.; Chakraborty, S.; Kolthur-Seetharam, U.; Rao, B.J. Chromosome territories reposition during DNA damage-repair response. Genome Biol. 2013, 14, R135. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.Q.; Bosco, G. Gene Positioning Effects on Expression in Eukaryotes. Annu. Rev. Genet. 2015, 49, 627–646. [Google Scholar] [CrossRef] [PubMed]
- Kinney, N.A.; Sharakhov, I.V.; Onufriev, A.V. Chromosome-nuclear envelope attachments affect interphase chromosome territories and entanglement. Epigenet. Chromatin 2018, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, M.; Cremer, C. The 4D Nucleome: Genome Compartmentalization in an Evolutionary Context. Biochemistry 2018, 83, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, M.; Hubner, B.; Strickfaden, H.; Smeets, D.; Popken, J.; Sterr, M.; Markaki, Y.; Rippe, K.; Cremer, C. The 4D nucleome: Evidence for a dynamic nuclear landscape based on co-aligned active and inactive nuclear compartments. FEBS Lett. 2015, 589, 2931–2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pederson, T. Dynamics and genome-centricity of interchromatin domains in the nucleus. Nat. Cell Biol. 2002, 4, E287–E291. [Google Scholar] [CrossRef] [PubMed]
- Pascarella, A.; Ferrandino, G.; Credendino, S.C.; Moccia, C.; D’Angelo, F.; Miranda, B.; D’Ambrosio, C.; Bielli, P.; Spadaro, O.; Ceccarelli, M.; et al. DNAJC17 is localized in nuclear speckles and interacts with splicing machinery components. Sci. Rep. 2018, 8, 7794. [Google Scholar] [CrossRef] [PubMed]
- Rieder, D.; Ploner, C.; Krogsdam, A.M.; Stocker, G.; Fischer, M.; Scheideler, M.; Dani, C.; Amri, E.Z.; Muller, W.G.; McNally, J.G.; et al. Co-expressed genes prepositioned in spatial neighborhoods stochastically associate with SC35 speckles and RNA polymerase II factories. Cell. Mol. Life Sci. 2014, 71, 1741–1759. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Chang, P.Y.; Chao, C.C. CITED2 silencing sensitizes cancer cells to cisplatin by inhibiting p53 trans-activation and chromatin relaxation on the ERCC1 DNA repair gene. Nucleic Acids Res. 2015, 43, 10760–10781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, K.S.; Casas-Delucchi, C.S.; Torok, T.; Cmarko, D.; Rapp, A.; Raska, I.; Cardoso, M.C. Peripheral re-localization of constitutive heterochromatin advances its replication timing and impairs maintenance of silencing marks. Nucleic Acids Res. 2018, 46, 6112–6128. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, P.; Castronovo, V.; Matheus, N.; Polese, C.; Peulen, O.; Gonzalez, A.; Boxus, M.; Verdin, E.; Thiry, M.; Dequiedt, F.; et al. HDAC5 is required for maintenance of pericentric heterochromatin, and controls cell-cycle progression and survival of human cancer cells. Cell Death Differ. 2012, 19, 1239–1252. [Google Scholar] [CrossRef] [PubMed]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Young, R.A. New Insights into Genome Structure: Genes of a Feather Stick Together. Mol. Cell 2017, 67, 730–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, X.; Dadon, D.B.; Powell, B.E.; Fan, Z.P.; Borges-Rivera, D.; Shachar, S.; Weintraub, A.S.; Hnisz, D.; Pegoraro, G.; Lee, T.I.; et al. 3D Chromosome Regulatory Landscape of Human Pluripotent Cells. Cell Stem Cell 2016, 18, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 2018, 33, 512–526.e8. [Google Scholar] [CrossRef] [PubMed]
- Hodges, H.C.; Stanton, B.Z.; Cermakova, K.; Chang, C.Y.; Miller, E.L.; Kirkland, J.G.; Ku, W.L.; Veverka, V.; Zhao, K.; Crabtree, G.R. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat. Struct. Mol. Biol. 2018, 25, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Sharakhov, I.V.; Bondarenko, S.M.; Artemov, G.N.; Onufriev, A.V. The Role of Chromosome-Nuclear Envelope Attachments in 3D Genome Organization. Biochemistry 2018, 83, 350–358. [Google Scholar] [CrossRef] [PubMed]
- De Magistris, P.; Antonin, W. The Dynamic Nature of the Nuclear Envelope. Curr. Biol. 2018, 28, R487–R497. [Google Scholar] [CrossRef] [PubMed]
- de Las Heras, J.I.; Schirmer, E.C. The nuclear envelope and cancer: A diagnostic perspective and historical overview. Adv. Exp. Med. Biol. 2014, 773, 5–26. [Google Scholar] [PubMed]
- Chow, K.H.; Factor, R.E.; Ullman, K.S. The nuclear envelope environment and its cancer connections. Nat. Rev. Cancer 2012, 12, 196–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- English, A.R.; Voeltz, G.K. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb. Perspect. Biol. 2013, 5, a013227. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.L.; Burke, B. LINC complexes and nuclear positioning. Semin. Cell Dev. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Meinke, P.; Schirmer, E.C. LINC’ing form and function at the nuclear envelope. FEBS Lett. 2015, 589, 2514–2521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bethmann, C.; Worth, N.F.; Davies, J.D.; Wasner, C.; Feuer, A.; Ragnauth, C.D.; Yi, Q.; Mellad, J.A.; Warren, D.T.; et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum. Mol. Genet. 2007, 16, 2816–2833. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.T.; Bottrill, A.; Prosser, S.L.; Jayaraman, S.; Straatman, K.; Fry, A.M.; Shackleton, S. Mitotic phosphorylation of SUN1 loosens its connection with the nuclear lamina while the LINC complex remains intact. Nucleus 2014, 5, 462–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camozzi, D.; Capanni, C.; Cenni, V.; Mattioli, E.; Columbaro, M.; Squarzoni, S.; Lattanzi, G. Diverse lamin-dependent mechanisms interact to control chromatin dynamics. Focus on laminopathies. Nucleus 2014, 5, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Athirasala, A.; Hirsch, N.; Buxboim, A. Nuclear mechanotransduction: Sensing the force from within. Curr. Opin. Cell Biol. 2017, 46, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Jahed, Z.; Soheilypour, M.; Peyro, M.; Mofrad, M.R. The LINC and NPC relationship—It’s complicated! J. Cell Sci. 2016, 129, 3219–3229. [Google Scholar] [CrossRef] [PubMed]
- Nikolova-Krstevski, V.; Leimena, C.; Xiao, X.H.; Kesteven, S.; Tan, J.C.; Yeo, L.S.; Yu, Z.Y.; Zhang, Q.; Carlton, A.; Head, S.; et al. Nesprin-1 and actin contribute to nuclear and cytoskeletal defects in lamin A/C-deficient cardiomyopathy. J. Mol. Cell. Cardiol. 2011, 50, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Gimpel, P.; Lee, Y.L.; Sobota, R.M.; Calvi, A.; Koullourou, V.; Patel, R.; Mamchaoui, K.; Nedelec, F.; Shackleton, S.; Schmoranzer, J.; et al. Nesprin-1alpha-Dependent Microtubule Nucleation from the Nuclear Envelope via Akap450 Is Necessary for Nuclear Positioning in Muscle Cells. Curr. Biol. 2017, 27, 2999–3009.e9. [Google Scholar] [CrossRef] [PubMed]
- Ketema, M.; Wilhelmsen, K.; Kuikman, I.; Janssen, H.; Hodzic, D.; Sonnenberg, A. Requirements for the localization of nesprin-3 at the nuclear envelope and its interaction with plectin. J. Cell Sci. 2007, 120, 3384–3394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Schneider, M.; Neumann, S.; Jaeger, V.M.; Taranum, S.; Munck, M.; Cartwright, S.; Richardson, C.; Carthew, J.; Noh, K.; et al. Nesprin interchain associations control nuclear size. Cell. Mol. Life Sci. 2012, 69, 3493–3509. [Google Scholar] [CrossRef] [PubMed]
- Jahed, Z.; Vu, U.T.; Fadavi, D.; Ke, H.; Rathish, A.; Kim, S.C.J.; Feng, W.; Mofrad, M.R.K. A Molecular Model for LINC complex regulation: Activation of SUN2 for KASH binding. Mol. Biol. Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Stumpf, M.; Muller, R.; Eichinger, L.; Glockner, G.; Noegel, A.A. The function of the inner nuclear envelope protein SUN1 in mRNA export is regulated by phosphorylation. Sci. Rep. 2017, 7, 9157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelkar, P.; Walter, A.; Papadopoulos, S.; Mross, C.; Munck, M.; Peche, V.S.; Noegel, A.A. Nesprin-2 mediated nuclear trafficking and its clinical implications. Nucleus 2015, 6, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Noegel, A.A. Inner nuclear envelope protein SUN1 plays a prominent role in mammalian mRNA export. Nucleic Acids Res. 2015, 43, 9874–9888. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, H.; Sato, K.; Nishihara, A.; Minami, K.; Koizumi, M.; Matsuura, N.; Hieda, M. X-ray-enhanced cancer cell migration requires the linker of nucleoskeleton and cytoskeleton complex. Cancer Sci 2018, 109, 1158–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, E.; Castagnino, A.; Ferrari, R.; Monteiro, P.; Aguera-Gonzalez, S.; Paul-Gilloteaux, P.; Domingues, M.J.; Maiuri, P.; Raab, M.; Shanahan, C.M.; et al. LINC complex-Lis1 interplay controls MT1-MMP matrix digest-on-demand response for confined tumor cell migration. Nat. Commun. 2018, 9, 2443. [Google Scholar] [CrossRef] [PubMed]
- Isermann, P.; Lammerding, J. Nuclear mechanics and mechanotransduction in health and disease. Curr. Biol. 2013, 23, R1113–R1121. [Google Scholar] [CrossRef] [PubMed]
- Clever, M.; Mimura, Y.; Funakoshi, T.; Imamoto, N. Regulation and coordination of nuclear envelope and nuclear pore complex assembly. Nucleus 2013, 4, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Kim, Y.; Shimi, T.; Goldman, R.D.; Zheng, Y. Concentration-dependent lamin assembly and its roles in the localization of other nuclear proteins. Mol. Biol. Cell 2014, 25, 1287–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osmanagic-Myers, S.; Dechat, T.; Foisner, R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015, 29, 225–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harr, J.C.; Luperchio, T.R.; Wong, X.; Cohen, E.; Wheelan, S.J.; Reddy, K.L. Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A-type lamins. J. Cell Biol. 2015, 208, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Zullo, J.M.; Demarco, I.A.; Pique-Regi, R.; Gaffney, D.J.; Epstein, C.B.; Spooner, C.J.; Luperchio, T.R.; Bernstein, B.E.; Pritchard, J.K.; Reddy, K.L.; et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell 2012, 149, 1474–1487. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turgay, Y.; Eibauer, M.; Goldman, A.E.; Shimi, T.; Khayat, M.; Ben-Harush, K.; Dubrovsky-Gaupp, A.; Sapra, K.T.; Goldman, R.D.; Medalia, O. The molecular architecture of lamins in somatic cells. Nature 2017, 543, 261–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.E.; Adam, S.A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell 2015, 26, 4075–4086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungricht, R.; Kutay, U. Establishment of NE asymmetry-targeting of membrane proteins to the inner nuclear membrane. Curr. Opin. Cell Biol. 2015, 34, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins A and C but not lamin B1 regulate nuclear mechanics. J. Biol. Chem. 2006, 281, 25768–25780. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Swift, J.; Irianto, J.; Shin, J.W.; Spinler, K.R.; Athirasala, A.; Diegmiller, R.; Dingal, P.C.; Ivanovska, I.L.; Discher, D.E. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 2014, 204, 669–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, A.D.; Banigan, E.J.; Adam, S.A.; Goldman, R.D.; Marko, J.F. Chromatin and lamin A determine two different mechanical response regimes of the cell nucleus. Mol. Biol. Cell 2017, 28, 1984–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilluy, C.; Osborne, L.D.; Van Landeghem, L.; Sharek, L.; Superfine, R.; Garcia-Mata, R.; Burridge, K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 2014, 16, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliara, S.; Franze, K.; McClain, C.R.; Wylde, G.; Fisher, C.L.; Franklin, R.J.M.; Kabla, A.J.; Keyser, U.F.; Chalut, K.J. Auxetic nuclei in embryonic stem cells exiting pluripotency. Nat. Mater. 2014, 13, 638–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiam, H.R.; Vargas, P.; Carpi, N.; Crespo, C.L.; Raab, M.; Terriac, E.; King, M.C.; Jacobelli, J.; Alberts, A.S.; Stradal, T.; et al. Perinuclear Arp2/3-driven actin polymerization enables nuclear deformation to facilitate cell migration through complex environments. Nat. Commun. 2016, 7, 10997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, P.; Wolf, K.; Lammerding, J. Bursting the Bubble—Nuclear Envelope Rupture as a Path to Genomic Instability? Trends Cell Biol. 2017, 27, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Capo-Chichi, C.D.; Yeasky, T.M.; Smith, E.R.; Xu, X.X. Nuclear envelope structural defect underlies the main cause of aneuploidy in ovarian carcinogenesis. BMC Cell Biol. 2016, 17, 37. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Cleveland, D.W. Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol. 2017, 27, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Utani, K.; Kawamoto, J.K.; Shimizu, N. Micronuclei bearing acentric extrachromosomal chromatin are transcriptionally competent and may perturb the cancer cell phenotype. Mol. Cancer Res. 2007, 5, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.H. The diagnostic pathology of the nuclear envelope in human cancers. Adv. Exp. Med. Biol. 2014, 773, 49–75. [Google Scholar] [PubMed]
- Chang, P.; Li, Y.; Li, D. Micronuclei levels in peripheral blood lymphocytes as a potential biomarker for pancreatic cancer risk. Carcinogenesis 2011, 32, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Edens, L.J.; White, K.H.; Jevtic, P.; Li, X.; Levy, D.L. Nuclear size regulation: From single cells to development and disease. Trends Cell Biol. 2013, 23, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Jevtic, P.; Edens, L.J.; Li, X.; Nguyen, T.; Chen, P.; Levy, D.L. Concentration-dependent Effects of Nuclear Lamins on Nuclear Size in Xenopus and Mammalian Cells. J. Biol. Chem. 2015, 290, 27557–27571. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.N.; Chen, P.; Levy, D.L. Recent advances in understanding nuclear size and shape. Nucleus 2016, 7, 167–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preston, C.C.; Wyles, S.P.; Reyes, S.; Storm, E.C.; Eckloff, B.W.; Faustino, R.S. NUP155 insufficiency recalibrates a pluripotent transcriptome with network remodeling of a cardiogenic signaling module. BMC Syst. Biol. 2018, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- von Appen, A.; Beck, M. Structure Determination of the Nuclear Pore Complex with Three-Dimensional Cryo electron Microscopy. J. Mol. Biol. 2016, 428, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- von Appen, A.; Kosinski, J.; Sparks, L.; Ori, A.; DiGuilio, A.L.; Vollmer, B.; Mackmull, M.T.; Banterle, N.; Parca, L.; Kastritis, P.; et al. In situ structural analysis of the human nuclear pore complex. Nature 2015, 526, 140–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, A.K.; Baker, L.A. Synthetic hydrogel mimics of the nuclear pore complex display selectivity dependent on FG-repeat concentration and electrostatics. Soft Matter 2016, 12, 9477–9484. [Google Scholar] [CrossRef] [PubMed]
- Frey, S.; Richter, R.P.; Gorlich, D. FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science 2006, 314, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, Y.; Hieda, M.; Nagoshi, E.; Miyamoto, Y. Nucleocytoplasmic protein transport and recycling of Ran. Cell Struct. Funct. 1999, 24, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A. Interplay between nuclear transport and ubiquitin/SUMO modifications in the regulation of cancer-related proteins. Semin. Cancer Biol. 2014, 27, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Mahipal, A.; Malafa, M. Importins and exportins as therapeutic targets in cancer. Pharmacol. Ther. 2016, 164, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Faustino, R.S.; Nelson, T.J.; Terzic, A.; Perez-Terzic, C. Nuclear transport: Target for therapy. Clin. Pharmacol. Ther. 2007, 81, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.Q.; Du, J.; Jiang, H.; Hou, J. Effect of nuclear receptor inhibitor importazole on the proliferation and apoptosis of multiple myeloma cells. Zhonghua Xue Ye Xue Za Zhi 2013, 34, 323–326. [Google Scholar] [PubMed]
- Soderholm, J.F.; Bird, S.L.; Kalab, P.; Sampathkumar, Y.; Hasegawa, K.; Uehara-Bingen, M.; Weis, K.; Heald, R. Importazole, a small molecule inhibitor of the transport receptor importin-beta. ACS Chem. Biol. 2011, 6, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Muqbil, I.; Azmi, A.S.; Mohammad, R.M. Nuclear Export Inhibition for Pancreatic Cancer Therapy. Cancers 2018, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Li, Y.; Muqbil, I.; Aboukameel, A.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Shacham, S.; Kauffman, M.G.; Philip, P.A.; et al. Exportin 1 (XPO1) inhibition leads to restoration of tumor suppressor miR-145 and consequent suppression of pancreatic cancer cell proliferation and migration. Oncotarget 2017, 8, 82144–82155. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, T.; Argueta, C.; Aboukameel, A.; Unger, T.J.; Klebanov, B.; Mohammad, R.M.; Muqbil, I.; Azmi, A.S.; Drolen, C.; Senapedis, W.; et al. Selinexor, a Selective Inhibitor of Nuclear Export (SINE) compound, acts through NF-kappaB deactivation and combines with proteasome inhibitors to synergistically induce tumor cell death. Oncotarget 2016, 7, 78883–78895. [Google Scholar] [CrossRef] [PubMed]
- Kazim, S.; Malafa, M.P.; Coppola, D.; Husain, K.; Zibadi, S.; Kashyap, T.; Crochiere, M.; Landesman, Y.; Rashal, T.; Sullivan, D.M.; et al. Selective Nuclear Export Inhibitor KPT-330 Enhances the Antitumor Activity of Gemcitabine in Human Pancreatic Cancer. Mol. Cancer Ther. 2015, 14, 1570–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurila, E.; Vuorinen, E.; Savinainen, K.; Rauhala, H.; Kallioniemi, A. KPNA7, a nuclear transport receptor, promotes malignant properties of pancreatic cancer cells in vitro. Exp. Cell Res. 2014, 322, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, E.M.; Rajala, N.K.; Rauhala, H.E.; Nurminen, A.T.; Hytonen, V.P.; Kallioniemi, A. Search for KPNA7 cargo proteins in human cells reveals MVP and ZNF414 as novel regulators of cancer cell growth. Biochim. Biophys. Acta 2017, 1863, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, E.M.; Rajala, N.K.; Ihalainen, T.O.; Kallioniemi, A. Depletion of nuclear import protein karyopherin alpha 7 (KPNA7) induces mitotic defects and deformation of nuclei in cancer cells. BMC Cancer 2018, 18, 325. [Google Scholar] [CrossRef] [PubMed]
- Fornerod, M.; Boer, J.; van Baal, S.; Jaegle, M.; von Lindern, M.; Murti, K.G.; Davis, D.; Bonten, J.; Buijs, A.; Grosveld, G. Relocation of the carboxyterminal part of CAN from the nuclear envelope to the nucleus as a result of leukemia-specific chromosome rearrangements. Oncogene 1995, 10, 1739–1748. [Google Scholar] [PubMed]
- Nakamura, T.; Largaespada, D.A.; Lee, M.P.; Johnson, L.A.; Ohyashiki, K.; Toyama, K.; Chen, S.J.; Willman, C.L.; Chen, I.M.; Feinberg, A.P.; et al. Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7;11)(p15;p15) in human myeloid leukaemia. Nat. Genet. 1996, 12, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Yaseen, N.R. Nucleoporins and nucleocytoplasmic transport in hematologic malignancies. Semin. Cancer Biol. 2014, 27, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Makise, M.; Nakamura, H.; Kuniyasu, A. The role of vimentin in the tumor marker Nup88-dependent multinucleated phenotype. BMC Cancer 2018, 18, 519. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, J.; Li, Y. Multiple biological processes may be associated with tumorigenesis under NUP88-overexpressed condition. Genes Chromosomes Cancer 2017, 56, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Naylor, R.M.; Jeganathan, K.B.; Cao, X.; van Deursen, J.M. Nuclear pore protein NUP88 activates anaphase-promoting complex to promote aneuploidy. J. Clin. Investig. 2016, 126, 543–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.N.; Rout, M.P. Cancer and the nuclear pore complex. Adv. Exp. Med. Biol. 2014, 773, 285–307. [Google Scholar] [PubMed]
- Shimozono, N.; Jinnin, M.; Masuzawa, M.; Masuzawa, M.; Wang, Z.; Hirano, A.; Tomizawa, Y.; Etoh-Kira, T.; Kajihara, I.; Harada, M.; et al. NUP160-SLC43A3 is a novel recurrent fusion oncogene in angiosarcoma. Cancer Res. 2015, 75, 4458–4465. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Liu, Y.; Feng, W.; Lei, L.; Du, Y.; Wu, J.; Wang, S. NUP37, a positive regulator of YAP/TEAD signaling, promotes the progression of hepatocellular carcinoma. Oncotarget 2017, 8, 98004–98013. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.R.; Zhang, L.J.; Wang, Y.Y.; Li, F.; Wang, M.W.; Sun, X.F. Increased serum level of Nup88 protein is associated with the development of colorectal cancer. Med. Oncol. 2012, 29, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, A.; Tanoshima, R.; Tsujimoto, S.I.; Yanagimachi, M.; Takeuchi, M.; Sasaki, K.; Ikeda, J.; Kajiwara, R.; Ito, S.; Takahashi, H. Crizotinib treatment for refractory pediatric acute myeloid leukemia with RAN-binding protein 2-anaplastic lymphoma kinase fusion gene. Blood Cancer J. 2016, 6, e456. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, Y.; Wang, B.; Lan, H.; Liu, Y.; Chen, F.; Zhang, J.; Luo, J. Sumoylation in p27kip1 via RanBP2 promotes cancer cell growth in cholangiocarcinoma cell line QBC939. BMC Mol. Biol. 2017, 18, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashikari, D.; Takayama, K.; Tanaka, T.; Suzuki, Y.; Obinata, D.; Fujimura, T.; Urano, T.; Takahashi, S.; Inoue, S. Androgen induces G3BP2 and SUMO-mediated p53 nuclear export in prostate cancer. Oncogene 2017, 36, 6272–6281. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, L.; Gambino, V.; Raaijmakers, J.; Schlicker, A.; Fumagalli, A.; Russo, M.; Villanueva, A.; Beerling, E.; Bartolini, A.; Mollevi, D.G.; et al. A Vulnerability of a Subset of Colon Cancers with Potential Clinical Utility. Cell 2016, 165, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Snow, C.J.; Paschal, B.M. Roles of the nucleoporin Tpr in cancer and aging. Adv. Exp. Med. Biol. 2014, 773, 309–322. [Google Scholar] [PubMed]
- Neureiter, D.; Jager, T.; Ocker, M.; Kiesslich, T. Epigenetics and pancreatic cancer: Pathophysiology and novel treatment aspects. World J. Gastroenterol. 2014, 20, 7830–7848. [Google Scholar] [CrossRef] [PubMed]
- Kind, J.; Pagie, L.; Ortabozkoyun, H.; Boyle, S.; de Vries, S.S.; Janssen, H.; Amendola, M.; Nolen, L.D.; Bickmore, W.A.; van Steensel, B. Single-cell dynamics of genome-nuclear lamina interactions. Cell 2013, 153, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Kind, J.; Pagie, L.; de Vries, S.S.; Nahidiazar, L.; Dey, S.S.; Bienko, M.; Zhan, Y.; Lajoie, B.; de Graaf, C.A.; Amendola, M.; et al. Genome-wide maps of nuclear lamina interactions in single human cells. Cell 2015, 163, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Du, Y.; Kong, X.; Li, Z.; Jia, Z.; Cui, J.; Gao, J.; Wang, G.; Xie, K. Lamin B1 is a novel therapeutic target of betulinic acid in pancreatic cancer. Clin. Cancer Res. 2013, 19, 4651–4661. [Google Scholar] [CrossRef] [PubMed]
- Camps, J.; Wangsa, D.; Falke, M.; Brown, M.; Case, C.M.; Erdos, M.R.; Ried, T. Loss of lamin B1 results in prolongation of S phase and decondensation of chromosome territories. FASEB J. 2014, 28, 3423–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somech, R.; Shaklai, S.; Geller, O.; Amariglio, N.; Simon, A.J.; Rechavi, G.; Gal-Yam, E.N. The nuclear-envelope protein and transcriptional repressor LAP2beta interacts with HDAC3 at the nuclear periphery, and induces histone H4 deacetylation. J. Cell Sci. 2005, 118, 4017–4025. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Song, J.; Wang, Z.; Dormann, H.L.; Casadio, F.; Li, H.; Luo, J.L.; Patel, D.J.; Allis, C.D. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 2009, 459, 847–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franks, T.M.; McCloskey, A.; Shokirev, M.N.; Benner, C.; Rathore, A.; Hetzer, M.W. Nup98 recruits the Wdr82-Set1A/COMPASS complex to promoters to regulate H3K4 trimethylation in hematopoietic progenitor cells. Genes Dev. 2017, 31, 2222–2234. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Yang, D.; Sabbatini, M.E.; Colby, A.H.; Grinstaff, M.W.; Oberlies, N.H.; Pearce, C.; Liu, K. Contrasting roles of H3K4me3 and H3K9me3 in regulation of apoptosis and gemcitabine resistance in human pancreatic cancer cells. BMC Cancer 2018, 18, 149. [Google Scholar] [CrossRef] [PubMed]
- Lomberk, G.A.; Urrutia, R. The Triple-Code Model for Pancreatic Cancer: Cross Talk Among Genetics, Epigenetics, and Nuclear Structure. Surg. Clin. N. Am. 2015, 95, 935–952. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Hieda, M.; Yokoyama, Y.; Nishioka, Y.; Yoshidome, K.; Tsujimoto, M.; Matsuura, N. Global loss of a nuclear lamina component, lamin A/C, and LINC complex components SUN1, SUN2, and nesprin-2 in breast cancer. Cancer Med. 2015, 4, 1547–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yajun, C.; Chen, Y.; Xiaosa, L.; Xiao, W.; Jia, C.; Zhong, W.; Bin, X. Loss of Sun2 promotes the progression of prostate cancer by regulating fatty acid oxidation. Oncotarget 2017, 8, 89620–89630. [Google Scholar] [CrossRef] [PubMed]
- Aymard, F.; Aguirrebengoa, M.; Guillou, E.; Javierre, B.M.; Bugler, B.; Arnould, C.; Rocher, V.; Iacovoni, J.S.; Biernacka, A.; Skrzypczak, M.; et al. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nat. Struct. Mol. Biol. 2017, 24, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Nakano, K.; Obchoei, S.; Setoguchi, K.; Matsumoto, M.; Yamamoto, T.; Obika, S.; Shimada, K.; Hiraoka, N. Small Nucleolar Noncoding RNA SNORA23, Up-Regulated in Human Pancreatic Ductal Adenocarcinoma, Regulates Expression of Spectrin Repeat-Containing Nuclear Envelope 2 to Promote Growth and Metastasis of Xenograft Tumors in Mice. Gastroenterology 2017, 153, 292–306.e2. [Google Scholar] [CrossRef] [PubMed]
- Janin, A.; Bauer, D.; Ratti, F.; Millat, G.; Mejat, A. Nuclear envelopathies: A complex LINC between nuclear envelope and pathology. Orphanet J. Rare Dis. 2017, 12, 147. [Google Scholar] [CrossRef] [PubMed]
- Cappello, P.; Curcio, C.; Mandili, G.; Roux, C.; Bulfamante, S.; Novelli, F. Next Generation Immunotherapy for Pancreatic Cancer: DNA Vaccination is Seeking New Combo Partners. Cancers 2018, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Hilmi, M.; Bartholin, L.; Neuzillet, C. Immune therapies in pancreatic ductal adenocarcinoma: Where are we now? World J. Gastroenterol. 2018, 24, 2137–2151. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Saif, M.W. T cell optimization for the treatment of pancreatic cancer. Expert Opin. Biol. Ther. 2017, 17, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.; Mahalingam, D. Immunotherapy in pancreatic adenocarcinoma-overcoming barriers to response. J. Gastrointest. Oncol. 2018, 9, 143–159. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Landscape of the nuclear envelope. Schematic cross-section of a nuclear envelope illustrating the LINC complex, the nuclear lamina, and the nuclear pore complexes as they relate to one another. The LINC complex is connected to the cytoskeleton by the N-termini of the KASH-domain proteins with their C-termini embedded in the outer nuclear membrane (ONM) of the nuclear envelope. Within the perinuclear space (PNS), the KASH-domain C-term interacts with the C-term of the SUN protein within the inner nuclear membrane (INM). From there, the nucleoplasmic portion of the LINC complex connects to the nuclear lamina that interacts with chromatin as well as the nuclear pore complex. The nuclear lamina is located adjacent to the INM on the nucleoplasmic face where it forms a dynamic meshwork that provides several structural properties to the nucleus, i.e., nuclear stiffness regulated by composition of the nuclear lamina (see text); it serves to anchor nuclear pore complexes (NPCs) through interactions with distinct nucleoporins (nups) in the nuclear basket portion of the NPC; and it functions as a repressive subnuclear compartment. The NPC is a large multiproteinaceous complex that spans the nuclear envelope where the ONM and INM meet and is the main transporter that facilitates nucleocytoplasmic transport between nuclear and cytoplasmic compartments (see text).
Figure 1.
Landscape of the nuclear envelope. Schematic cross-section of a nuclear envelope illustrating the LINC complex, the nuclear lamina, and the nuclear pore complexes as they relate to one another. The LINC complex is connected to the cytoskeleton by the N-termini of the KASH-domain proteins with their C-termini embedded in the outer nuclear membrane (ONM) of the nuclear envelope. Within the perinuclear space (PNS), the KASH-domain C-term interacts with the C-term of the SUN protein within the inner nuclear membrane (INM). From there, the nucleoplasmic portion of the LINC complex connects to the nuclear lamina that interacts with chromatin as well as the nuclear pore complex. The nuclear lamina is located adjacent to the INM on the nucleoplasmic face where it forms a dynamic meshwork that provides several structural properties to the nucleus, i.e., nuclear stiffness regulated by composition of the nuclear lamina (see text); it serves to anchor nuclear pore complexes (NPCs) through interactions with distinct nucleoporins (nups) in the nuclear basket portion of the NPC; and it functions as a repressive subnuclear compartment. The NPC is a large multiproteinaceous complex that spans the nuclear envelope where the ONM and INM meet and is the main transporter that facilitates nucleocytoplasmic transport between nuclear and cytoplasmic compartments (see text).

{kind=link}
Table 1.
Gene mutations associated with pancreatic adenocarcinoma [2].
Table 1.
Gene mutations associated with pancreatic adenocarcinoma [2].
| Types of Mutation | Gene Symbol | Gene Description | Entrez Gene ID | Ensembl Gene ID |
|---|---|---|---|---|
| Somatic | AKT2 | AKT serine/threonine kinase 2 | 208 | ENSG00000105221 |
| ARID1A | AT-rich interaction domain 1A | 8289 | ENSG00000117713 | |
| BRAF | B-Raf proto-oncogene, serine/threonine kinase | 673 | ENSG00000157764 | |
| CCSER1/FAM190A | Coiled-coil serine rich protein 1 | 401,145 | ENSG00000184305 | |
| CDKN2A | Cyclin dependent kinase inhibitor 2A | 1029 | ENSG00000147889 | |
| EP300 | E1A binding protein p300 | 2033 | ENSG00000100393 | |
| KMT2C/MLL3 | Lysine methyltransferase 2C | 58,508 | ENSG00000055609 | |
| KRAS | KRAS proto-oncogene, GTPase | 3845 | ENSG00000133703 | |
| MYB | MYB proto-oncogene, transcription factor | 4602 | ENSG00000118513 | |
| NCOA3/AIB1 | Nuclear receptor coactivator 3 | 8202 | ENSG00000124151 | |
| SMAD4 | SMAD family member 4 | 4089 | ENSG00000141646 | |
| TGFBR2 | Transforming growth factor beta receptor 2 | 7048 | ENSG00000163513 | |
| TP53 | Tumor protein p53 | 7157 | ENSG00000141510 | |
| USP9X | Ubiquitin specific peptidase 9, X-linked | 8239 | ENSG00000124486 | |
| Germline | ATM | ATM serine/threonine kinase | 472 | ENSG00000149311 |
| BRCA2 | BRCA2, DNA repair associated | 675 | ENSG00000139618 | |
| CDKN2A | Cyclin dependent kinase inhibitor 2A | 1029 | ENSG00000147889 | |
| PALB2 | Partner and localizer of BRCA2 | 79,728 | ENSG00000083093 | |
| PRSS1 | Protease, serine 1 | 5644 | ENSG00000204983 | |
| STK11 | Serine/threonine kinase 11 | 6794 | ENSG00000118046 |
Table 2.
Functional enrichment analysis of common somatic genes mutated in pancreatic adenocarcinoma.
Table 2.
Functional enrichment analysis of common somatic genes mutated in pancreatic adenocarcinoma.
| Cluster No. (E Score) | Database | Functional Term | Fold Enrichment | p Value | Benjamini |
|---|---|---|---|---|---|
| Cluster 1 (3.12) | UP_KEYWORDS | Acetylation | 4.723 | 0.0000028 | 0.0002505 |
| KEGG_PATHWAY | Cell cycle | 20.264 | 0.0006173 | 0.0033905 | |
| UP_KEYWORDS | Transcription | 4.904 | 0.0002617 | 0.0046472 | |
| UP_KEYWORDS | Transcription regulation | 5.043 | 0.0002192 | 0.0048656 | |
| GOTERM_BP_DIRECT | Positive regulation of transcription, DNA-templated | 15.049 | 0.0000176 | 0.0066071 | |
| BIOCARTA | First Multivalent Nuclear Factor | 28.889 | 0.0001714 | 0.0068348 | |
| UP_KEYWORDS | Nucleus | 2.803 | 0.0011438 | 0.0101343 | |
| UP_KEYWORDS | Phosphoprotein | 2.139 | 0.0013327 | 0.0107323 | |
| GOTERM_CC_DIRECT | Nuclear chromatin | 29.054 | 0.0002398 | 0.0112081 | |
| GOTERM_CC_DIRECT | Nucleoplasm | 4.028 | 0.0007497 | 0.0174706 | |
| GOTERM_CC_DIRECT | Nucleus | 2.589 | 0.0015626 | 0.0242026 | |
| GOTERM_MF_DIRECT | DNA binding | 5.430 | 0.0005138 | 0.0263695 | |
| Cluster 2 (2.95) | KEGG_PATHWAY | FoxO signaling pathway | 28.128 | 0.0000006 | 0.0000098 |
| KEGG_PATHWAY | Adherens junction | 26.543 | 0.0044414 | 0.0168064 | |
| KEGG_PATHWAY | TGF-beta signaling pathway | 22.435 | 0.0061682 | 0.0224314 | |
| Cluster 3 (2.86) | KEGG_PATHWAY | Chronic myeloid leukemia | 61.073 | 0.0000000 | 0.0000000 |
| KEGG_PATHWAY | Pancreatic cancer | 67.650 | 0.0000000 | 0.0000000 | |
| KEGG_PATHWAY | Colorectal cancer | 60.792 | 0.0000000 | 0.0000003 | |
| KEGG_PATHWAY | HTLV-I infection | 19.631 | 0.0000000 | 0.0000003 | |
| KEGG_PATHWAY | Pathways in cancer | 12.787 | 0.0000002 | 0.0000037 | |
| KEGG_PATHWAY | FoxO signaling pathway | 28.128 | 0.0000006 | 0.0000098 | |
| KEGG_PATHWAY | Non-small cell lung cancer | 56.088 | 0.0000008 | 0.0000111 | |
| KEGG_PATHWAY | Glioma | 48.322 | 0.0000014 | 0.0000178 | |
| KEGG_PATHWAY | Melanoma | 44.238 | 0.0000021 | 0.0000226 | |
| KEGG_PATHWAY | Prostate cancer | 35.692 | 0.0000049 | 0.0000482 | |
| KEGG_PATHWAY | Thyroid hormone signaling pathway | 27.552 | 0.0000137 | 0.0001230 | |
| KEGG_PATHWAY | Bladder cancer | 61.286 | 0.0000226 | 0.0001865 | |
| KEGG_PATHWAY | Hepatitis B | 21.661 | 0.0000354 | 0.0002697 | |
| KEGG_PATHWAY | Endometrial cancer | 48.322 | 0.0000465 | 0.0003286 | |
| KEGG_PATHWAY | Renal cell carcinoma | 38.657 | 0.0000910 | 0.0006002 | |
| UP_KEYWORDS | Disease mutation | 5.188 | 0.0000396 | 0.0011754 | |
| KEGG_PATHWAY | MAPK signaling pathway | 12.317 | 0.0003191 | 0.0019727 | |
| KEGG_PATHWAY | Neurotrophin signaling pathway | 20.939 | 0.0005607 | 0.0032611 | |
| KEGG_PATHWAY | Hepatitis C | 18.893 | 0.0007577 | 0.0037452 | |
| KEGG_PATHWAY | Thyroid cancer | 64.984 | 0.0007496 | 0.0038995 | |
| UP_KEYWORDS | Proto-oncogene | 24.916 | 0.0003911 | 0.0043423 | |
| KEGG_PATHWAY | Proteoglycans in cancer | 12.564 | 0.0024661 | 0.0115730 | |
| KEGG_PATHWAY | Viral carcinogenesis | 12.257 | 0.0026465 | 0.0118544 | |
| KEGG_PATHWAY | Acute myeloid leukemia | 33.653 | 0.0027845 | 0.0119306 | |
| KEGG_PATHWAY | Central carbon metabolism in cancer | 29.446 | 0.0036227 | 0.0148594 | |
| KEGG_PATHWAY | Long-term potentiation | 28.554 | 0.0038486 | 0.0151538 | |
| UP_SEQ_FEATURE | Mutagenesis site | 5.233 | 0.0001733 | 0.0202413 | |
| KEGG_PATHWAY | MicroRNAs in cancer | 8.817 | 0.0067131 | 0.0227320 | |
| KEGG_PATHWAY | ErbB signaling pathway | 21.661 | 0.0066041 | 0.0231553 | |
| KEGG_PATHWAY | Progesterone-mediated oocyte maturation | 21.661 | 0.0066041 | 0.0231553 | |
| GOTERM_CC_DIRECT | Cytosol | 3.383 | 0.0021984 | 0.0255285 | |
| UP_KEYWORDS | Isopeptide bond | 6.493 | 0.0043625 | 0.0274111 | |
| UP_KEYWORDS | Apoptosis | 10.971 | 0.0041342 | 0.0279639 | |
| KEGG_PATHWAY | PI3K-Akt signaling pathway | 7.283 | 0.0113878 | 0.0370899 | |
| KEGG_PATHWAY | Sphingolipid signaling pathway | 15.705 | 0.0122869 | 0.0387127 | |
| UP_KEYWORDS | Kinase | 8.000 | 0.0099232 | 0.0434085 | |
| KEGG_PATHWAY | Signaling pathways regulating pluripotency of stem cells | 13.461 | 0.0164870 | 0.0486500 | |
| KEGG_PATHWAY | Insulin signaling pathway | 13.656 | 0.0160424 | 0.0488026 | |
| Cluster 4 (2.60) | KEGG_PATHWAY | Cell cycle | 20.264 | 0.0006173 | 0.0033905 |
| KEGG_PATHWAY | Viral carcinogenesis | 12.257 | 0.0026465 | 0.0118544 | |
| KEGG_PATHWAY | MicroRNAs in cancer | 8.817 | 0.0067131 | 0.0227320 | |
| GOTERM_MF_DIRECT | p53 binding | 58.144 | 0.0009983 | 0.0340310 | |
| GOTERM_BP_DIRECT | Apoptotic process | 11.391 | 0.0005128 | 0.0470735 | |
| Cluster 5 (2.58) | KEGG_PATHWAY | Hepatitis B | 21.661 | 0.0000354 | 0.0002697 |
| KEGG_PATHWAY | Cell cycle | 20.264 | 0.0006173 | 0.0033905 | |
| UP_KEYWORDS | Ubl conjugation | 6.035 | 0.0003300 | 0.0048835 | |
| GOTERM_BP_DIRECT | Negative regulation of transcription from RNA polymerase II promoter | 10.764 | 0.0000880 | 0.0164117 | |
| UP_KEYWORDS | Isopeptide bond | 6.493 | 0.0043625 | 0.0274111 | |
| GOTERM_BP_DIRECT | Positive regulation of transcription from RNA polymerase II promoter | 7.900 | 0.0003776 | 0.0462357 | |
| KEGG_PATHWAY | Wnt signaling pathway | 13.656 | 0.0160424 | 0.0488026 | |
| Cluster 6 (1.94) | UP_KEYWORDS | Acyltransferase | 25.791 | 0.0050407 | 0.0295385 |
Functional enrichment analysis was performed with DAVID Bioinformatics Resources 6.8 using high stringency. Depicted are the functional clusters, with their enrichment scores (E scores), comprised of statistically significant terms (p < 0.05).
Table 3.
Functional enrichment analysis of germline genes mutated in pancreatic adenocarcinoma.
| Cluster No. (E Score) | Database | Functional Term | Fold Enrichment | p Value | Benjamini |
|---|---|---|---|---|---|
| Cluster 1 (4.25) | GOTERM_BP_DIRECT | Strand displacement | 322.923 | 0.0000230 | 0.0038316 |
| GOTERM_BP_DIRECT | DNA synthesis involved in DNA repair | 239.886 | 0.0000420 | 0.0035042 | |
| GOTERM_BP_DIRECT | Double-strand break repair via homologous recombination | 113.459 | 0.0001900 | 0.0105193 | |
| Cluster 2 (2.74) | UP_KEYWORDS | Tumor suppressor | 96.897 | 0.0000000 | 0.0000015 |
| GOTERM_CC_DIRECT | Nucleoplasm | 5.455 | 0.0023864 | 0.0557288 | |
| Cluster 3 (1.76) | GOTERM_BP_DIRECT | Cell cycle arrest | 59.546 | 0.0006886 | 0.0283490 |
| Cluster 4 (1.72) | UP_KEYWORDS | Cell cycle | 21.109 | 0.0002990 | 0.0056659 |
Functional enrichment analysis was performed with DAVID Bioinformatics Resources 6.8 using high stringency. Depicted are the functional clusters, with their enrichment scores (E scores), comprised of statistically significant terms (p < 0.05).
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Preston, C.C.; Faustino, R.S. Nuclear Envelope Regulation of Oncogenic Processes: Roles in Pancreatic Cancer. Epigenomes 2018, 2, 15. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes2030015
AMA Style
Preston CC, Faustino RS. Nuclear Envelope Regulation of Oncogenic Processes: Roles in Pancreatic Cancer. Epigenomes. 2018; 2(3):15. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes2030015
Chicago/Turabian StylePreston, Claudia C., and Randolph S. Faustino. 2018. "Nuclear Envelope Regulation of Oncogenic Processes: Roles in Pancreatic Cancer" Epigenomes 2, no. 3: 15. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes2030015