Epigenetic Dynamics of the Infant Immune System Reveals a Tumor Necrosis Factor Superfamily Signature in Early Human Life

Abstract
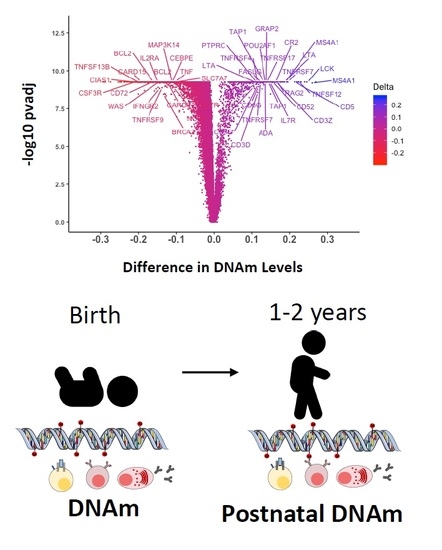
:
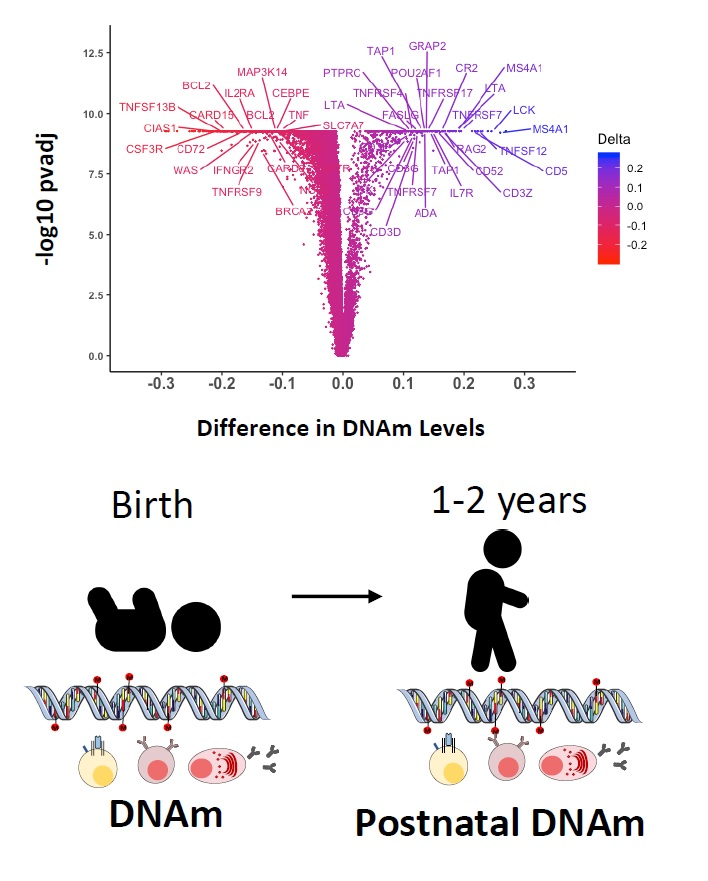{kind=link}
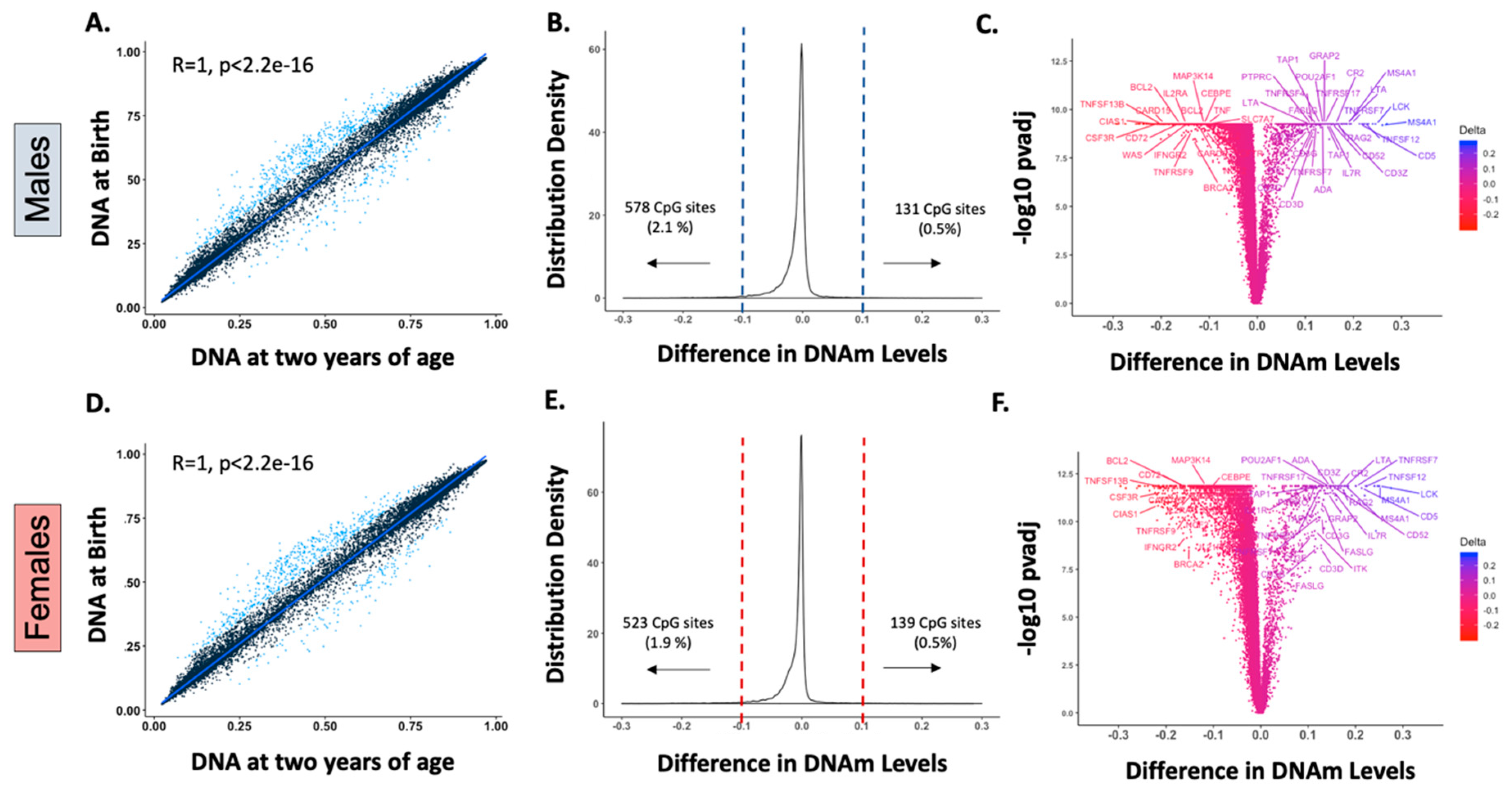{kind=link}
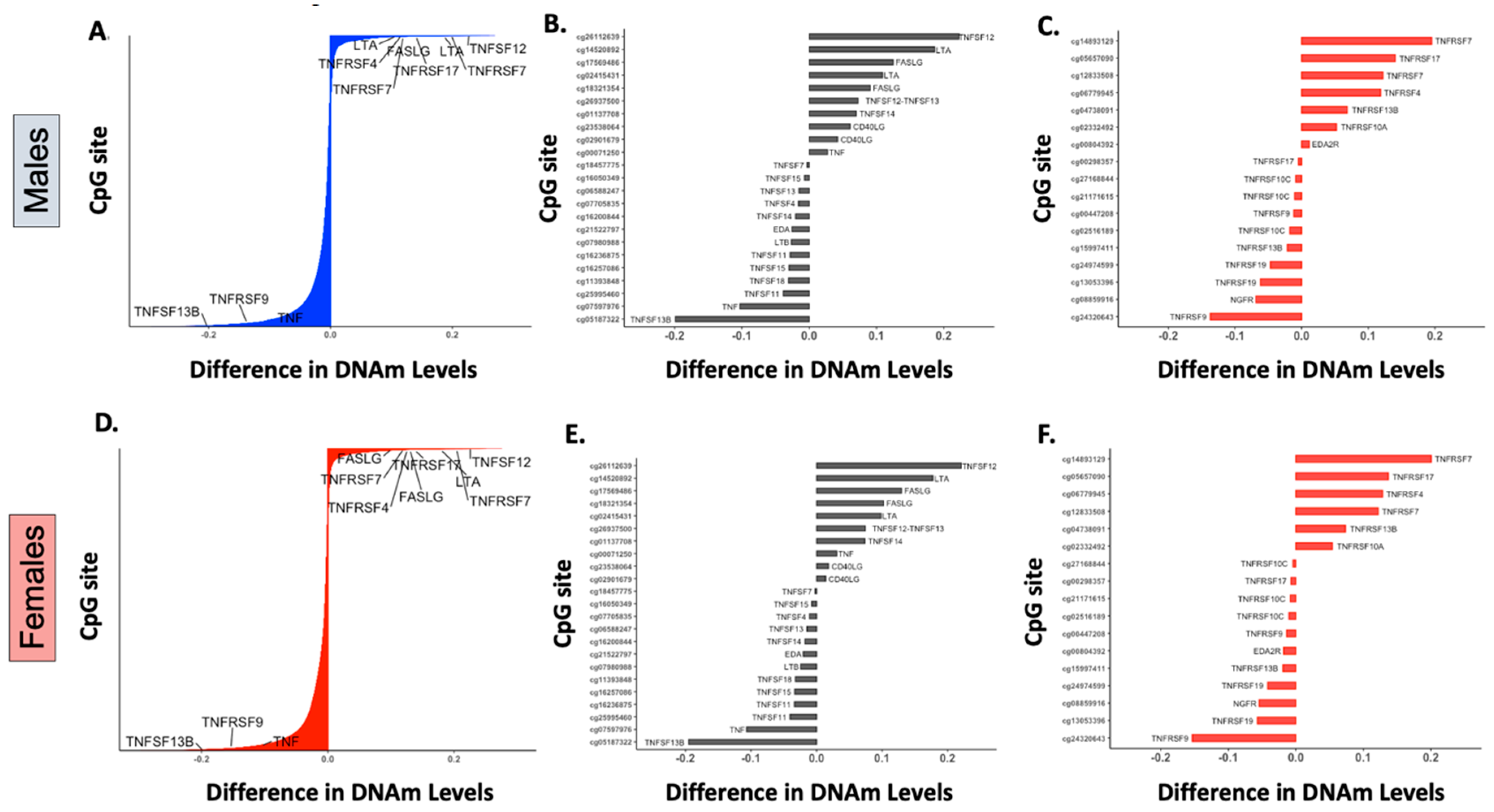{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Luo, C.; Hajkova, P.; Ecker, J.R. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Riggs, A.D. X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet. 1975, 14, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Field, A.E.; Robertson, N.A.; Wang, T.; Havas, A.; Ideker, T.; Adams, P.D. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol. Cell. 2018, 71, 882–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Gan, Y.; Zou, G.; Guan, J.; Zhou, S. Genome-wide analysis of epigenetic dynamics across human developmental stages and tissues. BMC Genomics 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Merid, S.K.; Novoloaca, A.; Sharp, G.C.; Küpers, L.K.; Kho, A.T.; Roy, R.; Gao, L.; Annesi-Maesano, I.; Jain, P.; Plusquin, M.; et al. Epigenome-wide meta-analysis of blood DNA methylation in newborns and children identifies numerous loci related to gestational age. Genome Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Liu, X.; Zhou, Y.; Xie, H.; Hong, X.; Tsai, H.J.; Wang, G.; Liu, R.; Wang, X. Individual variation and longitudinal pattern of genome-wide DNA methylation from birth to the first two years of life. Epigenetics 2012, 7, 594–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, D.J.; Tulic, M.K.; Gordon, L.; Hodder, M.; Richman, T.R.; Metcalfe, J.; Prescott, S.L.; Saffery, R. Evidence for age-related and individual-specific changes in DNA methylation profile of mononuclear cells during early immune development in humans. Epigenetics 2011, 6, 1085–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulis, M.; Merkel, A.; Heath, S.; Queirós, A.C.; Schuyler, R.P.; Castellano, G.; Beekman, R.; Raineri, E.; Esteve, A.; Clot, G.; et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat. Genet. 2015, 47, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Olin, A.; Henckel, E.; Chen, Y.; Lakshmikanth, T.; Pou, C.; Mikes, J.; Gustafsson, A.; Bernhardsson, A.K.; Zhang, C.; Bohlin, K.; et al. Stereotypic Immune System Development in Newborn Children. Cell 2018, 174, 1277–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miosge, L.A.; Goodnow, C.C. Genes, pathways and checkpoints in lymphocyte development and homeostasis. Immunol. Cell Biol. 2005, 83, 318–335. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Croft, M.; Siegel, R.M. Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 217–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward-Kavanagh, L.K.; Lin, W.W.; Sedy, J.R.; Ware, C.F. The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity 2016, 44, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef] [PubMed]
- Collette, Y.; Gilles, A.; Pontarotti, P.; Olive, D. A co-evolution perspective of the TNFSF and TNFRSF families in the immune system. Trends Immunol. 2003, 24, 387–394. [Google Scholar] [CrossRef]
- Yi, F.; Frazzette, N.; Cruz, A.C.; Klebanoff, C.A.; Siegel, R.M. Beyond Cell Death: New Functions for TNF Family Cytokines in Autoimmunity and Tumor Immunotherapy. Trends Mol. Med. 2018, 24, 642–653. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Bossen, C.; Schneider, P. BAFF, APRIL and their receptors: Structure, function and signaling. Semin. Immunol. 2006, 18, 263–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatuni, G.S.; Sciortino, S.; Currier, R.J.; Naides, S.J.; Church, J.A.; Puck, J.M. Reference intervals for lymphocyte subsets in preterm and term neonates without immune defects. J. Allergy Clin. Immunol. 2019, 144, 1674–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutierrez, M.J.; Nino, G.; Hong, X.; Wang, X. Epigenetic Dynamics of the Infant Immune System Reveals a Tumor Necrosis Factor Superfamily Signature in Early Human Life. Epigenomes 2020, 4, 12. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes4030012
Gutierrez MJ, Nino G, Hong X, Wang X. Epigenetic Dynamics of the Infant Immune System Reveals a Tumor Necrosis Factor Superfamily Signature in Early Human Life. Epigenomes. 2020; 4(3):12. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes4030012
Chicago/Turabian StyleGutierrez, Maria J., Gustavo Nino, Xiumei Hong, and Xiaobin Wang. 2020. "Epigenetic Dynamics of the Infant Immune System Reveals a Tumor Necrosis Factor Superfamily Signature in Early Human Life" Epigenomes 4, no. 3: 12. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes4030012