Implication of m6A mRNA Methylation in Susceptibility to Inflammatory Bowel Disease

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. IBD Associated SNPs Are Located Near m6A Motifs in Differentially Expressed Genes
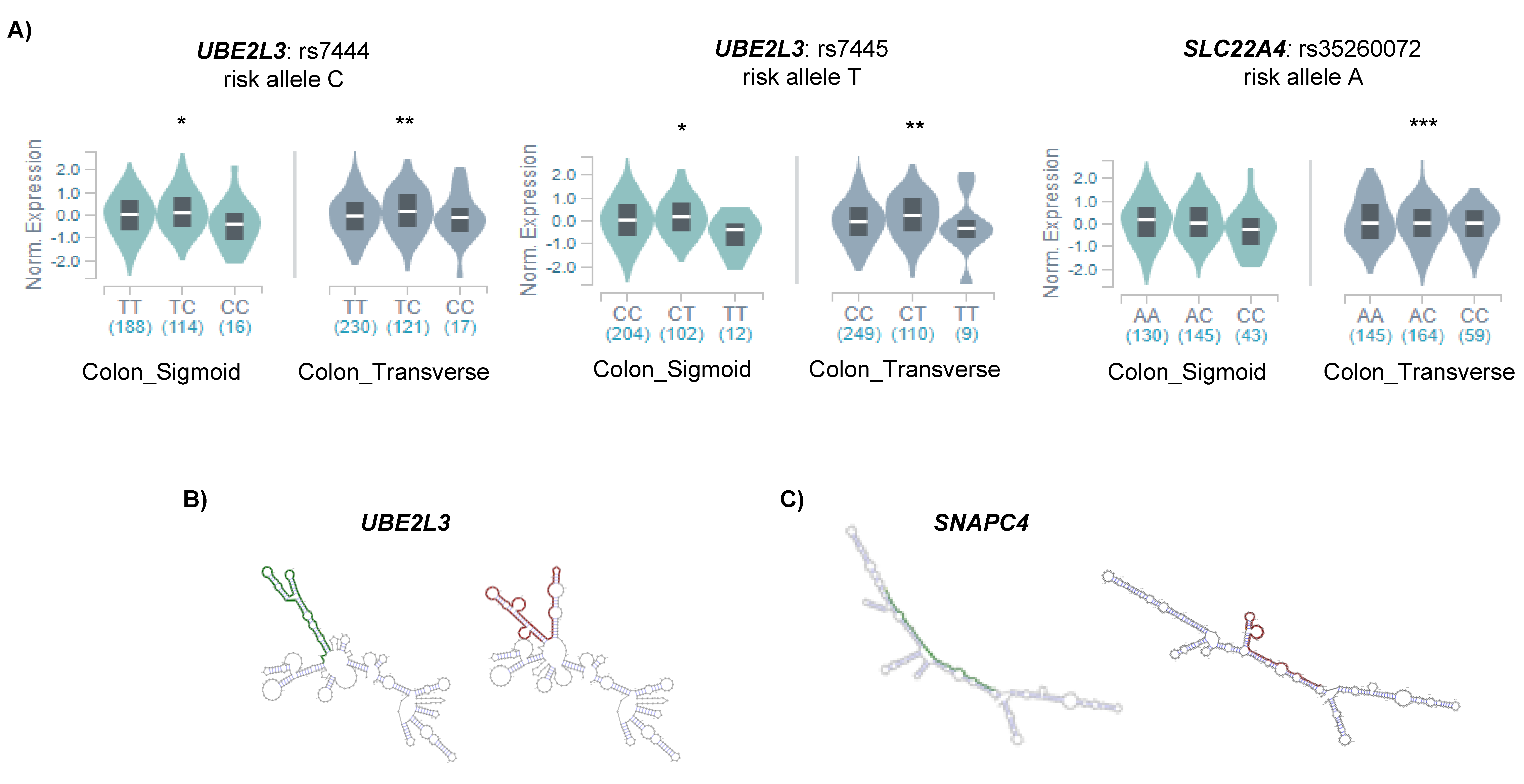
2.2. m6A SNPs Can Affect Different Layers of RNA Regulation
2.3. Genes Harboring m6A-SNPs Interact and Co-Express with m6A Machinery Proteins
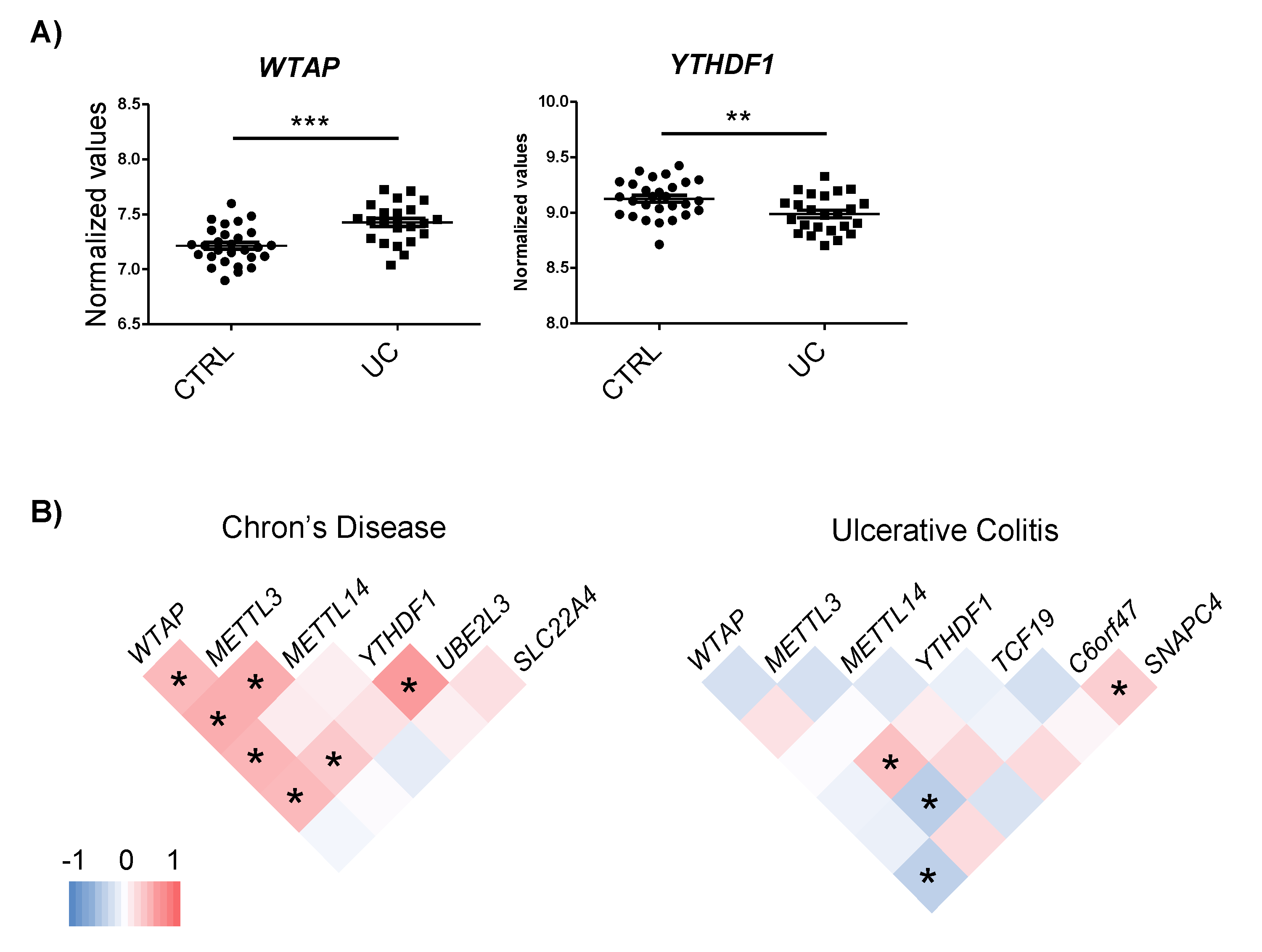
2.4. IBD-Related Inflammatory Conditions Alter m6A-Related Processes
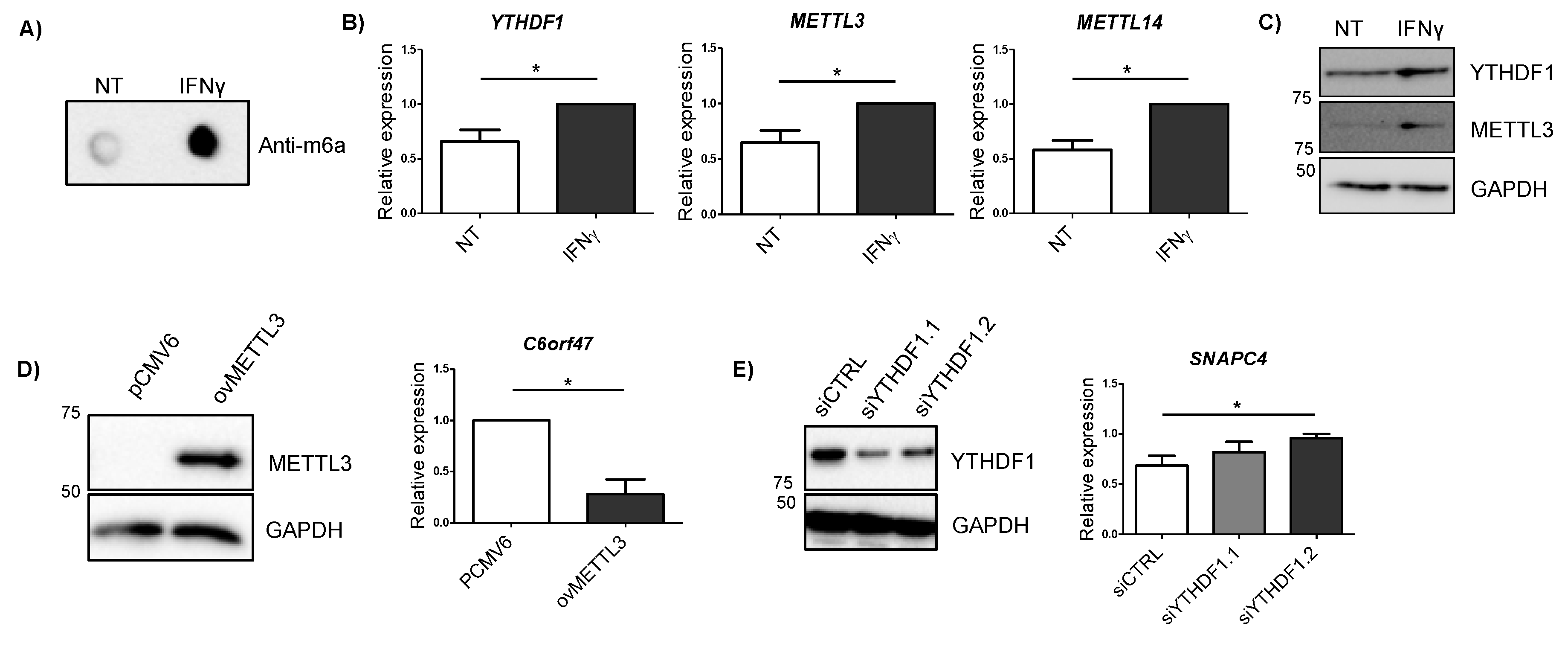
2.5. Alterations in METTL3 and YTHDF1 Affect Candidate Gene Expression
3. Discussion
4. Materials and Methods
4.1. Selection of Candidate Genes
4.2. Analysis of m6A-SNP Regulatory Capacity
4.3. Candidate Gene and m6A Machinery Protein Interaction Analysis
4.4. Candidate Gene and m6A Machinery Protein Co-expression Analysis
4.5. Cell Lines and Stimulations
4.6. Overexpression and Silencing
4.7. RNA and Protein Extractions
4.8. Dot Blot
4.9. Gene Expression Analysis
4.10. Western Blot
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fusunyan, D.; Sanderson, I.R. Inflammatory Bowel Disease. In Encyclopedia of Immunology; Elsevier: Amsterdam, The Netherlands, 1998; pp. 1375–1381. [Google Scholar]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 7351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckburg, P.B.; Relman, D.A. The Role of Microbes in Crohn’s Disease. Clin. Infect. Dis. 2007, 44, 256–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstock, J.V. Helminths and mucosal immune modulation. Ann. N. Y. Acad. Sci. 2006, 1072, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Verstockt, B.; Ferrante, M.; Vermeire, S.; van Assche, G. New treatment options for inflammatory bowel diseases. J. Gastroenterol. 2018, 53, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.; Duerr, R.; McGovern, D.; Hui, K.; Lee, J.; Philip Schumm, L.; Sharma, Y.; Anderson, C.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Ye, B.D.; McGovern, D.P.B. Genetic variation in IBD: Progress, clues to pathogenesis and possible clinical utility. Expert Rev. Clin. Immunol. 2016, 12, 1091–1107. [Google Scholar] [CrossRef] [Green Version]
- Karatzas, P.S.; Gazouli, M.; Safioleas, M.; Mantzaris, G.J. DNA methylation changes in inflammatory bowel disease. Ann. Gastroenterol. 2014, 27, 125–132. [Google Scholar]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m 6 A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef]
- Maity, A.; Das, B. N6-methyladenosine modification in mRNA: Machinery, function and implications for health and diseases. FEBS J. 2016, 283, 1607–1630. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Nie, P.; Peng, D.; He, Z.; Liu, M.; Xie, Y.; Miao, Y.; Zuo, Z.; Ren, J. M6AVar: A database of functional variants involved in m 6 A modification. Nucleic Acids Res. 2018, 46, D139–D145. [Google Scholar] [CrossRef]
- Jiang, S.; Xie, Y.; He, Z.; Zhang, Y.; Zhao, Y.; Chen, L.; Yueyuan, Z.; Miao, Y.; Zuo, Z.; Ren, J. m6ASNP: A tool for annotating genetic variants by m6A function. GigaScience 2018, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.; Wang, H.; Zhu, L.; Jin, H.; Wang, X. N6-methyladenosine links RNA metabolism to cancer progression review-article. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Ohsugi, M.; Sasako, T.; Awazawa, M.; Umehara, T.; Iwane, A.; Kobayashi, N.; Okazaki, Y.; Kubota, N.; Suzuki, R.; et al. The RNA Methyltransferase Complex of WTAP, METTL3, and METTL14 Regulates Mitotic Clonal Expansion in Adipogenesis. Mol. Cell. Biol. 2018, 38, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.B.; Tong, J.; Zhu, S.; Batista, P.; Duffy, E.; Zhao, J.; Bailis, W.; Cao, G.; Kroehling, L.; Chen, Y.; et al. M 6 A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 2017, 548, 338–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, R.; Gillis, E.; Lasman, L.; Safra, M.; Geula, S.; Soyris, C.; Nachshon, A.; Tai-Schmiedel, J.; Friedman, N.; Le-Trilling, K.; et al. m 6 A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 2019, 20, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, Y.; Konno, M.; Asai, A.; Koseki, J.; Kawamoto, K.; Miyoshi, N.; Takahashi, H.; Nishida, N.; Haraguchi, N.; Sakai, D.; et al. Oncogene c-Myc promotes epitranscriptome m6A reader YTHDF1 expression in colorectal cancer. Oncotarget 2018, 9, 7476–7486. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Wang, J.; Chen, D.; He, Y.; Meng, N.; Chen, M.; Lu, R.; Chen, X.; Zhang, X.; Yan, G. An oncopeptide regulates m6A recognition by the m6A reader IGF2BP1 and tumorigenesis. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Z.; Van Sommeren, S.; Huang, H.; Ng, S.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- Tanaka, M.; Sasaki, K.; Kamata, R.; Hoshino, Y.; Yanagihara, K.; Sakai, R. A Novel RNA-Binding Protein, Ossa/C9orf10, Regulates Activity of Src Kinases to Protect Cells from Oxidative Stress-Induced Apoptosis. Mol. Cell. Biol. 2009, 29, 402–413. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N 6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Bushkin, G.G.; Pincus, D.; Morgan, J.; Richardson, K.; Lewis, C.; Chan, S.; Bartel, D.; Fink, G. m6A modification of a 3′ UTR site reduces RME1 mRNA levels to promote meiosis. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hu, X.; Huang, M.; Liu, J.; Gu, Y.; Ma, L.; Zhou, Q.; Cao, X. Mettl3-mediated mRNA m 6 A methylation promotes dendritic cell activation. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.; Cao, G.; Zhang, T.; Sefik, E.; Vesely, M.; Broughton, J.; Zhu, S.; Li, H.; Li, B.; Chen, L.; et al. M6 A mRNA methylation sustains Treg suppressive functions. Cell Res. 2018, 28, 253–256. [Google Scholar] [CrossRef] [Green Version]
- Vagefi, P.A.; Longo, W.E. Colorectal cancer in patients with inflammatory bowel disease. Clin. Colorectal Cancer 2005, 4, 313–319. [Google Scholar] [CrossRef]
- Li, H.; Su, Q.; Li, B.; Lan, L.; Wang, C.; Li, W.; Wang, G.; Chen, W.; He, Y.; Zhang, C. High expression of WTAP leads to poor prognosis of gastric cancer by influencing tumour-associated T lymphocyte infiltration. J. Cell. Mol. Med. 2020, 24, 4452–4465. [Google Scholar] [CrossRef]
- Karatzas, P.S.; Mantzaris, G.J.; Safioleas, M.; Gazouli, M. DNA methylation profile of genes involved in inflammation and autoimmunity in inflammatory bowel disease. Medicine 2014, 93, e309. [Google Scholar] [CrossRef]
- Liu, H.; Wang, H.; Wei, Z.; Zhang, S.; Hua, G.; Zhang, S.; Zhang, L.; Gao, S.; Meng, J.; Chen, X.; et al. MeT-DB V2.0: Elucidating context-specific functions of N 6 -methyl-adenosine methyltranscriptome. Nucleic Acids Res. 2018, 46, D281–D287. [Google Scholar] [CrossRef] [Green Version]
- Weiser, M.; Simon, J.; Kochar, B.; Tovar, A.; Israel, J.; Robinson, A.; Gipson, G.; Schaner, M.; Herfarth, H.; Sartor, R.; et al. Molecular classification of Crohn’s disease reveals two clinically relevant subtypes. Gut 2018, 67, 36–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Vinayaga-Pavan, M.; Frampton, M.; Pontikos, N.; Levine, A.; Smith, P.; Jonasson, J.; Bjõrnsson, E.; Segal, A.; Smith, A. Elevation in Cell Cycle and Protein Metabolism Gene Transcription in Inactive Colonic Tissue from Icelandic Patients with Ulcerative Colitis. Inflamm. Bowel Dis. 2019, 25, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xu, Y.; Yang, Y.; Xu, Z.; Chen, X.; Shi, B.; Xie, D.; Lu, Z.; Wang, P. POSTAR2: Deciphering the post-Transcriptional regulatory logics. Nucleic Acids Res. 2019, 47, D203–D211. [Google Scholar] [CrossRef] [Green Version]
- Sabarinathan, R.; Tafer, H.; Seemann, S.E.; Hofacker, I.L.; Stadler, P.F.; Gorodkin, J. The RNAsnp web server: Predicting SNP effects on local RNA secondary structure. Nucleic Acids Res. 2013, 41, W475–W479. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Associated SNP | Disease | SNP Location | Functional Predictions | In Vitro Effect | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Structure | eQTL | Expression | m6A Binding Proteins | Other RBP | ovMETTL3 | siYTHDF1 | ||||
| UBE2L3 | rs7444, rs7445 | CD | 3′UTR | YES | YES | CD, up | YTHDF1, YTHDF2 | FAM120A | ns | ns |
| SLC22A4 | rs35260072 | CD | Intron | Not observed | YES | CD, up | WTAP, METTL3, METTL14, YTHDF1 | No | ns | ns |
| TCF19 | rs139102013 | UC | Intron | Not observed | ns | UC, up | YTHDF1 | No | ns | ns |
| C6orf47 | rs148844907 | UC/CD | 5′UTR | NO | ns | UC, down | YTHDF1, YTHDF2 | No | Yes, down | ns |
| SNAPC4 | rs3812565 | UC/CD | CS, Synonimous | NO | ns | UC, down | WTAP, METTL3, METTL14, YTHDF1 | No | ns | Yes, up |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sebastian-delaCruz, M.; Olazagoitia-Garmendia, A.; Gonzalez-Moro, I.; Santin, I.; Garcia-Etxebarria, K.; Castellanos-Rubio, A. Implication of m6A mRNA Methylation in Susceptibility to Inflammatory Bowel Disease. Epigenomes 2020, 4, 16. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes4030016
Sebastian-delaCruz M, Olazagoitia-Garmendia A, Gonzalez-Moro I, Santin I, Garcia-Etxebarria K, Castellanos-Rubio A. Implication of m6A mRNA Methylation in Susceptibility to Inflammatory Bowel Disease. Epigenomes. 2020; 4(3):16. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes4030016
Chicago/Turabian StyleSebastian-delaCruz, Maialen, Ane Olazagoitia-Garmendia, Itziar Gonzalez-Moro, Izortze Santin, Koldo Garcia-Etxebarria, and Ainara Castellanos-Rubio. 2020. "Implication of m6A mRNA Methylation in Susceptibility to Inflammatory Bowel Disease" Epigenomes 4, no. 3: 16. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes4030016