
Ultra-Low DNA Input into Whole Genome Methylation Assays and Detection of Oncogenic Methylation and Copy Number Variants in Circulating Tumour DNA

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Sample QC Metrics
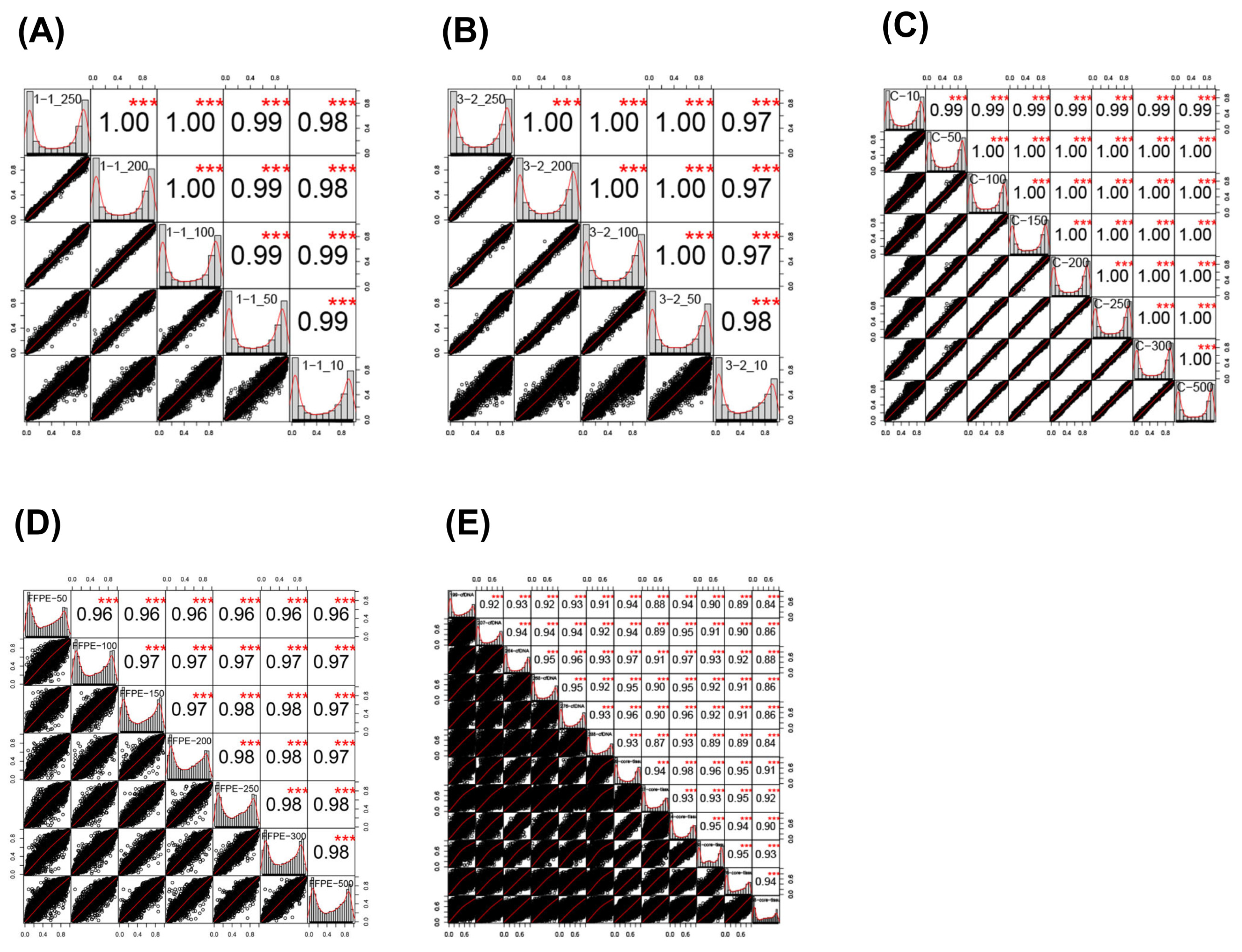
2.2. Sample Correlations
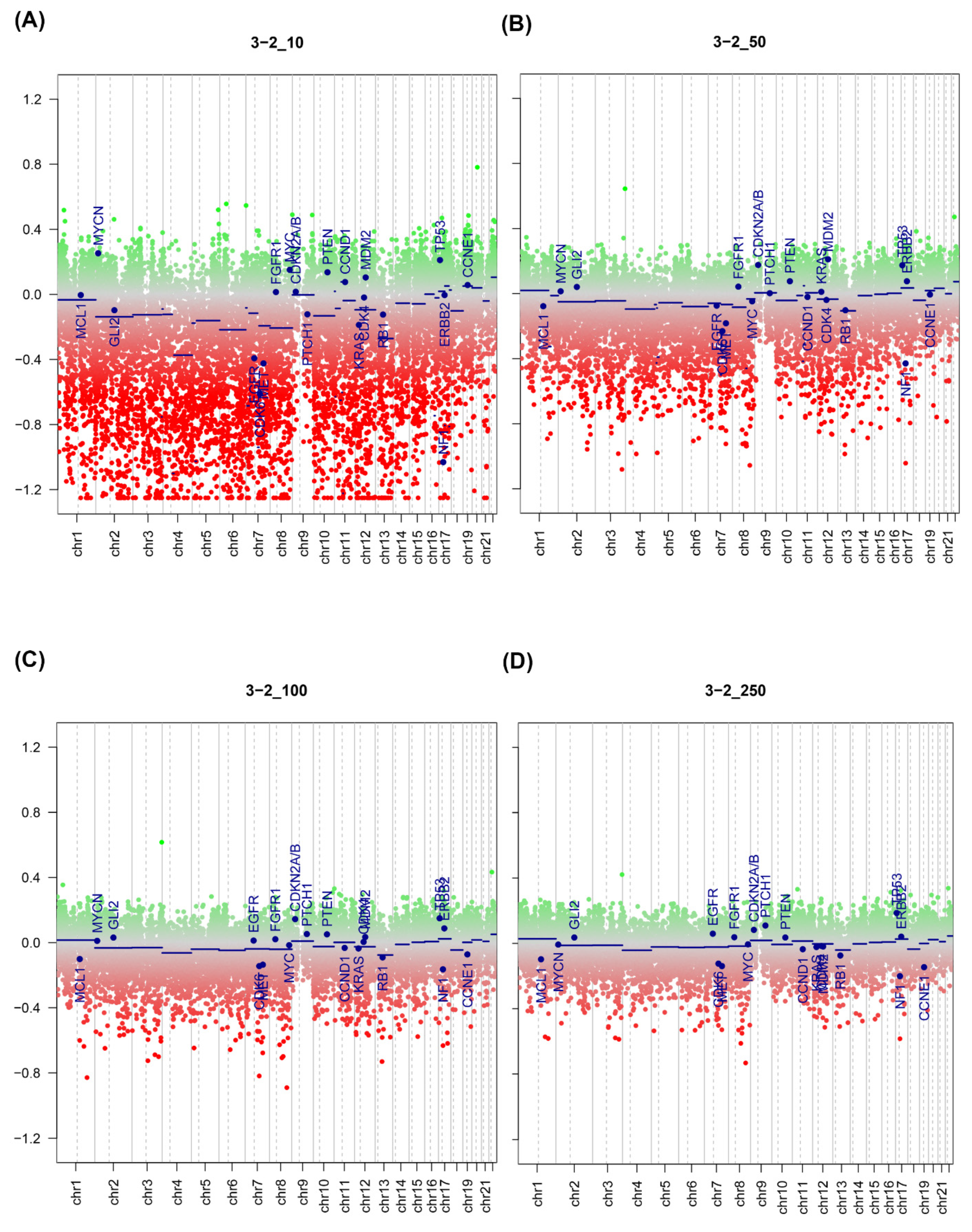
2.3. Effect of Input Amount on Results
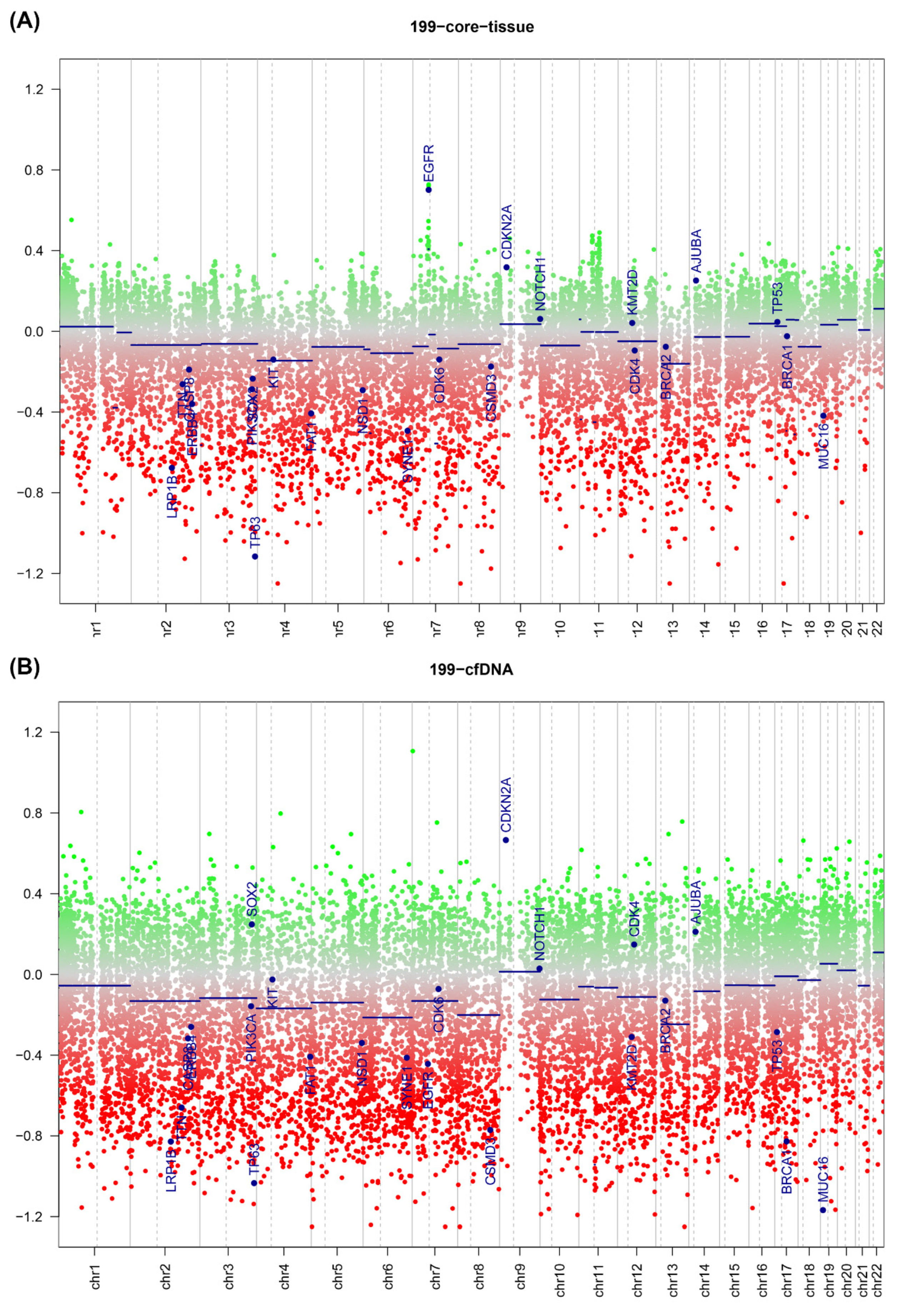
2.4. ctDNA Analysis
2.5. DNA Copy Number Calls
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. DNA Extraction
4.3. FFPE QC Assay
4.4. Bisulphite Conversion and Restoration of DNA
4.5. Infinium HD FFPE Methylation Assay
4.6. Downstream Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DNA | Deoxyribonucleic acid |
| ctDNA | Circulating tumour DNA |
| FFPE | Formalin fixed, paraffin embedded |
| WGA | Whole genome amplification |
| CIMP | CpG Island methylator phenotype |
| DMP | Differentially methylated position |
| DMR | Differentially methylated region |
References
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Issa, J.P. CpG island methylator phenotypes in aging and cancer. Semin. Cancer Biol. 1999, 9, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Cantor, M.; Kawasaki, T.; Brahmandam, M.; Kirkner, G.J.; Weisenberger, D.J.; Campan, M.; Laird, P.W.; Loda, M.; Fuchs, C.S. CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut 2006, 55, 1000–1006. [Google Scholar] [CrossRef]
- Puccini, A.; Berger, M.D.; Naseem, M.; Tokunaga, R.; Battaglin, F.; Cao, S.; Hanna, D.L.; McSkane, M.; Soni, S.; Zhang, W.; et al. Colorectal cancer: Epigenetic alterations and their clinical implications. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Ye, M.; Ni, S.; Li, Q.; Ye, D.; Li, J.; Shen, Z.; Deng, H. DNA methylation biomarkers for head and neck squamous cell carcinoma. Epigenetics 2018, 13, 398–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colacino, J.A.; Dolinoy, D.C.; Duffy, S.A.; Sartor, M.A.; Chepeha, D.B.; Bradford, C.R.; McHugh, J.B.; Patel, D.A.; Virani, S.; Walline, H.M.; et al. Comprehensive analysis of DNA methylation in head and neck squamous cell carcinoma indicates differences by survival and clinicopathologic characteristics. PLoS ONE 2013, 8, e54742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warton, K.; Mahon, K.L.; Samimi, G. Methylated circulating tumor DNA in blood: Power in cancer prognosis and response. Endocr. Relat. Cancer 2016, 23, R157–R171. [Google Scholar] [CrossRef] [Green Version]
- Razavi, P.; Li, B.T.; Brown, D.N.; Jung, B.; Hubbell, E.; Shen, R.; Abida, W.; Juluru, K.; De Bruijn, I.; Hou, C.; et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019, 25, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Feber, A.; Dhami, P.; Dong, L.; de Winter, P.; Tan, W.S.; Martinez-Fernandez, M.; Paul, D.S.; Hynes-Allen, A.; Rezaee, S.; Gurung, P.; et al. UroMark-a urinary biomarker assay for the detection of bladder cancer. Clin. Epigenetics 2017, 9, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olova, N.; Krueger, F.; Andrews, S.; Oxley, D.; Berrens, R.V.; Branco, M.R.; Reik, W. Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data. Genome Biol. 2018, 19, 33. [Google Scholar] [CrossRef] [Green Version]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, S.; Arribas, C.; Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 2016, 8, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyanchandani, R.; Kvam, E.; Heller, R.; Finehout, E.; Smith, N.; Kota, K.; Nelson, J.R.; Griffin, W.; Puhalla, S.; Brufsky, A.M.; et al. Whole genome amplification of cell-free DNA enables detection of circulating tumor DNA mutations from fingerstick capillary blood. Sci. Rep. 2018, 8, 17313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.E.; Triboulet, M.; Zia, A.; Vuppalapaty, M.; Kidess-Sigal, E.; Coller, J.; Natu, V.S.; Shokoohi, V.; Che, J.; Renier, C.; et al. Workflow optimization of whole genome amplification and targeted panel sequencing for CTC mutation detection. Npj Genom. Med. 2017, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Johansson, G.; Andersson, D.; Filges, S.; Li, J.; Muth, A.; Godfrey, T.E.; Stahlberg, A. Considerations and quality controls when analyzing cell-free tumor DNA. Biomol. Detect. Quantif 2019, 17, 100078. [Google Scholar] [CrossRef]
- Gallardo-Gomez, M.; Moran, S.; Paez de la Cadena, M.; Martinez-Zorzano, V.S.; Rodriguez-Berrocal, F.J.; Rodriguez-Girondo, M.; Esteller, M.; Cubiella, J.; Bujanda, L.; Castells, A.; et al. A new approach to epigenome-wide discovery of non-invasive methylation biomarkers for colorectal cancer screening in circulating cell-free DNA using pooled samples. Clin. Epigenetics 2018, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Alderdice, M.; Richman, S.D.; Gollins, S.; Stewart, J.P.; Hurt, C.; Adams, R.; McCorry, A.M.; Roddy, A.C.; Vimalachandran, D.; Isella, C.; et al. Prospective patient stratification into robust cancer-cell intrinsic subtypes from colorectal cancer biopsies. J. Pathol. 2018, 245, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Leygo, C.; Williams, M.; Jin, H.C.; Chan, M.W.Y.; Chu, W.K.; Grusch, M.; Cheng, Y.Y. DNA Methylation as a Noninvasive Epigenetic Biomarker for the Detection of Cancer. Dis. Markers 2017, 2017, 3726595. [Google Scholar] [CrossRef] [PubMed]
- Warton, K.; Samimi, G. Methylation of cell-free circulating DNA in the diagnosis of cancer. Front. Mol. Biosci. 2015, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Rosell, R.; Karachaliou, N. Lung cancer: Using ctDNA to track EGFR and KRAS mutations in advanced-stage disease. Nat. Rev. Clin. Oncol. 2016, 13, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Schrock, A.; Leisse, A.; de Vos, L.; Gevensleben, H.; Droge, F.; Franzen, A.; Wachendorfer, M.; Schrock, F.; Ellinger, J.; Teschke, M.; et al. Free-Circulating Methylated DNA in Blood for Diagnosis, Staging, Prognosis, and Monitoring of Head and Neck Squamous Cell Carcinoma Patients: An Observational Prospective Cohort Study. Clin. Chem. 2017, 63, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Mydlarz, W.K.; Hennessey, P.T.; Wang, H.; Carvalho, A.L.; Califano, J.A. Serum biomarkers for detection of head and neck squamous cell carcinoma. Head Neck 2016, 38, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, O.; MacIsaac, J.; Quach, H.; Tindula, G.; Kobor, M.S.; Huen, K.; Meaney, M.J.; Eskenazi, B.; Barcellos, L.F.; Holland, N. Comparison of DNA methylation measured by Illumina 450K and EPIC BeadChips in blood of newborns and 14-year-old children. Epigenetics 2018, 13, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Laird, P.W.; Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2017, 45, e22. [Google Scholar] [CrossRef]
- Nordlund, J.; Backlin, C.L.; Wahlberg, P.; Busche, S.; Berglund, E.C.; Eloranta, M.L.; Flaegstad, T.; Forestier, E.; Frost, B.M.; Harila-Saari, A.; et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013, 14, r105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Sample ID | DNA Input (ng) | Detected CpG (0.05) | % CpG Detection |
|---|---|---|---|
| C-10 | 10 | 483,570 | 99.59 |
| C-50 | 50 | 485,395 | 99.96 |
| C-100 | 100 | 485,475 | 99.98 |
| C-150 | 150 | 485,466 | 99.98 |
| C-200 | 200 | 485,473 | 99.98 |
| C-250 | 250 | 485,471 | 99.98 |
| C-300 | 300 | 485,455 | 99.97 |
| C-500 | 500 | 485,446 | 99.97 |
| 3-2_10 | 10 | 484,491 | 99.78 |
| 3-2_50 | 50 | 485,417 | 99.97 |
| 3-2_100 | 100 | 485,406 | 99.96 |
| 3-2_200 | 200 | 485,435 | 99.97 |
| 3-2_250 | 250 | 485,445 | 99.97 |
| 1-1_10 | 10 | 484,158 | 99.71 |
| 1-1_50 | 50 | 485,346 | 99.95 |
| 1-1_100 | 100 | 485,446 | 99.97 |
| 1-1_200 | 200 | 485,458 | 99.98 |
| 1-1_250 | 250 | 485,485 | 99.98 |
| 199-ctDNA | 10 | 457,792 | 94.28% |
| 207-ctDNA | 10 | 467,045 | 96.18% |
| 264-ctDNA | 10 | 480,752 | 99.01% |
| 268-ctDNA | 10 | 471,080 | 97.01% |
| 276-ctDNA | 10 | 471,041 | 97.01% |
| 288-ctDNA | 10 | 451,610 | 93.00% |
| 199-tissue | 250 | 485,046 | 99.89% |
| 207-tissue | 250 | 484,988 | 99.88% |
| 264-tissue | 250 | 484,727 | 99.82% |
| 268-tissue | 250 | 484,983 | 99.88% |
| 276-tissue | 250 | 484,442 | 99.77% |
| 288-tissue | 250 | 484,962 | 99.87% |
| Sample ID | qPCR Delta Cq Value < 5 (>5 Poor) | % CpG Detection (p = 0.05) 150 ng Input | % CpG Detection (p = 0.05) 100 ng Input | % CpG Detection (p = 0.05) 50 ng Input |
|---|---|---|---|---|
| SC00236A2 | 2.34 | 96.55 | 97.57 | 96.69 |
| SC00264A2 | 1.59 | 97.95 | 98.73 | 97.93 |
| SC00267A2 | 2.95 | 98.03 | 98.40 | 97.50 |
| SC00273A2 | 3.10 | 97.99 | 98.22 | 97.66 |
| SC00275A2 | 2.72 | 97.60 | 98.09 | 97.10 |
| SC00292A2 | 3.61 | 96.99 | 97.97 | 96.30 |
| SC00296A2 | 2.08 | 99.22 | 99.27 | 98.01 |
| SC00357A2 | 0.89 | 99.40 | 99.44 | 99.22 |
| SC00447A2 | 0.71 | 98.66 | 99.04 | 98.38 |
| SC00476A2 | 1.87 | 99.18 | 99.17 | 99.19 |
| SC00482A2 | 1.09 | 98.95 | 98.85 | 98.49 |
| SC00486A2 | 1.97 | 98.85 | 98.94 | 98.63 |
| SC00491A2 | 1.83 | 98.28 | 98.29 | 98.10 |
| SC00496A2 | 3.05 | 93.07 | 95.28 | 94.96 |
| SC00498A2 | 1.83 | 98.86 | 98.98 | 98.54 |
| SC00529A2 | 1.08 | 99.14 | 99.20 | 98.75 |
| Comparison | Correlation Coefficient | Lower 95% CI | Upper 95% CI | p-Value |
|---|---|---|---|---|
| C-10–C-50 | 0.99 | 0.99 | 0.99 | <0.01 |
| C-10–C-100 | 0.99 | 0.99 | 0.99 | <0.01 |
| C-10–C-150 | 0.99 | 0.99 | 0.99 | <0.01 |
| C-10–C-200 | 0.99 | 0.99 | 0.99 | <0.01 |
| C-10–C-250 | 0.99 | 0.99 | 0.99 | <0.01 |
| C-10–C-300 | 0.99 | 0.99 | 0.99 | <0.01 |
| C-10–C-500 | 0.99 | 0.99 | 0.99 | <0.01 |
| C-50–C-100 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-50–C-150 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-50–C-200 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-50–C-250 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-50–C-300 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-50–C-500 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-100–C-150 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-100–C-200 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-100–C-250 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-100–C-300 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-100–C-500 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-150–C-200 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-150–C-250 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-150–C-300 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-150–C-500 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-200–C-250 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-200–C-300 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-200–C-500 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-250–C-300 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-250–C-500 | 1.00 | 1.00 | 1.00 | <0.01 |
| C-300–C-500 | 1.00 | 1.00 | 1.00 | <0.01 |
| 1-1_25-1-1_20 | 1.00 | 1.00 | 1.00 | <0.01 |
| 1-1_25-1-1_100 | 1.00 | 1.00 | 1.00 | <0.01 |
| 1-1_25-1-1_5 | 0.99 | 0.99 | 0.99 | <0.01 |
| 1-1_25-1-1_10 | 0.98 | 0.98 | 0.98 | <0.01 |
| 1-1_20-1-1_100 | 1.00 | 1.00 | 1.00 | <0.01 |
| 1-1_20-1-1_5 | 0.99 | 0.99 | 0.99 | <0.01 |
| 1-1_20-1-1_10 | 0.98 | 0.98 | 0.98 | <0.01 |
| 1-1_100-1-1_5 | 0.99 | 0.99 | 0.99 | <0.01 |
| 1-1_100-1-1_10 | 0.99 | 0.99 | 0.99 | <0.01 |
| 1-1_5-1-1_10 | 0.99 | 0.99 | 0.99 | <0.01 |
| 3-2_25-3-2_20 | 1.00 | 1.00 | 1.00 | <0.01 |
| 3-2_25-3-2_100 | 1.00 | 1.00 | 1.00 | <0.01 |
| 3-2_25-3-2_5 | 1.00 | 1.00 | 1.00 | <0.01 |
| 3-2_25-3-2_10 | 0.97 | 0.97 | 0.97 | <0.01 |
| 3-2_20-3-2_100 | 1.00 | 1.00 | 1.00 | <0.01 |
| 3-2_20-3-2_5 | 1.00 | 1.00 | 1.00 | <0.01 |
| 3-2_20-3-2_10 | 0.97 | 0.97 | 0.97 | <0.01 |
| 3-2_100-3-2_5 | 1.00 | 1.00 | 1.00 | <0.01 |
| 3-2_100-3-2_10 | 0.97 | 0.97 | 0.97 | <0.01 |
| 3-2_5-3-2_10 | 0.97 | 0.98 | 0.98 | <0.01 |
| FFPE-50-FFPE-10 | 0.96 | 0.96 | 0.96 | <0.01 |
| FFPE-50-FFPE-15 | 0.96 | 0.96 | 0.96 | <0.01 |
| FFPE-50-FFPE-20 | 0.96 | 0.96 | 0.96 | <0.01 |
| FFPE-50-FFPE-250 | 0.96 | 0.96 | 0.96 | <0.01 |
| FFPE-50-FFPE-300 | 0.96 | 0.96 | 0.96 | <0.01 |
| FFPE-50-FFPE-500 | 0.96 | 0.96 | 0.96 | <0.01 |
| FFPE-10-FFPE-15 | 0.97 | 0.97 | 0.97 | <0.01 |
| FFPE-10-FFPE-20 | 0.97 | 0.97 | 0.97 | <0.01 |
| FFPE-10-FFPE-25 | 0.97 | 0.97 | 0.97 | <0.01 |
| FFPE-10-FFPE-3 | 0.97 | 0.97 | 0.97 | <0.01 |
| FFPE-10-FFPE-500 | 0.97 | 0.97 | 0.97 | <0.01 |
| FFPE-15-FFPE-20 | 0.98 | 0.98 | 0.98 | <0.01 |
| FFPE-15-FFPE-25 | 0.98 | 0.98 | 0.98 | <0.01 |
| FFPE-15-FFPE-3 | 0.98 | 0.98 | 0.98 | <0.01 |
| FFPE-15-FFPE-500 | 0.97 | 0.97 | 0.97 | <0.01 |
| FFPE-20-FFPE-25 | 0.98 | 0.98 | 0.98 | <0.01 |
| FFPE-20-FFPE-3 | 0.98 | 0.98 | 0.98 | <0.01 |
| FFPE-20-FFPE-500 | 0.97 | 0.97 | 0.97 | <0.01 |
| FFPE-25-FFPE-3 | 0.98 | 0.98 | 0.98 | <0.01 |
| FFPE-25-FFPE-500 | 0.97 | 0.98 | 0.98 | <0.01 |
| FFPE-3-FFPE-500 | 0.98 | 0.98 | 0.98 | <0.01 |
| FFPE-15-FFPE-20 | 0.98 | 0.98 | 0.98 | <0.01 |
| 199-ctDNA-207-ctDNA | 0.93 | 0.93 | 0.93 | <0.01 |
| 199-ctDNA-264-ctDNA | 0.93 | 0.93 | 0.94 | <0.01 |
| 199-ctDNA-268-ctDNA | 0.93 | 0.93 | 0.93 | <0.01 |
| 199-ctDNA-276-ctDNA | 0.94 | 0.94 | 0.94 | <0.01 |
| 199-ctDNA-288-ctDNA | 0.92 | 0.92 | 0.92 | <0.01 |
| 207-ctDNA-264-ctDNA | 0.94 | 0.94 | 0.95 | <0.01 |
| 207-ctDNA-268-ctDNA | 0.94 | 0.94 | 0.94 | <0.01 |
| 207-ctDNA-276-ctDNA | 0.94 | 0.94 | 0.95 | <0.01 |
| 207-ctDNA-288-ctDNA | 0.92 | 0.92 | 0.92 | <0.01 |
| 264-ctDNA-268-ctDNA | 0.95 | 0.95 | 0.95 | <0.01 |
| 264-ctDNA-276-ctDNA | 0.96 | 0.96 | 0.96 | <0.01 |
| 264-ctDNA-288-ctDNA | 0.93 | 0.93 | 0.93 | <0.01 |
| 268-ctDNA-276-ctDNA | 0.94 | 0.94 | 0.95 | <0.01 |
| 268-ctDNA-288-ctDNA | 0.92 | 0.92 | 0.92 | <0.01 |
| 276-ctDNA-288-ctDNA | 0.93 | 0.93 | 0.93 | <0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whalley, C.; Payne, K.; Domingo, E.; Blake, A.; Richman, S.; Brooks, J.; Batis, N.; Spruce, R.; S-CORT Consortium; Mehanna, H.; et al. Ultra-Low DNA Input into Whole Genome Methylation Assays and Detection of Oncogenic Methylation and Copy Number Variants in Circulating Tumour DNA. Epigenomes 2021, 5, 6. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes5010006
Whalley C, Payne K, Domingo E, Blake A, Richman S, Brooks J, Batis N, Spruce R, S-CORT Consortium, Mehanna H, et al. Ultra-Low DNA Input into Whole Genome Methylation Assays and Detection of Oncogenic Methylation and Copy Number Variants in Circulating Tumour DNA. Epigenomes. 2021; 5(1):6. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes5010006
Chicago/Turabian StyleWhalley, Celina, Karl Payne, Enric Domingo, Andrew Blake, Susan Richman, Jill Brooks, Nikolaos Batis, Rachel Spruce, S-CORT Consortium, Hisham Mehanna, and et al. 2021. "Ultra-Low DNA Input into Whole Genome Methylation Assays and Detection of Oncogenic Methylation and Copy Number Variants in Circulating Tumour DNA" Epigenomes 5, no. 1: 6. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes5010006