
Evolution of CG Methylation Maintenance Machinery in Plants


1
IPS, University of Bern, 3013 Bern, Switzerland
2
CIRAD, DIADE, IRD, University of Montpellier, 34393 Montpellier, France
*
Authors to whom correspondence should be addressed.
Epigenomes 2021, 5(3), 19; https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes5030019
Submission received: 19 July 2021
/
Revised: 6 September 2021
/
Accepted: 10 September 2021
/
Published: 14 September 2021
(This article belongs to the Special Issue Mechanisms of Plant Epigenome Dynamics)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Cytosine methylation is an epigenetic mark present in most eukaryotic genomes that contributes to the regulation of gene expression and the maintenance of genome stability. DNA methylation mostly occurs at CG sequences, where it is initially deposited by de novo DNA methyltransferases and propagated by maintenance DNA methyltransferases (DNMT) during DNA replication. In this review, we first summarize the mechanisms maintaining CG methylation in mammals that involve the DNA Methyltransferase 1 (DNMT1) enzyme and its cofactor, UHRF1 (Ubiquitin-like with PHD and RING Finger domain 1). We then discuss the evolutionary conservation and diversification of these two core factors in the plant kingdom and speculate on potential functions of novel homologues typically observed in land plants but not in mammals.
1. Introduction
DNA methylation is a highly conserved DNA modification, present across eukaryotes of the plant and animal kingdoms [1,2,3,4]. However, it is not universal, as certain non-plant eukaryotic genomes are devoid of DNA methylation [5,6]. DNA methylation is a covalent DNA modification affecting cytosine residues. It is typically involved in the regulation of gene expression and the silencing of transposable elements (TEs), by which it ensures genomic stability. In addition, DNA methylation is central to developmental processes such as genomic imprinting and X-chromosome inactivation [7,8].
Although DNA methylation occurs in CG and non-CG sites (CH, where H = A, T or C) in both mammals and plants, these two types of DNA methylation vary in terms of their genomic distribution and occurrence during development [3,9,10] as well as their dedicated enzymatic machinery [2]. In mammals, CG methylation is the main type of DNA methylation, and it covers the bodies of most genes and TEs [9,10,11,12]. In plants, CG methylation is detected only on a limited set of genes and TEs are covered by both CG and non-CG methylation [1,5,13,14]. CG methylation is ubiquitously detected during both plant and mammalian life cycle. However, in contrast to plants, non-CG methylation is only detected in specific mammalian tissues or cell types [15].
The establishment of a new DNA methylation pattern or de novo DNA methylation corresponds to the addition of a methyl group at the position C5 of an unmodified cytosine residue (5mC). As de novo methylation generates new DNA methylation patterns, the corresponding enzymatic machineries need to be highly regulated and precisely targeted. In mammals, de novo DNA methylation involves the de novo DNA methyltransferases 3 (DNMT3). DNMT3 enzymes methylate cytosine residues in all sequence contexts and are targeted by direct interaction with histone post-translational marks [16,17]. In contrast, de novo DNA methylation in plants involves the RNA-dependent DNA methylation (RdDM) pathway, which targets the DOMAINS REARRANGED METHYLTRANSFERASE 1 and 2 (DRM1, DRM2) to cytosines, guided by small RNA molecules [1,18,19].
After the establishment of novel DNA methylation marks, the newly created patterns must be faithfully transmitted by maintenance DNA methyltransferases during cell division [20,21]. CG methylation is maintained by two evolutionarily conserved core partners: (1) a maintenance DNA methyltransferase called DNMT1 in mammals and MET1 (DNA METHYLTRANSFERASE 1) in plants and (2) a cofactor named UHRF1 in mammals and VIM (VARIANT IN METHYLATION) in plants [2]. In mammals, the maintenance of non-CG methylation typically involves DNMT3 enzymes [15]. In plants, non-CG methylation is further divided in two classes of sequence—CHG and CHH [1,3,13,22]—and requires distinct enzymatic machineries. The maintenance of CHG sites relies on the plant-specific CHROMOMETHYLASE3 (CMT3) in cooperation with H3K9 histone methyltransferases [23,24,25,26,27,28]. Maintenance of the CHH context requires the combined action of the CHROMOMETHYLASE2 (CMT2) and the de novo methylation machinery, i.e., the RdDM pathway [26,27]. DNA methylation patterns can rapidly be lost by both passive and active DNA demethylation. Passive demethylation results from the absence of the recruitment of DNA methyltransferases during DNA replication while active DNA demethylation requires specific enzymes that differ between plants and mammals. In plants, active DNA demethylation is driven by DNA glycosylases that excise 5mC in all sequence contexts [29,30]. In mammals, ten-eleven translocation (TET) methylcytosine dioxygenases catalyze the conversion of 5mC to 5hmC (5-hydroxymethylcytosines) and further oxidation products. These modified cytosines can be retained or ultimately be replaced by naive cytosines [17,31,32].
As several recent reviews on non-CG methylation machineries and their evolution in plants are available [19,33,34,35], we focus here on the core actors of the maintenance of CG methylation. We first summarize the molecular mechanisms of the maintenance CG methylation in mammals and further discuss its conservation in plants. We then evaluate the diversification of the central actors in this process during plant evolution. Finally, we speculate on the potential roles of recently diversified factors in higher plants.
2. Molecular Mechanisms of the DNMT1/UHRF1 Pathway
During DNA replication, the parental DNA methylation pattern needs to be copied to newly synthesized daughter strands, which are devoid of DNA methylation. In mammals, multiple DNA replication-coupled methylation maintenance pathways are at play to faithfully propagate CG methylation throughout the genome and involve two main players: the DNMT1 enzyme and its cofactor UHRF1 (Figure 1) [36,37,38].
DNMT1 is the main CG maintenance DNA methyltransferase in mammals. It is recruited concomitantly to DNA replication at hemi-methylated CG sites (hemi-mCG) to methylate the cytosines on the newly synthetized DNA strands and is therefore key to maintaining symmetrical CG methylation patterns. DNMT1 typically combines a N-terminal Replication Foci Targeting Sequence (RFTS) domain responsible for its targeting to the replication foci, a CXXC Zinc-finger domain, two bromodomain-adjacent homology (BAH) domains and a large C-terminal methyltransferase (MTase) domain (Figure 2b) [39,40,41].
Biochemical studies on DNMT1 revealed a higher efficiency on hemi-methylated targets compared to unmethylated targets therefore ensuring the proper maintenance of DNA methylation [42,43]. The de novo methylation activity of DNMT1 is prevented by two auto-inhibitory regulations: (1) an intramolecular interaction between the RFTS domain with the MTase catalytic domain locks DNMT1 methyltransferase activity until needed [44,45] and (2) the binding of the CXXC domain to unmethylated cytosines prevents the DNMT1 catalytic cleft from accessing these sequences [39,40].
The recruitment of DNMT1 to replicated sites can occur through an interaction with PCNA (Proliferating cell nuclear antigen) [46]. However, PCNA-binding deficient dnmt1 mutants were still able to rescue dnmt1 ES cells suggesting that PCNA-dependent recruitment of DNMT1 is not essential in maintaining DNA methylation [46]. DNMT1 recruitment and activation at hemi-mCG sequences is intimately linked to its cofactor UHRF1. Uhrf1 loss-of-function leads to genome-wide demethylation as observed for dnmt1 knock-out [47]. UHRF1 is a multidomain protein with a ubiquitin-like (UBL) domain, two adjacent histone reader domains, a Tudor domain (TTD) followed by a PHD (Plant Homeodomain) finger that recognize, respectively, di/trimethylated lysine 9 on histone3 (H3K9me2/3) and unmodified arginine on H3 (H3R2), a su(var)3-9, enhancer-of-zeste–trithorax (SET)- and RING-associated (SRA) domain and a Really Interesting New Gene (RING) E3 ubiquitin ligase domain (Figure 1 and Figure 2a). UHRF1 can directly interact and recruit DNMT1 to hemi-mCG via its SRA methyl-binding domain (Figure 1, arrow 1) [48,49].
Interestingly, UHRF1 also provides a link between the maintenance of DNA methylation and histone or histone-like modifications. Indeed, the UHRF1 RING domain mono-ubiquitylates lysine residues in histone H3 and a H3 mimic domain present in the DNA replication factor PAF15 (PCNA-associated factor 15) (Figure 1, arrow 2 and 3). Each of these two modifications is recognized by the RFTS domain of DNMT1 and contributes to the maintenance of CG methylation [50,51]. Additionally, a methylated histone H3K9 mimic domain lying within the DNA ligase 1 (LIG1)—an enzyme that joins nicks in the lagging strand—is recognized by the UHRF1 histone reader TTD domain that ultimately favors maintenance methylation (Figure 1, arrow 5) [52,53]. The UHRF1 TTD domain also recognizes H3K9me2/me3 histone marks (Figure 1, arrow 4) [54,55] and contributes to DNMT1 recruitment through its H3K9me RFTS reader domain to heterochromatin regions [16]. Altogether the different domains of UHRF1 are thus essential to recruit and activate DNMT1 at hemi-methylated CG DNA therefore ensuring the proper maintenance of DNA methylation during DNA replication.
3. Molecular Mechanisms of the MET/VIM Pathway in Plants
In both plant and animal genomes, the presence of DNMT1/UHRF1 homologues coincides with the detection of CG methylation [5,56]. For example, Drosophila and C. elegans genomes typically lack both cytosine methylation and UHRF1 genes [3]. These observations suggest a conservation of core mechanisms involved in the maintenance of DNA methylation during evolution. DNMT1 and UHRF1 homologues have been identified in plants and are called MET1 and VIM, respectively. Similarly to mammals, mutations affecting those genes in Arabidopsis lead to a loss of CG methylation [9,57,58,59]. However, not enough is known at present in plants to conclude whether molecular mechanisms comparable to mammals (see above) are at play.
In Arabidopsis, the predicted MET proteins, including the functional MET1, share most of the domains present in mouse DNMT1 [60,61]. For example, all MET proteins in Arabidopsis have two RFTS domains (only one in DNMT1), two BAH domains and a C-terminal methyltransferase domain. The main difference is the absence of the CXXC domain in plant METs which, in DNMT1, reduces potential de novo activity [40]. Whether this activity is regulated for MET1 is currently unknown but a potential de novo activity of MET1 seems involved in de novo gene body methylation [62]. MET1 might, therefore, be more prone to induce de novo methylation than its mammalian counterpart due to the absence of the CXXC domain. Despite the presence of a conserved C-terminal methyltransferase domain in all Arabidopsis MET proteins, an enzymatic activity is only clear for MET1 and further experiments are needed to test whether MET1 paralogues have retained a functional methyltransferase activity.
In terms of domain structure, Arabidopsis VIMs show more differences than their mammalian counterpart, especially on their N-terminal part. They all have a N-terminal PHD domain and two RING domains flanking the SRA domain except VIM6 that lacks the PHD domain and C-terminal RING domain. Although each of the two RING domains of the tested VIM is sufficient to generate an E3 ligase activity [63] it is unclear whether VIM6 is still a functional enzyme. VIM proteins have retained most of the UHRF1 domains except the Tudor domain and the UBL domain localized on the N-terminal (Figure 2). The absence of Tudor domain in VIM proteins suggests a potential loss of a direct link between histone methylation and CG methylation maintenance. At present, biochemical analyses confirmed that all the Arabidopsis VIM tested have an E3 ubiquitin ligase activity [63,64] and preferentially bind to methylated CG in vitro but also to methylated CHG [25,65]. Some identified targets for ubiquitination by UHRF1 like LIG1 and histones H3 are well-conserved in plant genomes [66,67]. However, further experiments are needed to determine if these proteins are still targeted by VIM in plants.
4. Duplication of the MET and VIM Proteins in Plants
The mammalian genome (mouse and human) encodes only one DNMT1 gene but two UHRF genes (UHRF1, UHRF2). As both DNMT1 and UHRF1 are essential to maintain CG DNA methylation, dnmt1 and uhrf1 mutants suffer several defects and are embryo lethal. Interestingly, UHRF2 does not act redundantly with UHRF1 in maintaining CG methylation and uhrf2 does not complement the uhrf1 phenotype [41,68]. UHRF2 seems to be involved in cell cycle progression and possibly tumorigenesis via its binding to hydroxymethylated DNA [69,70]. This suggests that these two highly similar UHRF proteins have distinct functions in mammals.
To evaluate the degree of duplication as well as investigating the degree of conservation of MET and VIM protein in plants, we generated two phylogenetic trees (Figure 3a,b). We narrowed our analyses to a few well-annotated species representing major clades of the plant kingdom: A. lyrata, A. thaliana, C. rubella, E. salsugineum, P. trichocarpa, G. max, S. lycopersicum, Z. mays, O. sativa, S. moellendorfii, P. patens, C. reinhardtii, V. carteri, M. polymorpha, C. clementine, P. persica, B. distachyon. METs and VIMs protein sequences were obtained from the PHYTOZOME database [71] and filtered for the presence of specific PFAM protein domain: the C-5 cytosine-specific DNA methylase domain (PF00145) for MET proteins and the SET and Ring finger Associated, YDG motif protein domain (SAD_SRA, PF02182) for the VIM proteins. Details can be found in the legend of Figures S1 and S2.
The resulting trees show that both METs and VIMs clades are present in unicellular algae and have most likely be inherited from a common eukaryotic ancestor. Additionally, the SAD_SRA domain protein tree illustrates an early divergence of the VIM proteins with the other plant SAD_SRA proteins like other histone methyltransferases such as KRYPTONITE (KYP) proteins (H3K9 methyltransferases) (Figure S1). Similarly, the tree of plant proteins containing a DNA methylase domain shows a clear separation between the different classes of plant DNA methyltransferases: DRMs, CMTs and METs (Figure S2).
In our phylogenic analysis of UHRF homologues in plants, we can see that all plant genomes have at least one VIM protein (Figure 3a). In contrast to algae, which only possess one VIM protein, all other plants analyzed have at least two copies. The number of VIM homologs is particularly expanded in the Brassicaceae relative to other tested plants. While E. salsugineum only has two copies, A. lyrata has five copies and even six copies are present A. thaliana and C. rubella genomes. They are organized in three clades: a VIM2/3/4 clade, a VIM1/5 clade and a VIM6 clade (Figure 3a). In Arabidopsis, redundancy between VIM genes from different clades was observed. A reduction of CG methylation similar to the one observed in met1 mutant was obtained only in the triple vim1;vim2;vim3 mutants and not in single vim mutants [9,58,59]. RNA-seq analysis further showed an upregulation of a similar set of genes between met1 and vim1;vim2;vim3 mutants [72]. Altogether, these data suggest that VIM1, VIM2 and VIM3 proteins in Arabidopsis are the main contributors in maintaining CG methylation, potentially by recruiting MET1 as demonstrated for animal counterparts. Unexpectedly, VIM5 ubiquitin ligase activity targets MET1 for degradation rather than to recruit this enzyme to methylated sequences [64]. As no data are currently available for VIM from any other plants, it is unclear whether such novel function is present outside Arabidopsis.
Similarly to VIM proteins, at least one DNA methyltransferase homolog to DNMT1 is detected in all the selected species of algae and land plants (Figure 3b) [56]. Although only one MET copy can be detected in some species, the MET gene family has generally expanded in land plants. In Brassicaceae, three MET homologs are detected in Capsella rubella or Eutrema salsugineum and up to four in Arabidopsis thaliana (Figure 3b). Interestingly, they are divided in two separate groups: a MET1 group and a MET2/3 group, suggesting diverging function. Akin to mammals, knock-out mutants in the single MET gene in early land plants such as Marchantia or Physcomitrella, display a genomewide demethylation and pleiotropic developmental phenotypes [73,74]. In rice, mutations in each MET gene (MET1a, MET1b) lead to methylation pattern defects but only met1b generated a marked developmental defect [75,76]. This suggests that duplication of rice MET genes could have led to the emergence of a distinct non-overlapping function during development. The reason why the MET gene family expanded through evolution and why angiosperm plants are maintaining several copies of potentially functional methyltransferases remains unknown.
Altogether these phylogenetic relationships suggest that homologues of DNMT1 and UHRF1 are present in plant genomes surveyed displaying CG methylation and have been duplicated in some species during plant evolution. Much is still to be done to determine if these novel MET and VIM proteins are devoted to CG methylation maintenance or have evolved other specific functions.
5. Alternative MET/VIM Pathways during Reproduction
In flowering plants such as the model plant Arabidopsis, reproduction is initiated late in development when the flower generates organs producing the gametes after two successive phases [77]. During the first phase, called sporogenesis, a diploid germline precursor is selected to undergo meiosis and form the germ cells. During gametogenesis, the male and female germ cells undergo several mitoses to generate, respectively, two male gametes within a vegetative cell in the pollen grain and two female gametes (the egg cell and the central cell) and accessory cells in embryo embedded in the ovule. Upon fertilization, one sperm cell fuses with the egg cell, the second with the central cell to generate, respectively the embryo and the endosperm in a seed. The endosperm is a transient tissue supporting the growth of the embryo akin to the mammalian placenta. In contrast to the embryo, the endosperm does not contribute to the next generation [7,78].
Although DNA methylation patterns are mostly stable over many generations in plants [79,80,81,82], genome-wide DNA methylation profiling of reproductive cells (male meiocytes, sperm, egg and central cells) and fertilization products (embryo, endosperm) revealed highly dynamic DNA methylation patterns in reproductive tissues [83,84,85,86,87,88,89,90].
A genome-wide DNA demethylation was detected in isolated central cells, mainly at non-CG sequences in Arabidopsis [91] due to an active demethylation by the DNA demethylase DEMETER (DME) expressed in the central cell but barely in the endosperm [29,92]. A passive demethylation was proposed to contribute to this hypomethylated state as the main methyltransferase MET1 is downregulated before central cell differentiation [93,94]. However, expected CG hypomethylation in DME-independent target sequences was not observed, suggesting that the hypomethylated central cell genome only results from an active demethylation process [91]. As MET1—but not DME—remains expressed in the sperm cells [92,93], parental genomes are differentially methylated in the young endosperm. This asymmetry in DNA methylation can lead to a biased expression of genes depending on their parental origin, which corresponds to a phenomenon called imprinting also encountered in the mammalian placenta [7,95]. After fertilization, the initiated demethylation in the central cell is amplified in the endosperm at non-CG sequences but only slightly affected at CG sequences [91].
Interestingly, the three homologs of MET1 in Arabidopsis (MET2a, MET2b, MET3) are expressed in cell types where the main methyltransferase MET1 is not. MET2a and MET2b are detected in the central cell while MET3 is detected in the endosperm [94]. These three proteins could constitute alternative CG methylation maintenance pathways during sexual reproduction and potentially influence gene imprinting or seed development. Although the activity of MET1 homologs has not been assessed yet, met2a mutant has a limited reduction of methylation at selected transposons [96] and a met3 mutant called MATERNAL EFFECT EMBRYO ARREST 57 (MEE57) shows an arrest in endosperm development [97]. Expression data for MET proteins of other plants seem to suggest that duplicated MET genes also share a complementary expression pattern. In wheat, the nine MET1-like genes are members of three paralogous groups: MET2 (a, b and d), MET5 (a, b and d) and MET7 (a, b and d) [60]. Genes of MET2 group are enriched in vegetative tissues while genes of MET5 and MET7 are, respectively expressed in grains and reproductive tissues [60]. In Brassica rapa, BrMET1α is broadly expressed during plant development, while BrMET1β is only expressed in pistils [98].
On the other hand, the expression pattern of the VIM gene family in plants is very limited and restricted to Arabidopsis. The three canonical genes VIM1, 2 and 3 are expressed at least during the vegetative phase [65] and VIM5 is specifically expressed in Arabidopsis endosperm [99]. Further investigations are needed to clarify the contribution of the different MET/VIM proteins to CG methylation maintenance and understand why the MET/VIM gene family diversified during plant evolution but not in mammals.
6. Conclusions and Perspectives
Tremendous efforts have been concentrated towards the elucidation of the pathways contributing to non-CG methylation in plants and revealed that they differ from those acting in mammals. In contrast, the pathways maintaining CG methylation in plants remain poorly understood although the core players of CG methylation maintenance DNMT1/MET and UHRF1/VIM are well-conserved between mammals and plants, and that several distinct molecular mechanisms are now determined in mammals. Interestingly, the MET and VIM gene families have diversified during land plant evolution compared to the animal kingdom. The consequences of such an evolutionary trend that offer the potential for functional diversification in CG methylation pathways remain to be explored. This knowledge should also bring insights into whether the differences between the life cycles and lifestyles of animals and plants were key drivers towards the diversification of CG methylation machinery in plants.
Supplementary Materials
Supplementary Materials are available online at www.mdpi.com/article/10.3390/epigenomes5030019/s1.
Author Contributions
M.I. and L.T. wrote the manuscript, which was further edited by P.E.J.; M.I. contributed Figure 1, L.T. Figure 2 and P.E.J. Figure 3, Figures S1 and S2. All authors have read and agreed to the published version of the manuscript.
Funding
M.I. was supported by the French National Research Agency (ANR-15-CE12-0012). P.E.J. and L.T. are supported by an SNF professorship grant (no.163946) attributed to P.E.J., M.I. and P.E.J. are part of the European Cooperation in Science and Technology COST ACTION CA16212 INDEPTH.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Olivier Leblanc and Marie Mirouze for critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic reprogramming in plant and animal development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [Green Version]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Zilberman, D. Evolution of eukaryotic DNA methylation and the pursuit of safer sex. Curr. Biol. 2010, 20, R780–R785. [Google Scholar] [CrossRef] [Green Version]
- Raddatz, G.; Guzzardo, P.M.; Olova, N.; Fantappie, M.R.; Rampp, M.; Schaefer, M.; Reik, W.; Hannon, G.J.; Lyko, F. Dnmt2-dependent methylomes lack defined DNA methylation patterns. Proc. Natl. Acad. Sci. USA 2013, 110, 8627–8631. [Google Scholar] [CrossRef] [Green Version]
- Feil, R.; Berger, F. Convergent evolution of genomic imprinting in plants and mammals. Trends Genet. 2007, 23, 192–199. [Google Scholar] [CrossRef]
- Galupa, R.; Heard, E. X-Chromosome inactivation: A crossroads between chromosome architecture and gene regulation. Annu. Rev. Genet. 2018, 52, 535–566. [Google Scholar] [CrossRef]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [Green Version]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [Green Version]
- Brautigam, K.; Cronk, Q. DNA Methylation and the evolution of developmental complexity in Plants. Front. Plant Sci. 2018, 9, 1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Ecker, J.R. Non-CG Methylation in the Human Genome. Annu. Rev. Genom. Hum. Genet. 2015, 16, 55–77. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.; Fan, H.; Grimm, S.A.; Guo, Y.; Kim, J.J.; Yin, J.; Li, L.; Petell, C.J.; Tan, X.F.; Zhang, Z.M.; et al. Direct readout of heterochromatic H3K9me3 regulates DNMT1-mediated maintenance DNA methylation. Proc. Natl. Acad. Sci. USA 2020, 117, 18439–18447. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Jacobsen, S.E. Role of the arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 2002, 12, 1138–1144. [Google Scholar] [CrossRef] [Green Version]
- Matzke, M.A.; Kanno, T.; Matzke, A.J. RNA-Directed DNA methylation: The evolution of a complex epigenetic pathway in flowering plants. Annu. Rev. Plant Biol. 2015, 66, 243–267. [Google Scholar] [CrossRef]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef]
- Riggs, A.D. X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet. 1975, 14, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Kawakatsu, T.; Stuart, T.; Valdes, M.; Breakfield, N.; Schmitz, R.J.; Nery, J.R.; Urich, M.A.; Han, X.; Lister, R.; Benfey, P.N.; et al. Unique cell-type-specific patterns of DNA methylation in the root meristem. Nat. Plants 2016, 2, 16058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindroth, A.M.; Cao, X.; Jackson, J.P.; Zilberman, D.; McCallum, C.M.; Henikoff, S.; Jacobsen, S.E. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 2001, 292, 2077–2080. [Google Scholar] [CrossRef] [Green Version]
- Ebbs, M.L.; Bender, J. Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell 2006, 18, 1166–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.M.; Bostick, M.; Zhang, X.; Kraft, E.; Henderson, I.; Callis, J.; Jacobsen, S.E. The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr. Biol. 2007, 17, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Gehring, M.; Huh, J.H.; Hsieh, T.F.; Penterman, J.; Choi, Y.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 2006, 124, 495–506. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Galisteo, A.P.; Morales-Ruiz, T.; Ariza, R.R.; Roldan-Arjona, T. Arabidopsis DEMETER-LIKE proteins DML2 and DML3 are required for appropriate distribution of DNA methylation marks. Plant Mol. Biol. 2008, 67, 671–681. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Kendall, T.; Forsythe, E.S.; Dorantes-Acosta, A.; Li, S.; Caballero-Perez, J.; Chen, X.; Arteaga-Vazquez, M.; Beilstein, M.A.; Mosher, R.A. Ancient origin and recent innovations of RNA Polymerase IV and V. Mol. Biol. Evol. 2015, 32, 1788–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Xia, R.; Meyers, B.C.; Walbot, V. Evolution, functions, and mysteries of plant ARGONAUTE proteins. Curr. Opin. Plant Biol. 2015, 27, 84–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, R.J.; Lewis, Z.A.; Goll, M.G. DNA Methylation: Shared and divergent features across eukaryotes. Trends Genet. 2019, 35, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Zhang, Z.; Zou, Z.; Lv, C.; Dong, Q.; He, Q.; Yi, Y.; Li, Y.; Wang, H.; Zhu, B. Kinetics and mechanisms of mitotic inheritance of DNA methylation and their roles in aging-associated methylome deterioration. Cell Res. 2020, 30, 980–996. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.; Lu, C. The interplay between DNA and histone methylation: Molecular mechanisms and disease implications. EMBO Rep. 2021, 22, e51803. [Google Scholar] [CrossRef]
- Petryk, N.; Bultmann, S.; Bartke, T.; Defossez, P.A. Staying true to yourself: Mechanisms of DNA methylation maintenance in mammals. Nucleic Acids Res. 2021, 49, 3020–3032. [Google Scholar] [CrossRef]
- Svedruzic, Z.M.; Reich, N.O. cytosine C5 methyltransferase Dnmt1: Catalysis-dependent release of allosteric inhibition. Biochemistry 2005, 44, 9472–9485. [Google Scholar] [CrossRef]
- Song, J.; Rechkoblit, O.; Bestor, T.H.; Patel, D.J. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 2011, 331, 1036–1040. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Gao, Q.; Li, P.; Liu, X.; Jia, Y.; Wu, W.; Li, J.; Dong, S.; Koseki, H.; Wong, J. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res. 2011, 21, 1723–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowher, H.; Jeltsch, A. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: The enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J. Mol. Biol. 2001, 309, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Goyal, R.; Jeltsch, A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syeda, F.; Fagan, R.L.; Wean, M.; Avvakumov, G.V.; Walker, J.R.; Xue, S.; Dhe-Paganon, S.; Brenner, C. The replication focus targeting sequence (RFTS) domain is a DNA-competitive inhibitor of Dnmt1. J. Biol. Chem. 2011, 286, 15344–15351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, K.; Suetake, I.; Yamashita, E.; Suga, M.; Narita, H.; Nakagawa, A.; Tajima, S. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1). Proc. Natl. Acad. Sci. USA 2011, 108, 9055–9059. [Google Scholar] [CrossRef] [Green Version]
- Schermelleh, L.; Haemmer, A.; Spada, F.; Rosing, N.; Meilinger, D.; Rothbauer, U.; Cardoso, M.C.; Leonhardt, H. Dynamics of Dnmt1 interaction with the replication machinery and its role in postreplicative maintenance of DNA methylation. Nucleic Acids Res. 2007, 35, 4301–4312. [Google Scholar] [CrossRef] [Green Version]
- Sharif, J.; Muto, M.; Takebayashi, S.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Bostick, M.; Kim, J.K.; Esteve, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef] [Green Version]
- Bashtrykov, P.; Jankevicius, G.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The UHRF1 protein stimulates the activity and specificity of the maintenance DNA methyltransferase DNMT1 by an allosteric mechanism. J. Biol. Chem. 2014, 289, 4106–4115. [Google Scholar] [CrossRef] [Green Version]
- Arita, K.; Isogai, S.; Oda, T.; Unoki, M.; Sugita, K.; Sekiyama, N.; Kuwata, K.; Hamamoto, R.; Tochio, H.; Sato, M.; et al. Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc. Natl. Acad. Sci. USA 2012, 109, 12950–12955. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, A.; Mulholland, C.B.; Bultmann, S.; Kori, S.; Endo, A.; Saeki, Y.; Qin, W.; Trummer, C.; Chiba, Y.; Yokoyama, H.; et al. Two distinct modes of DNMT1 recruitment ensure stable maintenance DNA methylation. Nat. Commun. 2020, 11, 1222. [Google Scholar] [CrossRef] [Green Version]
- Ferry, L.; Fournier, A.; Tsusaka, T.; Adelmant, G.; Shimazu, T.; Matano, S.; Kirsh, O.; Amouroux, R.; Dohmae, N.; Suzuki, T.; et al. Methylation of DNA Ligase 1 by G9a/GLP recruits UHRF1 to replicating DNA and regulates DNA Methylation. Mol. Cell 2017, 67, 550–565.e5. [Google Scholar] [CrossRef] [Green Version]
- Kori, S.; Ferry, L.; Matano, S.; Jimenji, T.; Kodera, N.; Tsusaka, T.; Matsumura, R.; Oda, T.; Sato, M.; Dohmae, N.; et al. Structure of the UHRF1 Tandem Tudor domain bound to a methylated non-histone protein, LIG1, reveals rules for binding and regulation. Structure 2019, 27, 485–496.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rottach, A.; Frauer, C.; Pichler, G.; Bonapace, I.M.; Spada, F.; Leonhardt, H. The multi-domain protein Np95 connects DNA methylation and histone modification. Nucleic Acids Res. 2010, 38, 1796–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nady, N.; Lemak, A.; Walker, J.R.; Avvakumov, G.V.; Kareta, M.S.; Achour, M.; Xue, S.; Duan, S.; Allali-Hassani, A.; Zuo, X.; et al. Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J. Biol. Chem. 2011, 286, 24300–24311. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.-Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; Jeddeloh, J.A.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 2003, 163, 1109–1122. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Stroud, H.; Greenberg, M.V.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Pingault, L.; Poulet, A.; Duarte, J.; Throude, M.; Faure, S.; Pichon, J.P.; Paux, E.; Probst, A.V.; Tatout, C. Evolutionary history of Methyltransferase 1 genes in hexaploid wheat. BMC Genom. 2014, 15, 922. [Google Scholar] [CrossRef] [Green Version]
- Pei, L.; Zhang, L.; Li, J.; Shen, C.; Qiu, P.; Tu, L.; Zhang, X.; Wang, M. Tracing the origin and evolution history of methylation-related genes in plants. BMC Plant Biol. 2019, 19, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubko, E.; Gentry, M.; Kunova, A.; Meyer, P. De novo DNA methylation activity of methyltransferase 1 (MET1) partially restores body methylation in Arabidopsis thaliana. Plant J. 2012, 71, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Kraft, E.; Bostick, M.; Jacobsen, S.E.; Callis, J. ORTH/VIM proteins that regulate DNA methylation are functional ubiquitin E3 ligases. Plant J. 2008, 56, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.Q.; Zhao, J.H.; Chen, Q.; Zhang, Z.H.; Li, J.; Guo, Z.X.; Xie, Q.; Ding, S.W.; Guo, H.S. DNA geminivirus infection induces an imprinted E3 ligase gene to epigenetically activate viral gene transcription. Plant Cell 2020, 32, 3256–3272. [Google Scholar] [CrossRef]
- Woo, H.R.; Dittmer, T.A.; Richards, E.J. Three SRA-domain methylcytosine-binding proteins cooperate to maintain global CpG methylation and epigenetic silencing in Arabidopsis. PLoS Genet. 2008, 4, e1000156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunderland, P.A.; West, C.E.; Waterworth, W.M.; Bray, C.M. An evolutionarily conserved translation initiation mechanism regulates nuclear or mitochondrial targeting of DNA ligase 1 in Arabidopsis thaliana. Plant J. 2006, 47, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Ingouff, M.; Berger, F. Histone3 variants in plants. Chromosoma 2010, 119, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Pichler, G.; Wolf, P.; Schmidt, C.S.; Meilinger, D.; Schneider, K.; Frauer, C.; Fellinger, K.; Rottach, A.; Leonhardt, H. Cooperative DNA and histone binding by Uhrf2 links the two major repressive epigenetic pathways. J. Cell Biochem. 2011, 112, 2585–2593. [Google Scholar] [CrossRef] [Green Version]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.; Bauer, C.; Munzel, M.; Wagner, M.; Muller, M.; Khan, F.; et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Xiong, J.; Wang, M.; Yang, N.; Wong, J.; Zhu, B.; Xu, R.M. Structural basis for hydroxymethylcytosine recognition by the SRA domain of UHRF2. Mol. Cell 2014, 54, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Shook, M.S.; Richards, E.J. VIM proteins regulate transcription exclusively through the MET1 cytosine methylation pathway. Epigenetics 2014, 9, 980–986. [Google Scholar] [CrossRef] [Green Version]
- Yaari, R.; Noy-Malka, C.; Wiedemann, G.; Auerbach Gershovitz, N.; Reski, R.; Katz, A.; Ohad, N. DNA METHYLTRANSFERASE 1 is involved in (m)CG and (m)CCG DNA methylation and is essential for sporophyte development in Physcomitrella patens. Plant Mol. Biol. 2015, 88, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Nishihama, R.; Yamaoka, S.; Arteaga-Vazquez, M.A.; Aguilar-Cruz, A.; Grimanelli, D.; Pogorelcnik, R.; Martienssen, R.A.; Yamato, K.T.; Kohchi, T.; et al. Loss of CG Methylation in Marchantia polymorpha Causes disorganization of cell division and reveals unique DNA Methylation regulatory mechanisms of non-CG methylation. Plant Cell Physiol. 2018, 59, 2421–2431. [Google Scholar] [CrossRef]
- Yamauchi, T.; Johzuka-Hisatomi, Y.; Terada, R.; Nakamura, I.; Iida, S. The MET1b gene encoding a maintenance DNA methyltransferase is indispensable for normal development in rice. Plant Mol. Biol. 2014, 85, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Li, N.; Xu, C.; Zhong, S.; Lin, X.; Yang, J.; Zhou, T.; Yuliang, A.; Wu, Y.; Chen, Y.R.; et al. Mutation of a major CG methylase in rice causes genomewide hypomethylation, dysregulated genome expression, and seedling lethality. Proc. Natl. Acad. Sci. USA 2014, 111, 10642–10647. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, T.; Berger, F. Epigenetic reprogramming in plant sexual reproduction. Nat. Rev. Genet. 2014, 15, 613–624. [Google Scholar] [CrossRef]
- Li, J.; Berger, F. Endosperm: Food for humankind and fodder for scientific discoveries. New Phytol. 2012, 195, 290–305. [Google Scholar] [CrossRef]
- Teixeira, F.K.; Heredia, F.; Sarazin, A.; Roudier, F.; Boccara, M.; Ciaudo, C.; Cruaud, C.; Poulain, J.; Berdasco, M.; Fraga, M.F.; et al. A role for RNAi in the selective correction of DNA methylation defects. Science 2009, 323, 1600–1604. [Google Scholar] [CrossRef] [Green Version]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-Colombani, V.; Simon, M.; Agier, N.; Bulski, A.; Albuisson, J.; Heredia, F.; Audigier, P.; et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 2009, 5, e1000530. [Google Scholar] [CrossRef]
- Becker, C.; Hagmann, J.; Muller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 2011, 480, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehring, M.; Bubb, K.L.; Henikoff, S. Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 2009, 324, 1447–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, T.F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-wide demethylation of Arabidopsis endosperm. Science 2009, 324, 1451–1454. [Google Scholar] [CrossRef] [Green Version]
- Schoft, V.K.; Chumak, N.; Mosiolek, M.; Slusarz, L.; Komnenovic, V.; Brownfield, L.; Twell, D.; Kakutani, T.; Tamaru, H. Induction of RNA-directed DNA methylation upon decondensation of constitutive heterochromatin. EMBO Rep. 2009, 10, 1015–1021. [Google Scholar] [CrossRef] [Green Version]
- Calarco, J.P.; Borges, F.; Donoghue, M.T.; Van Ex, F.; Jullien, P.E.; Lopes, T.; Gardner, R.; Berger, F.; Feijo, J.A.; Becker, J.D.; et al. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 2012, 151, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Ibarra, C.A.; Feng, X.; Schoft, V.K.; Hsieh, T.F.; Uzawa, R.; Rodrigues, J.A.; Zemach, A.; Chumak, N.; Machlicova, A.; Nishimura, T.; et al. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 2012, 337, 1360–1364. [Google Scholar] [CrossRef] [Green Version]
- Pignatta, D.; Erdmann, R.M.; Scheer, E.; Picard, C.L.; Bell, G.W.; Gehring, M. Natural epigenetic polymorphisms lead to intraspecific variation in Arabidopsis gene imprinting. Elife 2014, 3, e03198. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Xia, H.; Zhang, Y.; Zhao, S.; Zhao, C.; Hou, L.; Li, C.; Li, A.; Ma, C.; Wang, X. Genome-wide high-resolution mapping of DNA methylation identifies epigenetic variation across embryo and endosperm in Maize (Zea mays). BMC Genom. 2015, 16, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.; Gao, H.; Zhang, J.; Aldridge, B.; Vickers, M.; Higgins, J.D.; Feng, X. Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat. Genet. 2018, 50, 130–137. [Google Scholar] [CrossRef]
- Park, K.; Kim, M.Y.; Vickers, M.; Park, J.S.; Hyun, Y.; Okamoto, T.; Zilberman, D.; Fischer, R.L.; Feng, X.; Choi, Y.; et al. DNA demethylation is initiated in the central cells of Arabidopsis and rice. Proc. Natl. Acad. Sci. USA 2016, 113, 15138–15143. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 2002, 110, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Jullien, P.E.; Mosquna, A.; Ingouff, M.; Sakata, T.; Ohad, N.; Berger, F. Retinoblastoma and its binding partner MSI1 control imprinting in Arabidopsis. PLoS Biol. 2008, 6, e194. [Google Scholar] [CrossRef] [PubMed]
- Jullien, P.E.; Susaki, D.; Yelagandula, R.; Higashiyama, T.; Berger, F. DNA methylation dynamics during sexual reproduction in Arabidopsis thaliana. Curr. Biol. 2012, 22, 1825–1830. [Google Scholar] [CrossRef] [Green Version]
- Gehring, M. Genomic imprinting: Insights from plants. Annu. Rev. Genet. 2013, 47, 187–208. [Google Scholar] [CrossRef]
- Quadrana, L.; Bortolini Silveira, A.; Mayhew, G.F.; LeBlanc, C.; Martienssen, R.A.; Jeddeloh, J.A.; Colot, V. The Arabidopsis thaliana mobilome and its impact at the species level. Elife 2016, 5, e15716. [Google Scholar] [CrossRef] [PubMed]
- Pagnussat, G.C.; Yu, H.J.; Ngo, Q.A.; Rajani, S.; Mayalagu, S.; Johnson, C.S.; Capron, A.; Xie, L.F.; Ye, D.; Sundaresan, V. Genetic and molecular identification of genes required for female gametophyte development and function in Arabidopsis. Development 2005, 132, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, R.; Sasaki, T.; Nishio, T. Characterization of DNA methyltransferase genes in Brassica rapa. Genes Genet. Syst. 2006, 81, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, T.F.; Shin, J.; Uzawa, R.; Silva, P.; Cohen, S.; Bauer, M.J.; Hashimoto, M.; Kirkbride, R.C.; Harada, J.J.; Zilberman, D.; et al. Regulation of imprinted gene expression in Arabidopsis endosperm. Proc. Natl. Acad. Sci. USA 2011, 108, 1755–1762. [Google Scholar] [CrossRef] [Green Version]
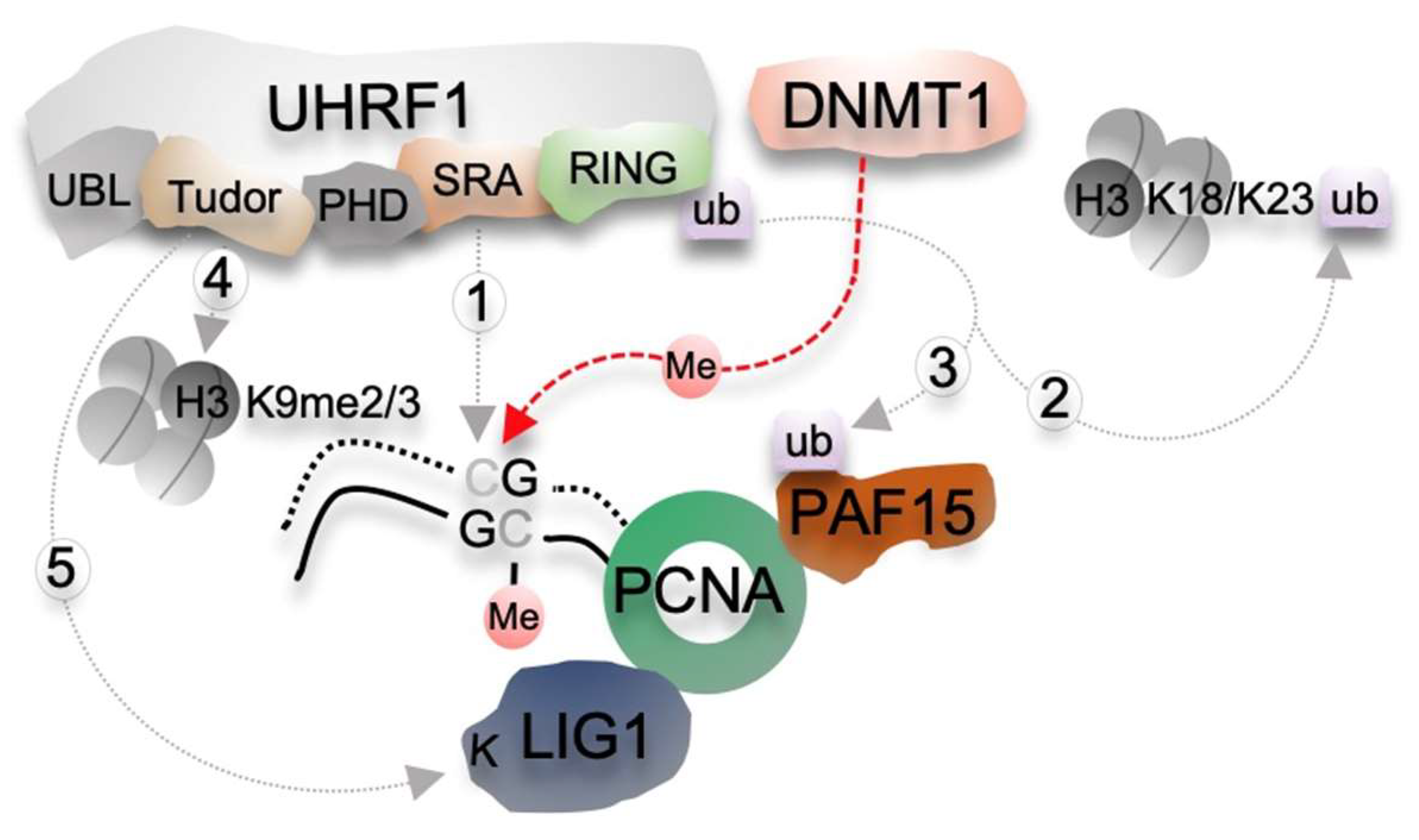
Figure 1.
Molecular mechanisms of CG methylation maintenance in mammals. CG methylation maintenance involves the maintenance DNA methyltransferase DNMT1 and distinct functional domains of UHRF1. UHRF1 is targeted to hemi-methylated DNA formed after DNA replication through its SRA domain (1) and ubiquitylates (ub) lysine (K) residues either on PAF15 (PCNA-associated factor 15) (2) or on histone H3 (3) through its E3 ubiquitin ligase RING domain. DNMT1 recognizes these ubiquitinated residues via its RFTS domain and restores symmetric CG methylation. In addition, The Tudor domain of UHRF1 binds H3K9me3 histone mark (4) and a methylated histone-like motif in DNA ligase 1 (K-LIG1) (5) enzyme that joins Okazaki fragments generated in the lagging strands. These interactions further facilitate the maintenance of CG methylation. Abbreviations: Me, methylated; PHD, Plant Homeodomain finger; RING, Really Interesting New Gene domain; SRA, su(var)3-9, enhancer-of-zeste–trithorax (SET) and RING-associated domain; TTD, Tudor domain; UBL, Ubiquitin-like domain.
Figure 1.
Molecular mechanisms of CG methylation maintenance in mammals. CG methylation maintenance involves the maintenance DNA methyltransferase DNMT1 and distinct functional domains of UHRF1. UHRF1 is targeted to hemi-methylated DNA formed after DNA replication through its SRA domain (1) and ubiquitylates (ub) lysine (K) residues either on PAF15 (PCNA-associated factor 15) (2) or on histone H3 (3) through its E3 ubiquitin ligase RING domain. DNMT1 recognizes these ubiquitinated residues via its RFTS domain and restores symmetric CG methylation. In addition, The Tudor domain of UHRF1 binds H3K9me3 histone mark (4) and a methylated histone-like motif in DNA ligase 1 (K-LIG1) (5) enzyme that joins Okazaki fragments generated in the lagging strands. These interactions further facilitate the maintenance of CG methylation. Abbreviations: Me, methylated; PHD, Plant Homeodomain finger; RING, Really Interesting New Gene domain; SRA, su(var)3-9, enhancer-of-zeste–trithorax (SET) and RING-associated domain; TTD, Tudor domain; UBL, Ubiquitin-like domain.

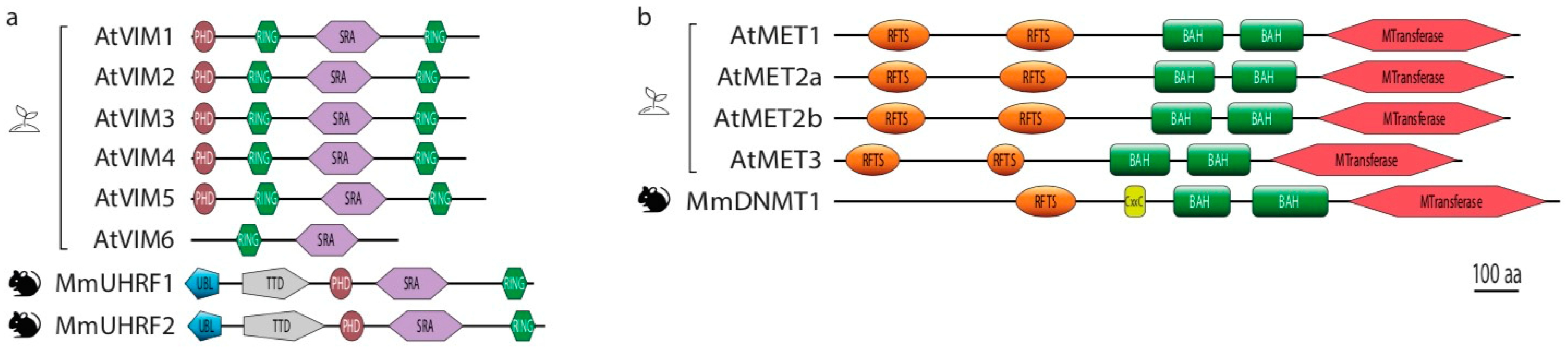
Figure 2.
Domain structures of the core players of CG methylation maintenance in plants (Arabidopsis) and mammals (mouse). (a). Domains identified in DNMT1 DNA methyltransferase and corresponding homologs MET in Arabidopsis. (b). Domains identified in DNMT1 cofactor UHRF1 and their corresponding homologs VIM in Arabidopsis. Abbreviations: BAH, Bromodomain-adjacent homology domain; CXXC, CXXC Zinc-finger domain; MTransferase, methyltransferase domain; PHD, Plant Homeodomain finger; RING, Really Interesting New Gene domain; RFTS, Replication Foci Targeting Sequence domain; SRA, su(var)3-9, enhancer-of-zeste–trithorax (SET) and RING-associated domain; Tudor, Tudor domain; UBL, Ubiquitinlike domain.
Figure 2.
Domain structures of the core players of CG methylation maintenance in plants (Arabidopsis) and mammals (mouse). (a). Domains identified in DNMT1 DNA methyltransferase and corresponding homologs MET in Arabidopsis. (b). Domains identified in DNMT1 cofactor UHRF1 and their corresponding homologs VIM in Arabidopsis. Abbreviations: BAH, Bromodomain-adjacent homology domain; CXXC, CXXC Zinc-finger domain; MTransferase, methyltransferase domain; PHD, Plant Homeodomain finger; RING, Really Interesting New Gene domain; RFTS, Replication Foci Targeting Sequence domain; SRA, su(var)3-9, enhancer-of-zeste–trithorax (SET) and RING-associated domain; Tudor, Tudor domain; UBL, Ubiquitinlike domain.
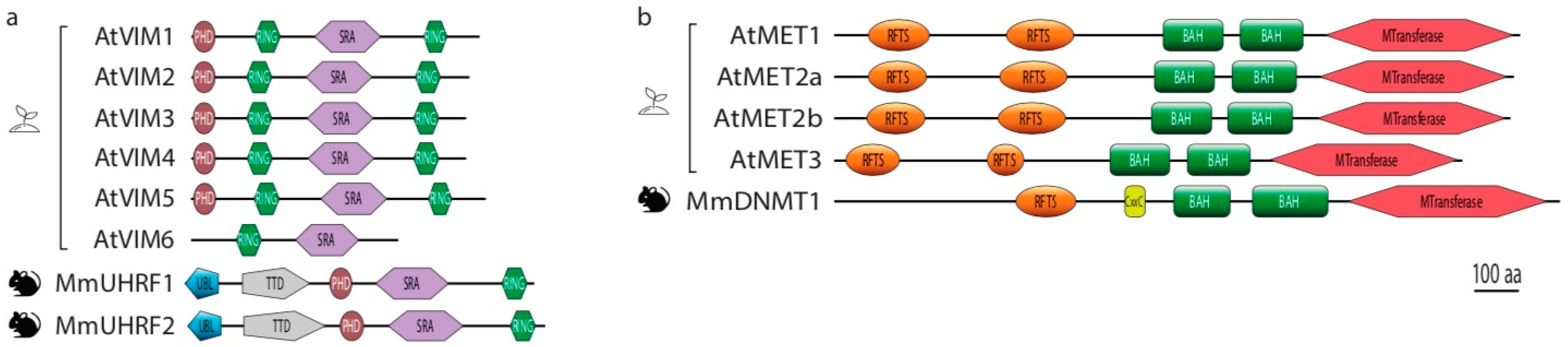
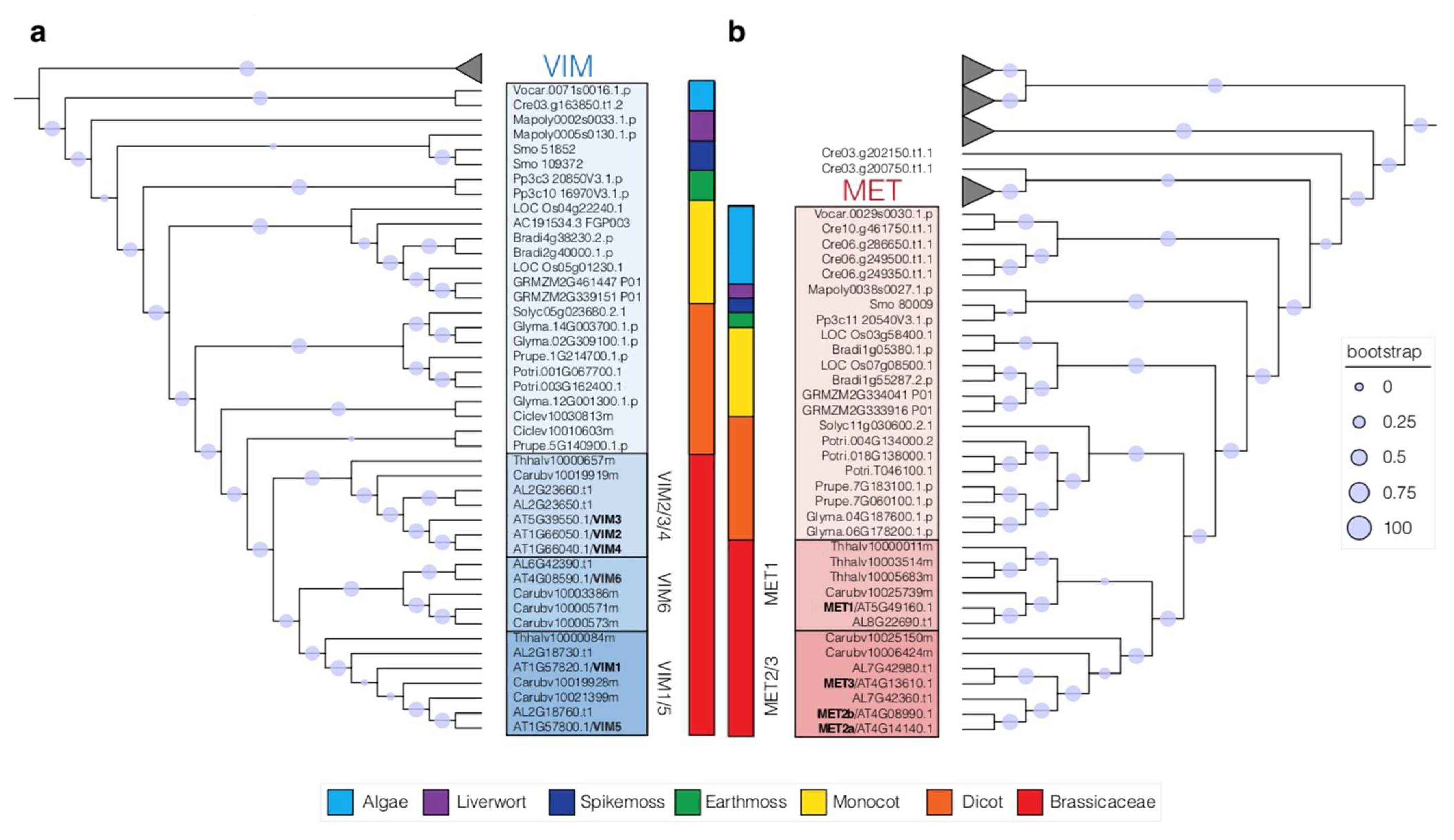
Figure 3.
Phylogenetic trees inferring phylogenetic relationships of VIM cofactors (a) and MET (b) DNA methyltransferases in green lineages. VIM proteins are shaded in blue (a) and MET proteins are shaded in red (b). Bootstrap values are represented by circles and their corresponding legends. Branches corresponding to additional clades of SAD_SRA domain proteins or DNA methylase proteins were collapsed. Genomes used for the phylogenetic analyses: Algae (C. reinhardtii, V. carteri), Livewort (M. polymorpha), Spikemoss (S. moellendorfii), Earthmoss (P. patens), Monocot (B. distachyon, O. sativa, Z. mays), Dicot (C. clementine, G. max, P. trichocarpa, P. persica, S. lycopersicum), Brassicacea (A. lyrata, A. thaliana, C. rubella, E. salsugineum). The full trees can be found in Figure S1 for the SAD_SRA domain proteins and in Figure S2 for the DNA methylase domain proteins.
Figure 3.
Phylogenetic trees inferring phylogenetic relationships of VIM cofactors (a) and MET (b) DNA methyltransferases in green lineages. VIM proteins are shaded in blue (a) and MET proteins are shaded in red (b). Bootstrap values are represented by circles and their corresponding legends. Branches corresponding to additional clades of SAD_SRA domain proteins or DNA methylase proteins were collapsed. Genomes used for the phylogenetic analyses: Algae (C. reinhardtii, V. carteri), Livewort (M. polymorpha), Spikemoss (S. moellendorfii), Earthmoss (P. patens), Monocot (B. distachyon, O. sativa, Z. mays), Dicot (C. clementine, G. max, P. trichocarpa, P. persica, S. lycopersicum), Brassicacea (A. lyrata, A. thaliana, C. rubella, E. salsugineum). The full trees can be found in Figure S1 for the SAD_SRA domain proteins and in Figure S2 for the DNA methylase domain proteins.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tirot, L.; Jullien, P.E.; Ingouff, M. Evolution of CG Methylation Maintenance Machinery in Plants. Epigenomes 2021, 5, 19. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes5030019
AMA Style
Tirot L, Jullien PE, Ingouff M. Evolution of CG Methylation Maintenance Machinery in Plants. Epigenomes. 2021; 5(3):19. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes5030019
Chicago/Turabian StyleTirot, Louis, Pauline E. Jullien, and Mathieu Ingouff. 2021. "Evolution of CG Methylation Maintenance Machinery in Plants" Epigenomes 5, no. 3: 19. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes5030019