
Fighting HIV-1 Persistence: At the Crossroads of “Shoc-K and B-Lock”
by
 , ,
, ,
Chiara Acchioni
1,†, 
Enrico Palermo
2,†,
Silvia Sandini
1,
Marta Acchioni
1,
John Hiscott
2 and
Marco Sgarbanti
1,* 1
Department of Infectious Diseases, Istituto Superiore di Sanità, Viale Regina Elena 299, 00161 Rome, Italy
2
Istituto Pasteur Italia—Cenci Bolognetti Foundation, Viale Regina Elena 291, 00161 Rome, Italy
*
Author to whom correspondence should be addressed.
†
Equal contribution.
Pathogens 2021, 10(11), 1517; https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10111517
Submission received: 6 October 2021
/
Revised: 10 November 2021
/
Accepted: 17 November 2021
/
Published: 20 November 2021
(This article belongs to the Special Issue Pathogenesis, Molecular Epidemiology, and Immune Response to Lentiviral Infections)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Despite the success of highly active antiretroviral therapy (HAART), integrated HIV-1 proviral DNA cannot be eradicated from an infected individual. HAART is not able to eliminate latently infected cells that remain invisible to the immune system. Viral sanctuaries in specific tissues and immune-privileged sites may cause residual viral replication that contributes to HIV-1 persistence. The “Shock or Kick, and Kill” approach uses latency reversing agents (LRAs) in the presence of HAART, followed by cell-killing due to viral cytopathic effects and immune-mediated clearance. Different LRAs may be required for the in vivo reactivation of HIV-1 in different CD4+ T cell reservoirs, leading to the activation of cellular transcription factors acting on the integrated proviral HIV-1 LTR. An important requirement for LRA drugs is the reactivation of viral transcription and replication without causing a generalized immune activation. Toll-like receptors, RIG-I like receptors, and STING agonists have emerged recently as a new class of LRAs that augment selective apoptosis in reactivated T lymphocytes. The challenge is to extend in vitro observations to HIV-1 positive patients. Further studies are also needed to overcome the mechanisms that protect latently infected cells from reactivation and/or elimination by the immune system. The Block and Lock alternative strategy aims at using latency promoting/inducing agents (LPAs/LIAs) to block the ability of latent proviruses to reactivate transcription in order to achieve a long term lock down of potential residual virus replication. The Shock and Kill and the Block and Lock approaches may not be only alternative to each other, but, if combined together (one after the other), or given all at once [namely “Shoc-K(kill) and B(block)-Lock”], they may represent a better approach to a functional cure.
Keywords:
HIV-1 latency; CD4+ T cell reservoirs; LRAs; LPAs; Shock and Kill; Block and Lock; immunostimulatory agents; LTR enhancer; NF-κB; p50; c-Jun; Tat1. Introduction
Infection with the human immunodeficiency virus (HIV-1), the lentivirus responsible for Acquired Immunodeficiency Syndrome (AIDS), appeared 40 years ago and continues to be a never-ending global threat for the world population. The death toll has reached an estimate of 34.7 million since the start of the pandemic, with approximately 37.6 million people living with HIV in 2020 (https://www.unaids.org/en/resources/fact-sheet, accessed on 6 October 2021). Since the mid-1990s, Highly Active Antiretroviral Therapy (HAART), a combination of antiretroviral drugs targeting different steps of the HIV-1 life cycle, has been the standard of care for patients living with HIV-1. More than 30 antiretroviral drugs have been approved by the Food and Drug Administration (FDA), including non-nucleoside reverse transcriptase inhibitors, nucleoside reverse transcriptase inhibitors, integrase inhibitors, protease inhibitors, and virus entry inhibitors (https://hivinfo.nih.gov/understanding-hiv/fact-sheets/fda-approved-hiv-medicines, accessed on 6 October 2021). While the original HAART regime was difficult to adhere to because of the complexities associated with multiple pills per day, currently, a single-tablet regimen (STRs) [1] or long-acting, injectable treatments have become available [2]. Moreover, by the end of 2020, an estimated number 27.4 million people had access to antiretroviral therapy (https://www.unaids.org/en/resources/fact-sheet, accessed on 6 October 2021).
HAART effectively decreased the viral load in HIV-1 infected patients to undetectable levels, and contributes to a partial reconstitution of the immune system, as demonstrated by an increase in the number and function of circulating CD4+ T cells [3,4]. Therefore, HAART blocks or slows the progression of AIDS, and HIV transmission, especially when started at higher CD4+ T cell counts [5]. Moreover, HAART proved effective in preventing HIV-1 infection among discordant couples [6,7], while Pre-Exposure Prophylaxis (PrEp), consisting of a combination of two drugs, provides a variable degree of protection in people at a high risk of HIV-1 infection [8].
Despite the undisputed achievement of HAART, HIV-1 patients undergoing HAART treatment experience non-AIDS-related comorbidities (NARC), especially above the age of 50 years. This particular seropositive population tends to age more rapidly than non-HIV-1 infected individuals, with the earlier onset of morbidities typically associated with aging, like non-AIDS-related malignancies [9], arterial hypertension [10], cardiovascular diseases [11], diabetes mellitus [12], kidney diseases [13], dyslipidemia [14], liver-related diseases [15], and low bone mineral density [16].
HIV-1 infection itself, despite the suppression of the viral load by HAART, is responsible for specific morbidities like HIV-related pulmonary arterial hypertension (HRPAH), caused by HIV-mediated chronic inflammation, immune activation, and HIV-mediated pulmonary vascular remodeling [17]. This scenario is worsened in the presence of co-infections with either hepatitis C or B viruses (HCV/HBV), or with mycobacterium tuberculosis (Mtb) [18]. It is presently unclear if the global COVID-19 pandemic will contribute to worsening outcomes in people living with HIV (PLWH). In this regard, immune suppression in PLWH could increase susceptibility to the SARS-CoV-2 infection; while some studies suggest that HIV patients with low CD4+ T cell counts display less severe SARS-CoV-2 infection symptoms [19], other results indicate an increased risk of death, after adjusting for age, sex, ethnicity, and comorbidities [20]. Interestingly, randomized clinical trials have demonstrated that the administration of HAART drugs, such as the protease inhibitor lopinavir together with ritonavir (LPV/r), did not improve COVID-19 outcomes or symptoms compared to the standard of care [21].
It is now widely accepted that HAART is unable to eradicate an established HIV infection [22,23,24], and therapy interruption results in a rebound in viremia and systemic infection [25,26,27,28]. A rebound after treatment interruption is due to the existence of long-lived stable HIV-1 cell reservoirs, generated early during the primary infection [29]. In these reservoirs, the integrated HIV-1 provirus is largely transcriptionally silent and not targeted by HAART. Moreover, latently infected cells express very limited levels of viral proteins, thus remaining invisible to the host immune system. The fact that large numbers of latently infected cells harbor defective proviruses indicates that only a few cells actually contribute to HIV-1 persistence [30]. Different cell types can serve as an HIV-1 reservoir, including macrophages, dendritic cells (DC), and microglial cells [31,32], but the long-lived resting CD4+ T cells are the most abundant and relevant cell type responsible for maintaining HIV-1 latency [33,34,35,36,37,38,39,40,41,42].
In the absence of a protective vaccine [43] and the failure of intensified HAART to eradicate the infection [44,45,46], novel therapeutic approaches are still required to eliminate HIV-1 provirus. Two main strategies are currently being pursued in the search for an HIV cure: eradication of the latent virus reservoirs or “sterilizing cure” and HAART-free control of HIV-1 replication, or a “functional cure”, in which the restoration of effective immune functions reduces the reservoir size, HIV-induced immune activation, and inflammation [47,48]. The sterilizing cure approach has important examples in the cases of the “Berlin patient” and the “London patient”, where HIV-1 was undetectable after allogenic bone marrow transplantations from homozygous CCR5∆32 donors [49,50]. Nevertheless, such an approach is not applicable on a large scale due to the high risk of the procedure and the paucity of donors bearing the HIV-1 resistant homozygous CCR5∆32 mutation. Therefore, the engineering of patient hematopoietic stem cells (HSC) to obtain a CCR5 deletion, or other gene modifications able to render CD4+ T cells resistant to HIV-1 infection, followed by an autologous infusion/transplantation has been proposed [51,52].
Due to the fact that HAART is most efficient at reducing the reservoir size when initiated early after HIV infection [53], a functional cure approach may come from the initiation of therapy during the very early primary stage of acute HIV infection. Post-treatment controllers (PTCs) from the VISCONTI cohort, where 14 HIV patients following HAART discontinuation maintained long lasting control of viremia, showed the potential efficacy of the approach [54]. Another approach is represented by the depletion of discrete T cell subsets carrying the integrated HIV-1 DNA based on the use of drugs with pro-oxidant activities taking advantage of metabolic imbalances occurring in latently infected cells [55,56,57,58].
To date, the “Shock or Kick, and Kill” approach has been the most studied of the “functional cure” approaches, with both promising and disappointing results [59,60]. The approach consists of the use of pharmacological stimulators known as latency reversing agents (LRAs), with the capacity to transcriptionally reactivate the latent provirus (the shock/kick phase) in the presence of HAART (to block reinfection of susceptible cells by the reactivated virus), followed by cell-killing due to viral cytopathic effects and/or immune-mediated clearance [61,62,63]. The LRA-mediated reactivation from latency is dependent on the engagement of signal transduction pathways typically involved in innate as well adaptive immune responses, culminating with the activation of cellular transcription factors and co-activators, responsible for the initiation/elongation of HIV-1 transcription, acting at the level of the 5′ long terminal repeats (LTR) of the integrated provirus. Several classes of LRAs have been employed for this purpose, including epigenetic drugs such as histone deacetylase inhibitors (HDIs), as well as protein kinase C agonists, bromodomain and extra-terminal motif (BET) protein inhibitors (BETis), activators of the Akt pathway, STAT5 sumoylation inhibitors, SMAC mimetics, and immunomodulators [64]. The alternative approach, named “Block and Lock” [65,66], is based on the rationale of blocking the reactivation of HIV-1 transcription from latently infected cells through pharmacologic inhibitors, for example, targeting Tat [67,68,69]; the final goal is a stable lock of proviral DNA due to an increase in permanent epigenetic silencing overtime, resulting in a HIV functional cure [70]. These next sections of this review will describe the current shock/kick and kill approaches and related immune pathway stimulation proposed to promote HIV-1 release from latency and subsequent cell-killing, as well as the apparently opposed Block and Lock approach.
2. Molecular Mechanisms of HIV-1 Latency Reversal
2.1. Mechanism of HIV-1 DNA Latency
Many factors contribute to HIV-1 silencing [71,72,73,74]. Retrotranscribed double stranded HIV-1 DNA can be present in infected cells in the form of linear, autointegrated (generated by the so called suicidal integration), 1- or 2-LTR circles, or proviral integrated DNA, with linear HIV-1 DNA serving as a precursor for the other forms [75,76]. Unintegrated circles are subjected to pre-integration mechanisms of latency, including epigenetic silencing through the assembly of histones with heterochromatin signatures [77]. These episomal HIV-1 genomes are unable to be replicated and mostly persist in non-dividing, or slowly dividing cells in vivo, as for example, naïve CD4+ T-cells, resting memory CD4+ T cells, and macrophages [76]. Post-integration latency represents the most important form of persistent HIV-1 DNA, able to replicate even in proliferating cells. HIV-1 mostly integrates into intronic regions of actively transcribed genes [78]. It was demonstrated that the expression of HIV-1 depends on the insertion site, with active transcription related to cellular promoter and enhancer proximity, while latent proviruses were mostly detected in genomic positions far from cellular promoters and enhancers [79]. However, when cellular promoter/enhancers are too close to the 5′ LTR, steric hindrance may occur if proviral integration is in the same transcriptional orientation as the cellular gene, resulting in the displacement of transcription factors from the LTR [80]. If the HIV-1 provirus is in an opposite orientation compared to the cellular gene, RNA-pol II from the two promoters (cellular and viral) can collide, resulting in transcriptional shut off, of both, the cellular gene and the HIV-1 genome, or at least of the genes bearing the promoter possessing a weaker activity (promoter occlusion) [80]. Another mechanism that may promote latency is the sequestration of the HIV-1 enhancer by a proximal cellular promoter, therefore failing at serving as a crucial sequence for HIV-1 LTR transcription initiation (enhancer trapping) [80]. Despite this scenario, latency can be reversed, if non-defective genomes are integrated, simply by the cooperative action of the viral transactivator Tat and cellular transcription factors and co-factors, resulting in a strong binding to the LTR and subsequent resistance to the transcription started at the cellular gene promoters. Other factors contribute to HIV-1 silencing like 3′ LTR antisense transcripts, the shortage of available cellular cofactors, suboptimal amounts of the viral trans-activator Tat, the presence of microRNA inhibiting nuclear export of viral transcripts, and chromatin modifications at the viral promoter [71]. Indeed, the epigenetic silencing of the integrated provirus also participates in shutting off viral transcription, thus contributing to HIV-1 latency. DNA hypermethylation at two CpG islands positioned close to the 5′ and 3′ of the LTR enhancer region, hypoacetylation, and methylation of histones, assembled in nucleosomes surrounding the HIV-1 LTR, and crotonylation represent hallmarks of heterochromatin at the integration site [75]. In this context, the role of chromatin environment in maintaining viral latency is widely described [74,81,82]. Structurally, chromatin is organized in nucleosomes, consisting of a section of 146 base pairs of DNA wrapped around an octamer of histone proteins, formed by two molecules of each core histone (H2A, H2B, H3 and H4) [83], whose activity can be regulated by post-translational modification (PTMs) resulting in a chromatin state transition and altered accessibility of DNA to transcription factors [83]. Interestingly, two nucleosomes, nuc-0 and nuc-1, have been found to be positioned in the 5′ LTR of HIV-1 [81], indicating a role in the modulation of viral promoter activity.
The positioning of nucleosomes within the genome is crucial for the control of gene expression and is regulated by different mechanisms. First, the activity of ATP-dependent chromatin remodeling complexes, which possess an ATPase domain to hydrolyze ATP and exploit the energy to move or remove nucleosomes [84]. Second, PTMs affecting the histone N-terminal tails, mainly acetylation and methylation, alter the interaction histones-DNA or histones-histones generating a “histone-code”, which serves as a docking site for proteins with chromatin remodeling activity [83]. Another mechanism of latency regulation is represented by the DNA CpG methylation, which, in the context of HIV-1 LTR, led to contrasting observations, with some studies supporting the thesis of an association of this modification with viral latency [85,86,87] and some confuting it [88,89].
Histone acetylation and methylation are the most described PTMs involved in the maintenance of HIV-1 latency [90]; histone acetylation results in a more accessible chromatin conformation and is catalyzed by Histone Acetyl Transferases (HATs), whereas Histone Deacetylases (HDACs) remove the acetyl groups leading to transcriptional silencing [91]. The recruitment of different HATs, such as p300/CBP, PCAF (P300 CBP-associated factor), Tip60, and GCN5 to the 5′ LTR of HIV-1 is regulated by the presence of Tat or by distinct stimuli involving the NF-κB signaling [92]. On the other hand, the activity of HDACs at the viral promoter is regulated by other cellular factors, including LSF-1/YY1, NF-κB p50/p50 homodimer, and CBF-1 [92]. In this case, several transcription factors such as NF-κB, NFAT, and c-Jun are unable to initiate transcription at the 5′ LTR, promoting viral latency [73]. In addition to HDACs, the activity of Histone Methyltransferases (HMTs) contributes to determining the transcriptional fate of latent provirus: the HMTs SUV39H1, a subunit of the polycomb repressive complex 2 (PRC2), and EZH2, catalyze the formation of H3K9me3 and H3K27me3, respectively [93,94], and are associated with maintenance of HIV-1 latency. Furthermore, the HMT G9a is involved in this process by catalyzing dimethylation of H3K9 [95].
Considering the critical contribution of PTMs in the induction or preservation of HIV-1 latency, pharmacological interventions have been pursued with the aim to perturbate the latent viral state and induce latency reversal.
An interesting feature of 0.5% of PLWH that do not require HAART to keep viral replication under control (also defined as elite controllers) is the presence of a high percentage of intact proviral HIV-1 DNA, potentially able to reactivate transcription [96,97]. Nevertheless, elite controllers’ integrated proviral DNAs express ten times less viral RNAs compared to people under HAART because of a deep state of latency, possibly due to a selective pressure exerted over time by a powerful anti-HIV-1 cellular response eliminating cells harboring HIV-1 sequences more prone to transcriptional reactivation [96,97]. Such residual deep latency is related to integration in non-protein coding areas of the host genome, such as chromosomal centromeric regions or in Krüppel-associated box domain-containing zinc finger genes on chromosome 19, characterized by a strong repression of transcription, with one patient almost considered functionally cured despite at least one intact proviral sequence [96,97]. Finally, non-intact defective proviral sequences do not contribute to a viral rebound after HAART interruption, and can suggest that a sterilizing cure is possible when they are the only traces left of an HIV-1 infection [97].
2.2. Cell Types Supporting HIV-1 Latency
CD4+ T lymphocytes represent the main target of HIV-1 latency [33,39,98]. Naïve T cells (TN), central memory T cells (TCM), transitional memory T cells (TTM), effector memory T cells (TEM), and stem cell like memory T cells (TSCM), all represent distinct stages of differentiation that are capable of becoming latently infected HIV reservoirs possessing different relevance as reservoirs due to their respective different half-life and their self-renewal ability [71]. Latently infected CD4+ T cells can also be found among other functional subsets, including regulatory T cells (Treg), Th17 memory cells, T follicular helper cells (Tfh), and resident memory CD4+ T cells (TRM) [71].
Other sources of viral rebound upon HAART treatment interruption are represented by non-T cell reservoirs like macrophages, dendritic cells (DCs) and microglial cells, while follicular dendritic cells (fDCs) can trap and transfer HIV-1 to CD4+ T cells in secondary lymphoid organs [71].
HIV-1 latency reduces to a minimum HIV-1 transcription initiation and elongation, post-transcriptional processing, and translation of viral proteins. LRA-mediated HIV-1 escape from latency involves the activation of immune-related signaling pathways, particularly those involving the interaction between CD4+ T cells and antigen presenting cells (APC). An important consideration for LRA drugs is reactivation of viral transcription and replication without causing a generalized immune activation [99] and life threatening toxicity, as is seen with anti-CD3 antibodies [100].
2.3. Cellular Transcription Factors Mediating Viral Transcription Initiation in HIV-1 Latently Infected CD4+ T Cells
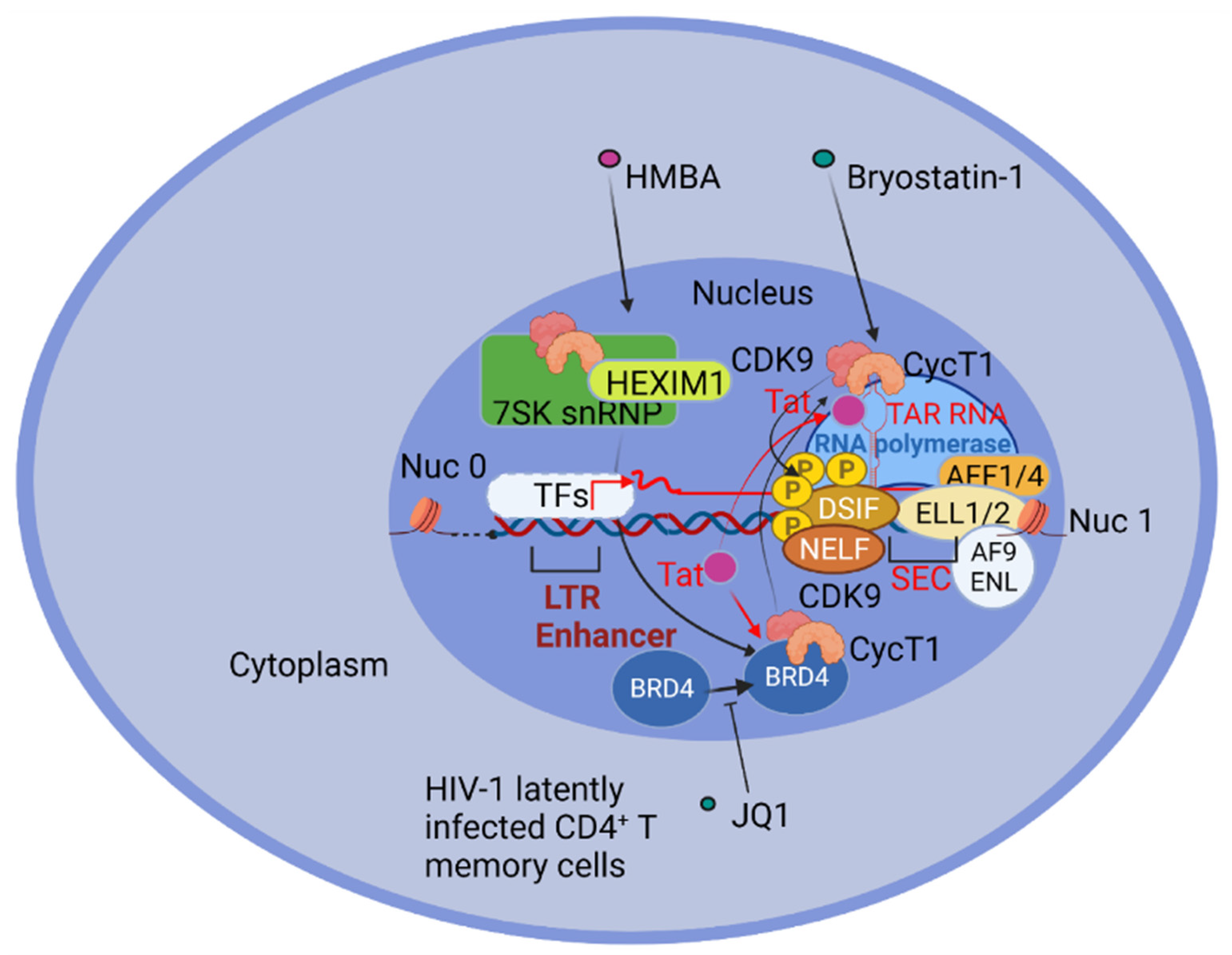
An initial HIV-1 transcription depends upon cellular transcription factors, before the viral transactivator protein Tat accumulates [80]. Once Tat becomes available, it binds the CycT1 subunit of the P-TEFb elongation complex, also formed by the cyclin-dependent kinase (CDK9), together with the transactivation-responsive element (TAR) at the 5′ end of HIV transcripts, dramatically increasing the rate of transcription [101,102]. The HIV-1 LTR promoter region contains several cellular transcription factor binding sites: (i) nuclear factor of activated T cells (NFAT); (ii) the activator protein-1 (AP1), composed of c-Jun homodimers or c-Jun/c-Fos heterodimers; (iii) the nuclear factor κB kappa-light-chain-enhancer of activated B cells (NF-κB), a family of transcription factors able to dimerize in different combinations; (iv) the interferon regulatory factors (IRFs); (v) the signal transducer and activator of transcription (STAT)-5; and several others [80]. Despite this plethora of potential sites and factors, only the enhancer binding NF-κB sites are essential for the inducible reactivation in a primary CD4+ T central memory (TCM) cell latency model [103].
The limited availability of these transcriptional regulators may determine a strong reduction in viral transcription, as in the case of viral shut-off during the transition from activated to resting memory T cells [104]. In these cells, cellular factors can be hijacked in the cytosol, synthesized and/or activated at suboptimal amounts, or sequestered into inactive protein complexes. This cellular localization of NF-κB family members, particularly the RelA transcription activator, is restricted to the cytoplasm by the binding to the IκB inhibitory proteins. Moreover, in HIV-1-infected resting cells, the inhibitory p50/p50 homodimer, lacking the transactivation domain present in the p50/RelA NF-κB complex, binds the enhancer sequence repressing transcription, additionally due to the recruitment of HDAC1 to the 5′ LTR [105]. Interferon regulatory factor 8 (IRF-8), described by our group in Jurkat [106], and by others in U1 cells [107] as an inhibitor of LTR-mediated transcription, has been recently suggested to induce HIV-1 latency in macrophages following the interactions with commensal and pathogenic bacteria [108]. Other negative regulators of HIV-1 transcription were also recently identified by using -omics and functional approaches, suggesting a role for them in latency maintenance. Such factors include the Krüppel-like C2H2 zinc finger DNA-binding proteins KLF2 and KLF3, that recognize GC-rich regions conserved in HIV-1 and HIV-2 LTRs; KLF2 and KLF3 are both able to repress HIV-1 and HIV-2 transcription in CD4+ T cells [109]. A whole-genome CRISPR knockout screen in infected T cells identified the zinc-finger protein 304 (ZNF304) as a binder of the HIV LTR around the enhancer NF-κB sites, and also acted as a promoter of latency by recruiting histone methyltransferases in association with TRIM28 [110]. Unrecognized host factors promoting HIV-1 latency, like FTSJ3, TMEM178A, NICN1, and the Integrator Complex, have been also identified by using the “Reiterative Enrichment and Authentication of CRISPRi Targets” (REACT) novel screening strategy [111].
CD4+ T cell activation triggers the phosphorylation and the subsequent ubiquitination and degradation of IκB-α, resulting in the nuclear accumulation of the heterodimer p50/RelA and the subsequent displacement of the p50/p50 homodimer from the enhancer sequence. The recruitment of histone acetyl transferases (HATs) remodels Nuc1 to a conformation that allows viral transcription to proceed [80]. Moreover, the acetylation of RelA by p300/CBP HATs at lysine 218, 221, and 310 affects NF-κB transcriptional activity; the acetylation of lysine 221 enhances DNA binding and impairs interaction with IκB-α, while the acetylation of lysine 310 promotes full NF-κB transcriptional activity [112,113].
The Ca2+/Calcineurin (CaN)-dependent transcription factor NFAT also binds the κB sites present in the enhancer region [114,115], recruits HATs [116], and promotes viral transcription [117]. Nevertheless, the role of NFAT in the reactivation of HIV-1 replication from latency with activators of the Ca2+ pathway and PKC activators has been questioned [118]. Other transcription factors, like AP1 and IRF-1, have been shown to bind the LTR enhancer in association with activated NF-κB, further promoting HIV-1 transcription [119,120]. The diverse cellular environment, corresponding to distinct differentiation stages of CD4+ T cells, seems to determine the relative importance of each of these factors in promoting HIV-1 escape from latency [121]. For example, the use of specific inhibitors in the NF-κB pathway prevents the blocking of HIV-1 reactivation in an HIV-1 latency model resembling TCM cells, stimulated by TCR engagement or phytohemagglutinin (PHA) [103].
Another important factor driving HIV-1 into the latent, transcriptionally silent state is the availability of the Tat protein; low transcription initiation rates or mutated Tat protein can also contribute to the emergence of the latent state [122]. The resting state of CD4+ T cells is crucial for HIV-1 to enter into latency, and the activation of these cells is essential to promote latency escape. Nevertheless, stochastic fluctuations of the Tat protein appear to be sufficient to determine the fate of viral transcription independently from the cellular activation state [123]. Furthermore, RNA polymerase II pausing, another process potentially impacting HIV-1 latency reversal, appears to be a stochastic phenomenon, with only a small percentage of transcripts pausing even in the absence of Tat, therefore further sustaining the hypothesis of randomly occurring events regulating HIV-1 transition from latency to active replication [124].
2.4. Elongation of Viral Transcripts during HIV-1 Latency Reversal in CD4+ T Cells
RNA Pol II is initially paused at position +30 to +60 from the initial transcriptional start site, but is able to proceed to position +60–+90 [125], where it pauses again, possibly due to a secondary structure, the pause hairpin, that forms alternatively to the TAR RNA element [126], and to the elongation repressing activity of the negative elongation factor (NELF) [127] and the DRB sensitivity inducing factor (DSIF) [128], even if the role of NELF in RNA pol II pausing is questioned [125]. The efficient elongation of viral transcripts requires the transcription elongation factor b (P-TEFb), composed of cyclin T1 and CDK9 [129,130,131]. In resting CD4+ T cells, Cyclin T1 and activated CDK9 are expressed at low levels, and accumulate upon T cell receptor engagement [132]. P-TEFb is sequestered in a ribonucleoprotein complex (RNP) comprising 7SK RNA and 7SK binding proteins as EXIM-1 or HEXIM-2, 7SK methylphosphate capping enzyme (MePCE), and La ribonucleoprotein domain family, member-7 (LARP7) [133,134,135,136]. In the absence of Tat, efficient PTEF-b recruitment and the LTR transcriptional elongation of a latent provirus are detected after TCR-mediated activation, with the release of PTEF-b from the inhibitory complex containing 7SK RNPs [137]. The released P-TEFb is recruited by Bromodomain-containing protein 4 (BRD4) to acetylated histones and also to Lys 310 acetylated RelA, to promote transcription elongation, particularly of NF-κB regulated genes [121].
Once available, Tat competes with BRD4 for P-TEFb binding, as demonstrated by using the BRD4 inhibitor JQ1 that displaces BRD4 from the LTR region of HIV-1 chromatin and increases the association of P-TEFb with Tat [138]. The Tat displacement of BRD4 from P-TEFb acts to hijack P-TEFb away from cellular promoters [139,140]. In the presence of Tat, P-TEFb interacts with AFF1/4, ELL1/2, and AF9/ENL to form the multi-subunit complex termed the “super elongation complex” (SEC). AFF1/AFF4 are molecular scaffolds, ENL/AF9 interact with Pol II on chromatin, and ELL1/2 prevents the backtracking of RNA polymerase II [141,142,143]. Therefore, Tat is able to recruit SEC to the nascent viral transcripts causing the largest subunit of RNA polymerase II to be phosphorylated at Ser2 and Ser5 by CDK9 and the multi subunit TFIIH transcription factor, respectively. CDK9 also phosphorylates negative transcription elongation factors, like NELF, causing its release, and DSIF, turning it into a positive elongating factor, thus determining the transition from the initiation to the fully elongated stage of transcription [143,144,145], (Figure 1).
3. To “Shock” and to “Kill”
3.1. Latency Reversing Agents (LRAs) Promoting HIV-1 Transcription Initiation and Elongation in CD4+ T Cells
Among different LRAs than can be used for therapeutic intervention, the PKC agonists appear to be the most efficient in driving the initiation of HIV-1 transcription and inducing escape from latency in J-Lat cell lines, latent primary CD4+ T cells, and in ex vivo T cells derived from aviremic patients [146]. Bryostatin-1, Prostratin, and Ingenol—all represent PKC agonists tested as LRAs. Nevertheless, the activation of multiple pathways using combinations of LRAs was most efficient as a reactivation protocol [71], while use of single LRA compounds resulted in poor reactivation. For example, Ingenol, together with the HDI Romidepsin, resulted in consistent HIV-1 reactivation in most subsets of latently infected CD4+ T cells from HAART-treated patients [147]. Synergistic reactivation was also obtained by the combination of Ingenol plus Romidepsin [147].
In other studies, Hexamethylenebisacetamide (HMBA), which causes the release of P-TEFb from HEXIM1 and the 7SK snRNPs [148], when combined with PKC agonists produced a synergistic re-activation, mediated by P-TEFb and NF-κB induction [149]. Similarly, combined treatment of the Ca2+ ionophore ionomycin with PKC activators resulted in a full transactivation on HIV-1 and nuclear translocation of RelA [118].
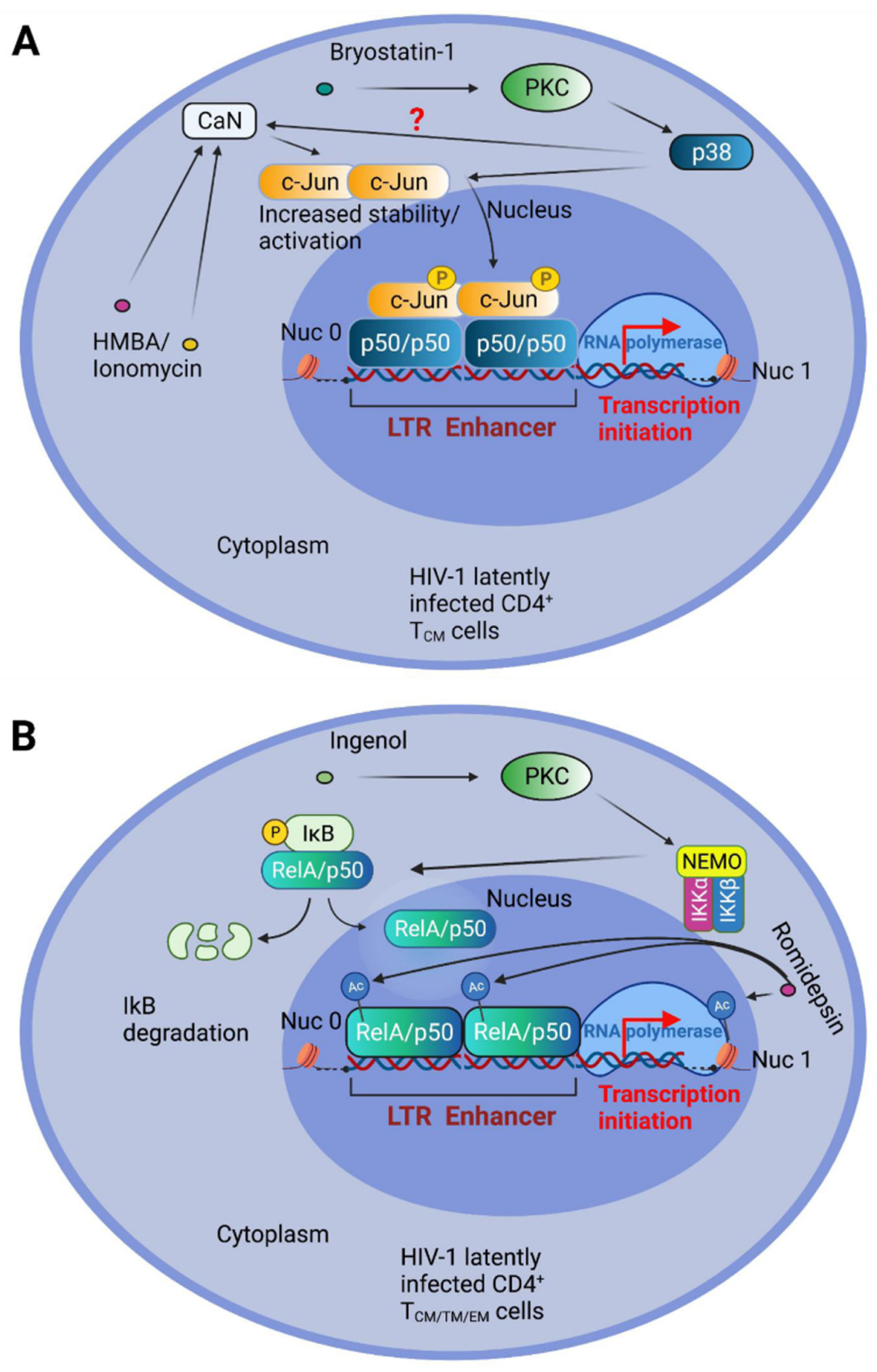
The NF-κB RelA/p50 heterodimer is crucial for the reactivation of the latent HIV-1 LTR in primary CD4+ T cells stimulated by PKC and Ca2+ pathway activators [118]. Nevertheless, prolonged NF-κB activation can lead to autoimmune, inflammatory, and malignant disorders. Using the stable expression of an NF-κB super-repressor (IκB-α 2NΔ4), our group has demonstrated that a synergistic reactivation from latency is achieved by the concomitant engagement of MAP kinase, and Ca2+/CaN, pathways in the J-Lat latency model using the PKC activator Bryostatin-1, together with the differentiating agent HMBA or the calcium ionophore Ionomycin [150]. We have also demonstrated that such combinations display the accumulation and activation of c-Jun. Indeed, CaN-mediated dephosphorylation of c-Jun at Ser-243 resulted in increased c-Jun protein stability, or nuclear accumulation when combined with phorbol 12-myristate 13-acetate (PMA) treatment [151,152]. Moreover, we demonstrated that c-Jun is crucial for latency reactivation by indirectly binding to the LTR enhancer κB sites in association with the p50 transcription factor [150] (Figure 2A). We also obtained comparable results using a primary CD4+ T central memory (TCM) cell latency model in the presence of acetylsalicylic acid (ASA), a potent inhibitor of the NF-κB kinase IKK-β. The combined treatment of Bryostatin-1 and the HDI Panobinostat effectively reversed latency in TSCM cells [147]. On the other hand, Panobinostat, when tested in combination with Ingenol, had strong antagonistic effects on viral reactivation in CD4+ T cells [147]. These different outcomes, obtained with diverse PKC agonists, may be explained by a preferential IKK-NF-κB activation capacity of Ingenol (Figure 2B).
The so-called non-canonical NF-κB pathway is triggered following TNF receptor family members, like FN14, the lymphotoxin β receptor (LTβR), and CD40, and involves the activation of the NF-κB inducing kinase (NIK), which subsequently phosphorylates the NF-κB family member p100, causing its cleavage by the proteasome to generate the p52 subunit; p52, bound to the other NF-κB family member RelB, migrates to the nucleus to activate transcription [153,154]. The inhibitors of apoptosis cIAP E3 ubiquitine ligases can determine the degradation of NIK, and therefore are inhibitors of the NF-κB non-canonical pathway [154]. c-IAPs have been implicated as negative regulators of HIV-1 transcription, and therefore as latency promoters, while small molecules able to bind and antagonize c-IAPs, mimicking the endogenous c-IAP antagonist, second mitochondrial activator of caspases (SMAC), have been proposed as a novel class of LRAs [155,156,157] capable of reversing latency in animal models [158], and also promoters of the killing of latently infected cells [159].
IL-15 stimulation has been at the center of new studies, promoting its use or the exploitation of the IL-15 activated pathway, for either the shock, or the kill phase of the Shock and Kill approach. In this respect, IL-15 super agonist N-803 was used and was effective to determine latency reversal in HAART-treated macaques and humanized mice infected with simian immunodeficiency virus (SIV) and HIV-1, respectively, when combined with the depletion of CD8+ T lymphocytes [160], also showing the role of these cells in contrasting HIV-1 latency reversal in vitro [160]. Soluble recombinant human IL-15 (rhIL-15) has also been shown to stimulate natural killer (NK) cells to clear HIV-1-infected cells following latency reversal ex vivo, obtained by using the HDi Vorinostat [161].
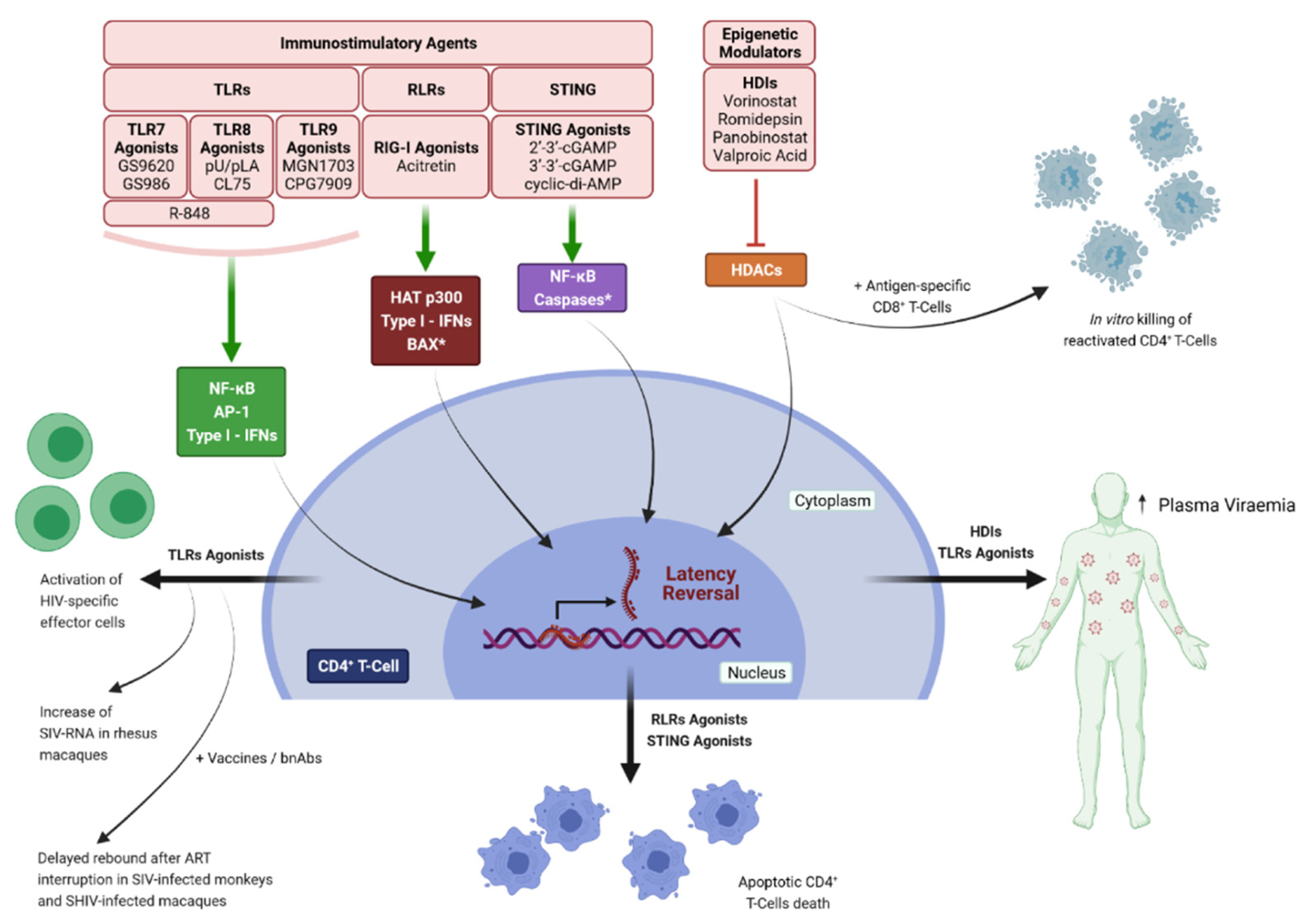
Histone deacetylase inhibitors (HDIs) represent the most studied epigenetic LRA drugs and Romidepsin, Panobinostat, Vorinostat, and Valproic acid have been extensively studied in clinical trials. Despite initial promising results, these LRAs failed in clearing or eliminating the viral reservoir [162,163,164,165,166,167,168]. In vivo administration of Vorinostat increased the levels of cell-associated unspliced (CA US) HIV-RNA, but not plasma viral loads in HIV-positive patients [163,169,170,171]. In contrast, treatment with Panobinostat or Romidepsin enhanced both CA US HIV-RNA and plasma viremia [164,165]. However, none of these treatments resulted in a reduction in the viral reservoir, indicating that latency reversal alone is not sufficient to obtain viral clearance. Some studies suggest that a combined treatment could lead to the killing of HIV reactivated cells, at least in vitro; in fact, the addition of immune effector cells such as antigen-specific CD8+ T-cells, or the recruitment of NK cells induced by HIV-specific antibodies, resulted in the elimination of infected cells after reactivation [172,173]. Despite this encouraging results, no measurable reduction in the viral reservoir has been achieved in vivo [174,175] (Figure 3).
3.2. Innate Immunity Signaling Pathways Exploited for “Shock and Kill”
To obtain the elimination of reactivated cells, new classes of LRAs with the ability to modulate the innate immune response are currently being tested. These molecules have the potential to reactivate latent provirus and stimulate the immune system to target and kill infected cells. During acute infection, pattern recognition receptors (PRR), including the Toll-like receptors (TLRs), the cGAS–STING cytosolic DNA-sensing pathway, and the retinoic acid-inducible gene I (RIG-I) pathway, recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), and subsequently induce apoptosis in virus-infected cells [176,177,178,179]. Different ex vivo and in vivo studies demonstrated that PRRs agonists reactivate latent HIV and simian immunodeficiency virus (SIV) and, at the same time, stimulate a specific anti-HIV CD8+-T and NK cell response [180,181,182,183,184].
Among the various PRRs, TLRs—mainly TLR 7, 8 and 9—have been extensively studied as LRAs. TLRs are expressed on different cells of the immune system including natural killer (NK) cells, macrophages, B cells, dendritic cells (DCs), and T cells, as well as epithelial and endothelial cells. TLR 7 and 8 senses single-stranded RNA, while TLR 9 senses unmethylated CpG-oligodeoxynucleotide-containing DNA on the endosomal membranes; this stimulation leads to the activation of several transcription factors, including NF-κB, AP-1, and interferon regulatory factors (IRFs) that induce the expression of inflammatory cytokines and type I interferons (IFNs) to protect the host from microbial infection. TLR7 agonists were reported to induce latency reversal in cells obtained from HIV-1 positive individuals and in SIV-infected rhesus macaques as well as a reduction in viral reservoir in the animals [181,182]. An increase in extracellular HIV-RNA levels in the ex vivo stimulation of PBMCs from HIV-1 infected patients undergoing ART was observed with the TLR 7 agonist GS-9620; this effect relied on type I IFN signaling activation in plasmacytoid dendritic cells and resulted in the activation of HIV-specific effector cells [182]. In an in vivo study on SIV-infected rhesus macaques on suppressive ART, it has been demonstrated that the TLR 7 agonists GS-986 and GS-9620 were able to induce the expression of SIV-RNA along with the activation of innate and adaptive immune response. This treatment also resulted in the reduction in SIV-DNA in ex vivo cell cultures. Furthermore, two out of nine animals did not show any viral rebound for more than 2 years after HAART interruption, and adoptive cell transfer from aviremic animals to naïve macaques did not result in de novo infection [181]. Unfortunately, these encouraging results were not reproduced in other studies involving non-human primates [185,186,187].
To improve the efficacy of TLR 7 agonists, different combinations with therapeutic vaccines and broadly neutralizing antibodies (bnAbs) have been tested. The combination of GS-9620 with the vaccine AD26/MVA led to innate and adaptive immune stimulation, a delay in viral rebound after ART cessation, and reduced viral DNA in lymph nodes and the peripheral blood of SIV-infected monkeys [187]. Similar results were obtained in rhesus macaques infected with Simian Human Chimeric Immunodeficiency virus (SHIV), receiving GS-986 together with two bnAbs, N6-LS and PGT121, where a modest delay in the viral rebound after ART interruption was observed [188]. However, in a double-blind, multicenter, placebo-controlled trial, no changes in plasma viral loads were observed in HIV-1 positive patients under ART treated with GS-9620, despite a consistent induction in IFN response and lymphocyte activation [189].
The ability of TLR 8 agonists to stimulate provirus transcriptional reactivation was assessed in resting CD4+ T cells isolated from the blood of nine virologically suppressed patients, in the presence or not of gamma-irradiated PBMCs and PHA [190]. The two synthetic ligands, pU/pLA and CL75, reversed latency in all patients and in three patients, respectively; in addition, these molecules were able to increase cytokine production and induce the upregulation of T cells’ surface activation markers [190]. Furthermore, the TLR7/8 agonist R-848 induced HIV-1 reactivation from latency in promonocytic cell lines [191], and in a subsequent work, it was demonstrated that the stimulation of TLR8 in monocyte-derived myeloid dendritic cells (MoMDCs) resulted in TNF-α—mediated—NF-κB—dependent reactivation of HIV, in an autocrine and paracrine manner [192]. TLR9 agonists can reactivate HIV from latently infected cells in vitro [193,194]. Among TLR9 agonists currently tested in vivo, MGN1703 was shown to increase plasma HIV-RNA levels in 6 out of 15 virologically suppressed patients on HAART [195]; this phenomenon was accompanied by the enhancement of immune responses, as indicated by the activation of plasmacytoid dendritic cells (pDCs), production of IFN-α2, upregulated transcription of IFN stimulated genes (ISGs) in CD4+ T cells, and activation of effectors cells [195]. However, despite a significant T-cell activation and an HIV-specific T-cell response, no changes in reservoir were observed in a following study [196]. A slight decrease in proviral DNA was observed in 31 HIV-positive patients treated with the pneumococcal vaccine, using the TLR9 agonist CPG7909 as an adjuvant [197]; this decrease correlated with the upregulation of HIV-specific CD8+ T cells, while no HIV-specific CD4+ T cells or HIV-specific antibodies were detected [197]. Collectively, TLRs agonists have shown promising activity as LRAs, especially when used in combination with bnAbs or as vaccine adjuvants; however, in vivo studies highlighted a lack of reproducibility in reducing the viral reservoir, suggesting that further improvements are needed.
In addition to TLRs, other PRRs have shown a potential as LRAs. The cytosolic sensors, RIG-I-like receptors (RLRs), and stimulator of interferon genes (STING) reactivate HIV from latency through p300 and NF-κB, respectively [198,199]. The RIG-I agonist Acitretin, a retinoic acid derivative, reactivated latent HIV and concomitantly induced apoptosis of cells actively transcribing HIV, in an IRF-3-dependent manner [198]. Moreover, a significant reduction in HIV-DNA levels was observed in an ex vivo stimulation with acitretin of CD4+ T cells obtained from 12 ART-suppressed HIV-positive patients, and the combination with the HDI Vorinostat further increase the magnitude of reactivation, the rate of apoptotic cell death, and the reduction in HIV-DNA [198]. However, a subsequent study failed to replicate these data in cell lines or in patient-derived samples [200]. A phase-I clinical trial to evaluate the effect of acitretin on immune cells and the expression of surface markers on CD4+ T cells has been planned (NCT03753867).
Another study demonstrated that STING agonists 2′-3′-cGAMP and cyclic-di-AMP induced SIV Gag RNA and reduced SIV Gag DNA in PBMCs from cynomolgus monkeys and demonstrated natural SIV control at 40 weeks post-infection [201]; furthermore, cyclic-di-AMP increased the frequency of SIV Gag-specific CD8+ T cells and stimulated reactivation in human PBMCs in vitro [201]. Our group also reported the ability of 2′-3′-cGAMP to reactivate HIV in an NF-κB dependent manner, and to induce the selective apoptosis of latently infected cells in vitro [199]. These effects were potentiated when cGAMP was used in combination with the HDI Resminostat. In addition, the cGAMP stimulation of a primary model of latency based on CD4+ TCM cells resulted in the selective death of HIV-1 harboring cells even in the absence of reactivation. cGAMP was also able to induce high levels of cell-associated HIV RNA in PBMCs and CD4+ T cells obtained from HIV-1 positive patients on HAART with undetectable levels of viremia [199]. Recently, 2′3′-cGAMP and 3′3′-cGAMP were found to be potent adjuvants for the induction of antigen-specific CD8+ T cells in HIV-1 Gag p24 vaccinated mice; this response was dependent on the induction of type I IFNs [202], (Figure 3).
4. To “Block” and to “Lock”
In an apparent contrast to what the Shock and Kill approach tries to achieve, some of the research focusing on a functional cure aims to definitively silence the HIV-1 integrated provirus. By targeting viral and cellular factors, playing an important role in HIV transcription and silencing, and the pathways they are involved in, it could be possible to implement the so called “Block-and-Lock” approaches by using small molecules called latency-promoting, or latency inducing, agents (LPAs/LIAs). Among these, one of the most advanced employs didehydro-cortistatin A (dCA), a Tat inhibitor that affects the function of the viral transactivator crucial in HIV-1 transcriptional elongation [203]. Moreover, the suppression of HIV-1 LTR transcription by the dCA inhibition of Tat function (“block”) has been associated with a progressive accumulation of chromatin repressive features (“lock”), specifically occurring at the HIV-1 LTR and especially involving Nuc1 [204], thus reinforcing the hypothesis that a permanent silencing of HIV-1 transcription could be achieved by a prolonged treatment with LPAs. In this respect, Triptolide, which stimulates the proteasomal degradation of Tat, could be useful in the context of such an approach [205], and a clinical trial aimed at studying the impact of Triptolide on HIV-1 reservoir in acute HIV-1 infection is on-going (https://clinicaltrials.gov/ct2/show/NCT02219672, accessed on 10 November 2021).
Inhibitors of the interaction between HIV integrase (IN) and the cellular chromatin-tethering factor LEDGF/p75 (LEDGINs) were suggested to implement a block-and-lock approach. As a consequence of treatment with LEDGINs in cell culture, the integrating viruses are repositioned out of active genes. Retargeted proviruses are predominantly in a latent state, and are refractory to reactivation by LRAs [206]. Such a class of small molecules could be employed during chronic infection to reduce the pool of reactivatable HIV-1 genomes in case residual viral replication occurs, or after treatment interruption if viral rebound takes place, coupled with HAART [207].
Curaxin CBL0100-mediated inhibition of the facilitates chromatin transcription complex (FACT), a newly identified regulator of HIV-1 transcription [208], acting as a histone chaperone capable of destabilizing the nucleosome structure and, as a consequence, promoting RNA pol II-driven transcription, represents an additional option to suppress HIV transcription [209].
An alternative Block and Lock approach involves the use of siRNA or shRNA to prevent the binding of specific transcription factors to the 5 ‘LTR, with the purpose of maintaining heterochromatin in a repressive state at the HIV-1 promoter. This approach is known as RNA-Induced Epigenetic Silencing of HIV transcription [210,211].
The Heat shock protein 90 (Hsp90) plays a role in HIV-1 reactivation due to its ability to stimulate NF-κB-dependent latency reversal mediated by PKC-activation [212]. Therefore, HSP90 inhibitors are being investigated to suppress viral transcription. In this respect, the administration of specific Hsp90 inhibitors in clinical development, like tanespimycin (17-(allylamino)-17-demethoxygeldanamycin) and AUY922, prevented viral rebound in HIV-infected humanized NOD scid IL-2Rγ−/−bone marrow-liver-thymus mice up to 11 weeks after treatment interruption [213].
The involvement of the Jak-STAT pathways in latency/latency reversal has also been hypothesized, since two FDA approved Jak inhibitors, Ruxolitinib and Tofacitinib, thanks to their anti-inflammatory effects and regulatory action on HIV transcription, are able to prevent the reactivation of HIV-1 replication by LRAs in primary CD4+ T cells [214]. Moreover, Ruxolitinib and Tofacitinib target the signal transduction pathways downstream of γ-C cytokine (IL-2, IL-7 and IL-15) receptors engagement, thus having an impact on the HIV reservoir in all memory CD4+T cell subsets in vitro and ex vivo [215]. A clinical trial is on-going to evaluate the safety and tolerability of Ruxolitinib in antiretroviral-treated HIV-infected adults (https://clinicaltrials.gov/ct2/show/NCT02475655, accessed on 10 November 2021).
It has been experimentally demonstrated in in vitro and ex vivo cell models that ZL0580, a novel and more specific inhibitor of BRD4, induced HIV-1 suppression. Moreover, in cells from aviremic patients under HAART, ZL0580 determines a delay in the rebound of viral replication after the drug combination was removed from cells [216]. These results are apparently in contrast with those obtained with the BET/BRD4 pan-inhibitor JQ1, which was instead shown to promote HIV-1 escape from latency [217]. Nevertheless, BRD4 is versatile in regulating target gene expression, and could exert positive or negative effects on HIV proviral transcription depending on its partner proteins [218]. Moreover, many lines of evidence suggest a crucial role for BRD4 in the early phase of HIV-1 transcription when Tat is not yet available [121].
Dual mTORC1 and mTORC2 inhibitors, like Torin1 and pp242, have been shown to be effective in suppressing HIV reactivation from latency by decreasing CDK9 phosphorylation induced by TCR co-stimulation in CD4+ T cells and possibly resulting in NF-κB blockade, and more potently inhibit HIV-1 compared to the more specific mTORC1 inhibitor Rapamycin [219]. Therefore, the use of such inhibitors, together with other latency promoting agents, could be useful to implement a combined Block and Lock strategy [220].
Finally, several kinase inhibitors are under investigation to evaluate their potential use for the “Block and Lock”, and have shown some effectiveness in preventing reactivation by LRAs [221].
5. Summary & Perspectives
Many hurdles remain on the road to an HIV-1 cure. The presence of viral sanctuaries in specific tissues, characterized by low penetration of HAART and immune-privileged sites, may be the source of potential residual viral replication and persistence [71].
Another challenge to latency reversal strategies is the heterogeneous nature of cell subtypes harboring a replication competent integrated HIV-1 provirus. The Shock and Kill approach to an HIV cure is further complicated by the observation that only a fraction of HIV-1 latently infected cells harbor replication competent proviruses [222], as well as the deep latency state of non-defective HIV-1 proviruses [70]. Moreover, different LRAs and combinations appear to be required for the in vivo reactivation of HIV-1 in different latent cell types [147].
The efficient reactivation of HIV-1 transcription in cell reservoirs following specific LRA treatments does not necessarily result in the production of viral proteins [147] or cell death, due to a reduction in HIV-1 specific cytotoxic T lymphocyte (CTL) responses [222]. The overexpression of the prosurvival factor B cell lymphoma 2 (BCL-2) in CD4+ T cells after co-culture with the corresponding HIV-CTL argues that BCL-2 may be a potential therapeutic target for the shock/kick and kill approach [223].
The importance of the anti-apoptotic Bcl-2 protein in promoting the prolonged survival of cells replicating HIV-1 is demonstrated by the use of Venetoclax, a Bcl-2 antagonist, that leads to HIV-1 productively infected primary T-cells selective killing [224], and also reduces the frequency of latently-infected T-cells from HAART patients when combined with anti-CD3/CD28 stimulation [225]. Furthermore, the role of the pro-survival pathway engaged by the activation of the Pi3K-AKT kinase axis for the replicating HIV-1 is evidenced following the activation of this pathway by the HIV-1 Nef and Tat proteins, thus preventing the premature apoptosis of HIV-1 infected and replicating T-cells [226]. AKT inhibitors work in HIV-1 infected macrophages, and their function in T cells has to be confirmed [226].
The identification of markers in latently infected cells would allow the specific targeting of these cells by LRAs stimulation. However, to date, the identification of T cell specific markers of latency—such as CD32a—is still debated [71].
Despite the enormous diversity of HIV-1 subtypes, most studies have been performed using subtype B virus sequences. In this regard, the virus subtype may have an impact on both the establishment of and reactivation from latency [227].
A promising strategy derives from the use of immunostimulatory agents such as LRAs to induce an adaptive response against latently infected cells. In animal models, TLR agonists induced the expression of viral RNA, and activation of both innate and adaptive immune response [181]. These effects were further enhanced when combining the agonists with therapeutic vaccines or bnAbs [187,188]. Recently, RLRs and STING agonists have emerged as a promising new class of LRAs, with the capacity to induce the latent provirus and stimulate the selective apoptosis of reactivated cells [199]. Despite a robust immune activation elicited by these compounds, it remains a challenge to extend the in vitro results to HIV-1 positive patients. Further studies are needed to identify and subvert the mechanisms that protect latently infected cells from reactivation and/or elimination by immune system in vivo.
One of the problems related to the shock phase of the Shock and Kill approach is also related to the activation, by different LRAs, of signal-transduction pathways associated with increased cell survival, such as in the case of the canonical NF-κB activation pathway, thus posing an additional challenge to the following “kill” phase. The use of SMAC mimetics may be one of the possible solutions, exerting a negative regulatory effect on the canonical pathway, while activating the non-canonical one and promoting apoptosis [154]. A down side of the non-canonical NF-κB pathway, however, is that it also modulates the effector function of differentiated T cells, potentially promoting TH17 cell-mediated neuroinflammation [228].
The Block and Lock approach is moving forward, offering an alternative to the many hurdles the “Shock and Kill” is facing. The idea of blocking LTR-mediated transcription with LPAs and to induce, as a consequence, a long-lasting repressive chromatin signature is intriguing and reminiscent of human endogenous retroviruses (HERVs) that are apparently transcriptionally silent [229]. HERVs can be stimulated by environmental stress and pathogenic infections to reactivate, thus producing envelope (ENV) proteins that have been associated with neuropsychological diseases [230]. Moreover, the HIV-1 reservoir present in the central nervous system (CNS) may be pushed by LRAs to produce viral proteins, like Tat, able to be released from infected cells and to exert a detrimental effect on neurons and in general on the CNS, arguing against the “Shock and Kill”. On the other hand, the fact that HERV lock-down may not be permanent suggests that a long lasting “lock” of the latent HIV-1 may not be easily achievable. The “Block and Lock” may work better with an HIV-1 genome integrated in transcriptionally silent chromatin regions, but poorly with proviruses close to actively transcribed genes and, on top of that, will not reduce the size of the reservoir.
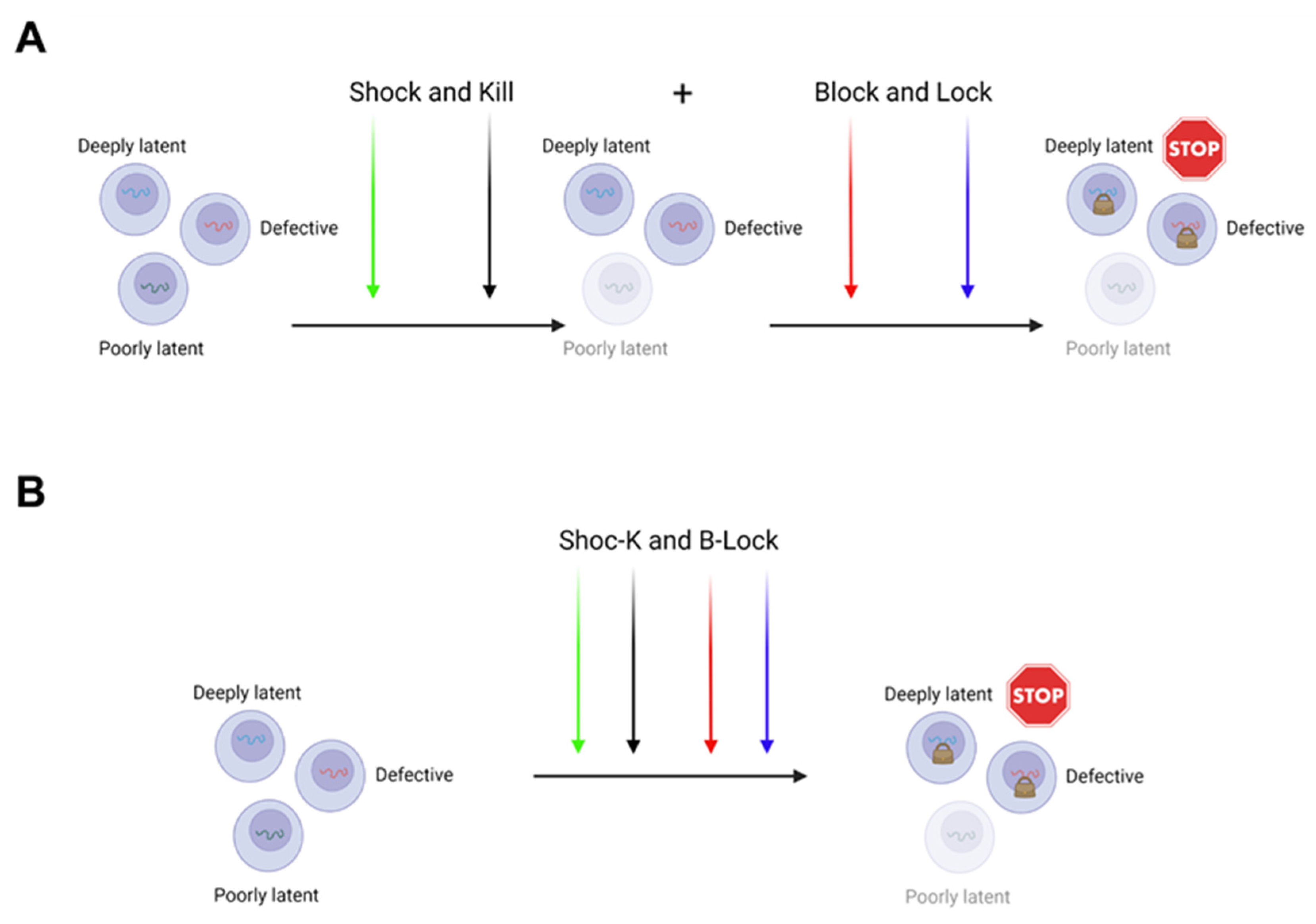
Therefore, the “Shock and Kill” and the “Block and Lock” may not only be alternative to each other, but in fact, can be merged and proposed together to achieve a functional cure (Figure 4).
Two ways can be envisioned to do so. In the first, a sequential application of both approaches (“Shock and Kill” first, followed by “Block and Lock”) could eliminate those cells where the virus is easy to be targeted for reactivation by LRAs, and subsequently a deeper silencing could be achieved by LPAs in deep seated latent residual proviruses (Figure 4A). The second possibility is to realize the “Shoc-K(Kill) and B(Block)-Lock” by using a unique all-in-one cocktail of compounds possessing tailored activities to promote both latency reversal and latency induction, resulting in a reduced and deeper silenced reservoir (Figure 4B). In this respect, we have demonstrated that the combination of activators of MAPKs and Calcineurin, like Bryostatin-1 and HMBA, used with acetylsalicylic acid (ASA) at a high dosage, the latter capable to strongly inhibit canonical NF-κB activation, is able to determine a synergistic reactivation of HIV-1 transcription from the JLat 10.6 cell line and a strong reactivation of transcription in a CD4+ TCM cell primary latency model [150]. Considering the role of the canonical NF-κB pathway activation in cell survival, its inhibition by Aspirin from one side could inhibit the CD8+ T cell-mediated clearance of cells that reactivate viral replication, due to the Aspirin inhibition of T-cell activation and proliferation; on the other hand, it could promote apoptosis in those cells, where activated c-Jun/p50 complex is enough to drive efficient transcription initiation, by binding to the LTR enhancer, also avoiding a potentially toxic effect related to a separated and sequential application of both approaches.
While intriguing, this theoretical approach awaits experimental testing and an experimental setting able to test the efficacy of each step of the “Shoc-K and B-Lock”.
Author Contributions
C.A. and E.P. equally contributed in conceiving and writing the manuscript, and in drawing figures. S.S. and M.A. participated in the writing of the manuscript, and gave suggestions for the writing of the manuscript. J.H. provided supervision, wrote and edited the manuscript. M.S. conceived and wrote the manuscript, provided supervision, contributed in drawing figures and edited the manuscript. C.A. and M.S. formulated the “Shoc-K and B-Lock” idea. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
All figures were created with BioRender.com (accessed on 6 October 2021 and 10 November 2021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Truong, W.R.; Schafer, J.J.; Short, W.R. Once-Daily, Single-Tablet Regimens For the Treatment of HIV-1 Infection. Pharm. Ther. 2015, 40, 44–55. [Google Scholar]
- Overton, E.T.; Richmond, G.; Rizzardini, G.; Jaeger, H.; Orrell, C.; Nagimova, F.; Bredeek, F.; Garcia Deltoro, M.; Swindells, S.; Andrade-Villanueva, J.F.; et al. Long-acting cabotegravir and rilpivirine dosed every 2 months in adults with HIV-1 infection (ATLAS-2M), 48-week results: A randomised, multicentre, open-label, phase 3b, non-inferiority study. Lancet 2021, 396, 1994–2005. [Google Scholar] [CrossRef]
- O’Brien, W.A.; Hartigan, P.M.; Daar, E.S.; Simberkoff, M.S.; Hamilton, J.D. Changes in plasma HIV RNA levels and CD4+ lymphocyte counts predict both response to antiretroviral therapy and therapeutic failure. VA Cooperative Study Group on AIDS. Ann. Intern. Med. 1997, 126, 939–945. [Google Scholar] [CrossRef]
- Pakker, N.G.; Notermans, D.W.; de Boer, R.J.; Roos, M.T.; de Wolf, F.; Hill, A.; Leonard, J.M.; Danner, S.A.; Miedema, F.; Schellekens, P.T. Biphasic kinetics of peripheral blood T cells after triple combination therapy in HIV-1 infection: A composite of redistribution and proliferation. Nat. Med. 1998, 4, 208–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Primers 2015, 1, 15035. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Del Romero, J.; Hernando, V.; Marincovich, B.; Garcia, S.; Rodriguez, C. Effectiveness of highly active antiretroviral therapy in reducing heterosexual transmission of HIV. J. Acquir. Immune Defic. Syndr. 2005, 40, 96–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, S.J.; Makumbi, F.; Nakigozi, G.; Kagaayi, J.; Gray, R.H.; Wawer, M.; Quinn, T.C.; Serwadda, D. HIV-1 transmission among HIV-1 discordant couples before and after the introduction of antiretroviral therapy. Aids 2011, 25, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.D.; Herbst, J.H.; Koppenhaver, R.T.; Smith, D.K. Antiretroviral Prophylaxis for Sexual and Injection Drug Use Acquisition of HIV. Am. J. Prev. Med. 2013, 44, S63–S69. [Google Scholar] [CrossRef]
- Spano, J.P.; Poizot-Martin, I.; Costagliola, D.; Boue, F.; Rosmorduc, O.; Lavole, A.; Choquet, S.; Heudel, P.E.; Leblond, V.; Gabarre, J.; et al. Non-AIDS-related malignancies: Expert consensus review and practical applications from the multidisciplinary CANCERVIH Working Group. Ann. Oncol. 2016, 27, 397–408. [Google Scholar] [CrossRef]
- Davis, K.; Perez-Guzman, P.; Hoyer, A.; Brinks, R.; Gregg, E.; Althoff, K.N.; Justice, A.C.; Reiss, P.; Gregson, S.; Smit, M. Association between HIV infection and hypertension: A global systematic review and meta-analysis of cross-sectional studies. BMC Med. 2021, 19, 105. [Google Scholar] [CrossRef]
- Alonso, A.; Barnes, A.E.; Guest, J.L.; Shah, A.; Shao, I.Y.; Marconi, V. HIV Infection and Incidence of Cardiovascular Diseases: An Analysis of a Large Healthcare Database. J. Am. Heart Assoc. 2019, 8, e012241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florescu, D.; Kotler, D.P. Insulin resistance, glucose intolerance and diabetes mellitus in HIV-infected patients. Antivir. Ther. 2007, 12, 149–162. [Google Scholar]
- Alfano, G.; Cappelli, G.; Fontana, F.; Di Lullo, L.; Di Iorio, B.; Bellasi, A.; Guaraldi, G. Kidney Disease in HIV Infection. J. Clin. Med. 2019, 8, 1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, J. Dyslipidemia and lipid management in HIV-infected patients. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 144–147. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, M.B.; Sterling, R.K. Mechanisms of liver disease in patients infected with HIV. BMJ Open Gastroenterol. 2017, 4, e000166. [Google Scholar] [CrossRef] [Green Version]
- Hileman, C.O.; Eckard, A.R.; McComsey, G.A. Bone loss in HIV: A contemporary review. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 446–451. [Google Scholar] [CrossRef] [Green Version]
- Almodovar, S.; Cicalini, S.; Petrosillo, N.; Flores, S.C. Pulmonary hypertension associated with HIV infection: Pulmonary vascular disease: The global perspective. Chest 2010, 137, 6S–12S. [Google Scholar] [CrossRef] [Green Version]
- McArdle, A.J.; Turkova, A.; Cunnington, A.J. When do co-infections matter? Curr. Opin. Infect. Dis. 2018, 31, 209–215. [Google Scholar] [CrossRef] [PubMed]
- SeyedAlinaghi, S.; Karimi, A.; MohsseniPour, M.; Barzegary, A.; Mirghaderi, S.P.; Fakhfouri, A.; Saeidi, S.; Razi, A.; Mojdeganlou, H.; Tantuoyir, M.M.; et al. The clinical outcomes of COVID-19 in HIV-positive patients: A systematic review of current evidence. Immun. Inflamm. Dis. 2021, 9, 1160–1185. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Yap, S.F.; Ngeow, Y.F.; Lye, M.S. COVID-19 in People Living with HIV: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2021, 18, 3554. [Google Scholar] [CrossRef] [PubMed]
- Group, R.C. Lopinavir-ritonavir in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2020, 396, 1345–1352. [Google Scholar] [CrossRef]
- Gulick, R.M.; Mellors, J.W.; Havlir, D.; Eron, J.J.; Gonzalez, C.; McMahon, D.; Richman, D.D.; Valentine, F.T.; Jonas, L.; Meibohm, A.; et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N. Engl. J. Med. 1997, 337, 734–739. [Google Scholar] [CrossRef] [Green Version]
- Hammer, S.M.; Squires, K.E.; Hughes, M.D.; Grimes, J.M.; Demeter, L.M.; Currier, J.S.; Eron, J.J., Jr.; Feinberg, J.E.; Balfour, H.H., Jr.; Deyton, L.R.; et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N. Engl. J. Med. 1997, 337, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Perelson, A.S.; Essunger, P.; Cao, Y.; Vesanen, M.; Hurley, A.; Saksela, K.; Markowitz, M.; Ho, D.D. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 1997, 387, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Fauci, A.S. HIV reservoirs: Pathogenesis and obstacles to viral eradication and cure. AIDS 2012, 26, 1261–1268. [Google Scholar] [CrossRef]
- Davey, R.T., Jr.; Bhat, N.; Yoder, C.; Chun, T.W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, F.; Plana, M.; Vidal, C.; Cruceta, A.; O’Brien, W.A.; Pantaleo, G.; Pumarola, T.; Gallart, T.; Miro, J.M.; Gatell, J.M. Dynamics of viral load rebound and immunological changes after stopping effective antiretroviral therapy. AIDS 1999, 13, F79–F86. [Google Scholar] [CrossRef]
- Mata, R.C.; Viciana, P.; de Alarcon, A.; Lopez-Cortes, L.F.; Gomez-Vera, J.; Trastoy, M.; Cisneros, J.M. Discontinuation of antiretroviral therapy in patients with chronic HIV infection: Clinical, virologic, and immunologic consequences. AIDS Patient Care STDS 2005, 19, 550–562. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Abbas, W.; Herbein, G. HIV-1 latency in monocytes/macrophages. Viruses 2014, 6, 1837–1860. [Google Scholar] [CrossRef] [Green Version]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [Green Version]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef]
- Chun, T.W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.K.; Hezareh, M.; Gunthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Hill, B.J.; Ambrozak, D.R.; Price, D.A.; Guenaga, F.J.; Casazza, J.P.; Kuruppu, J.; Yazdani, J.; Migueles, S.A.; Connors, M.; et al. T-cell subsets that harbor human immunodeficiency virus (HIV) in vivo: Implications for HIV pathogenesis. J. Virol. 2004, 78, 1160–1168. [Google Scholar] [CrossRef] [Green Version]
- Shan, L.; Deng, K.; Gao, H.; Xing, S.; Capoferri, A.A.; Durand, C.M.; Rabi, S.A.; Laird, G.M.; Kim, M.; Hosmane, N.N.; et al. Transcriptional Reprogramming during Effector-to-Memory Transition Renders CD4(+) T Cells Permissive for Latent HIV-1 Infection. Immunity 2017, 47, 766–775. [Google Scholar] [CrossRef] [Green Version]
- Chun, T.W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef]
- Venanzi Rullo, E.; Cannon, L.; Pinzone, M.R.; Ceccarelli, M.; Nunnari, G.; O’Doherty, U. Genetic Evidence That Naive T Cells Can Contribute Significantly to the Human Immunodeficiency Virus Intact Reservoir: Time to Re-evaluate Their Role. Clin. Infect. Dis. 2019, 69, 2236–2237. [Google Scholar] [CrossRef]
- Zerbato, J.M.; McMahon, D.K.; Sobolewski, M.D.; Mellors, J.W.; Sluis-Cremer, N. Naive CD4+ T Cells Harbor a Large Inducible Reservoir of Latent, Replication-competent Human Immunodeficiency Virus Type 1. Clin. Infect. Dis. 2019, 69, 1919–1925. [Google Scholar] [CrossRef]
- Lopez Angel, C.J.; Tomaras, G.D. Bringing the path toward an HIV-1 vaccine into focus. PLoS Pathog. 2020, 16, e1008663. [Google Scholar] [CrossRef] [PubMed]
- Dinoso, J.B.; Kim, S.Y.; Wiegand, A.M.; Palmer, S.E.; Gange, S.J.; Cranmer, L.; O’Shea, A.; Callender, M.; Spivak, A.; Brennan, T.; et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 9403–9408. [Google Scholar] [CrossRef] [Green Version]
- Ananworanich, J.; Chomont, N.; Fletcher, J.L.; Pinyakorn, S.; Schuetz, A.; Sereti, I.; Rerknimitr, R.; Dewar, R.; Kroon, E.; Vandergeeten, C.; et al. Markers of HIV reservoir size and immune activation after treatment in acute HIV infection with and without raltegravir and maraviroc intensification. J. Virus Erad. 2015, 1, 116–122. [Google Scholar] [CrossRef]
- Markowitz, M.; Evering, T.H.; Garmon, D.; Caskey, M.; La Mar, M.; Rodriguez, K.; Sahi, V.; Palmer, S.; Prada, N.; Mohri, H. A randomized open-label study of 3- versus 5-drug combination antiretroviral therapy in newly HIV-1-infected individuals. J. Acquir. Immune Defic. Syndr. 2014, 66, 140–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International AIDS Society Scientific Working Group on HIV Cure; Deeks, S.G.; Autran, B.; Berkhout, B.; Benkirane, M.; Cairns, S.; Chomont, N.; Chun, T.W.; Churchill, M.; Di Mascio, M.; et al. Towards an HIV cure: A global scientific strategy. Nat. Rev. Immunol. 2012, 12, 607–614. [Google Scholar] [CrossRef]
- Trono, D.; Van Lint, C.; Rouzioux, C.; Verdin, E.; Barre-Sinoussi, F.; Chun, T.W.; Chomont, N. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science 2010, 329, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.K.; Peppa, D.; Hill, A.L.; Galvez, C.; Salgado, M.; Pace, M.; McCoy, L.E.; Griffith, S.A.; Thornhill, J.; Alrubayyi, A.; et al. Evidence for HIV-1 cure after CCR5Delta32/Delta32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: A case report. Lancet HIV 2020, 7, e340–e347. [Google Scholar] [CrossRef] [Green Version]
- Beard, B.C.; Trobridge, G.D.; Ironside, C.; McCune, J.S.; Adair, J.E.; Kiem, H.P. Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. J. Clin. Investig. 2010, 120, 2345–2354. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Holmes, M.C. Engineering hematopoietic stem cells toward a functional cure of human immunodeficiency virus infection. Cytotherapy 2016, 18, 1370–1381. [Google Scholar] [CrossRef]
- Hocqueloux, L.; Avettand-Fenoel, V.; Jacquot, S.; Prazuck, T.; Legac, E.; Melard, A.; Niang, M.; Mille, C.; Le Moal, G.; Viard, J.P.; et al. Long-term antiretroviral therapy initiated during primary HIV-1 infection is key to achieving both low HIV reservoirs and normal T cell counts. J. Antimicrob. Chemother. 2013, 68, 1169–1178. [Google Scholar] [CrossRef] [Green Version]
- Saez-Cirion, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef]
- Lewis, M.G.; DaFonseca, S.; Chomont, N.; Palamara, A.T.; Tardugno, M.; Mai, A.; Collins, M.; Wagner, W.L.; Yalley-Ogunro, J.; Greenhouse, J.; et al. Gold drug auranofin restricts the viral reservoir in the monkey AIDS model and induces containment of viral load following ART suspension. AIDS 2011, 25, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Shytaj, I.L.; Chirullo, B.; Wagner, W.; Ferrari, M.G.; Sgarbanti, R.; Corte, A.D.; LaBranche, C.; Lopalco, L.; Palamara, A.T.; Montefiori, D.; et al. Investigational treatment suspension and enhanced cell-mediated immunity at rebound followed by drug-free remission of simian AIDS. Retrovirology 2013, 10, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, R.S.; Shytaj, I.L.; Giron, L.B.; Obermaier, B.; Della Libera, E., Jr.; Galinskas, J.; Dias, D.; Hunter, J.; Janini, M.; Gosuen, G.; et al. Potential impact of the antirheumatic agent auranofin on proviral HIV-1 DNA in individuals under intensified antiretroviral therapy: Results from a randomised clinical trial. Int. J. Antimicrob. Agents 2019, 54, 592–600. [Google Scholar] [CrossRef]
- Shytaj, I.L.; Procopio, F.A.; Tarek, M.; Carlon-Andres, I.; Tang, H.Y.; Goldman, A.R.; Munshi, M.; Kumar Pal, V.; Forcato, M.; Sreeram, S.; et al. Glycolysis downregulation is a hallmark of HIV-1 latency and sensitizes infected cells to oxidative stress. EMBO Mol. Med. 2021, 13, e13901. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef] [Green Version]
- Deeks, S.G.; Lewin, S.R.; Ross, A.L.; Ananworanich, J.; Benkirane, M.; Cannon, P.; Chomont, N.; Douek, D.; Lifson, J.D.; Lo, Y.R.; et al. International AIDS Society global scientific strategy: Towards an HIV cure 2016. Nat. Med. 2016, 22, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Deng, K.; Pertea, M.; Rongvaux, A.; Wang, L.; Durand, C.M.; Ghiaur, G.; Lai, J.; McHugh, H.L.; Hao, H.; Zhang, H.; et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature 2015, 517, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Cartwright, E.K.; Spicer, L.; Smith, S.A.; Lee, D.; Fast, R.; Paganini, S.; Lawson, B.O.; Nega, M.; Easley, K.; Schmitz, J.E.; et al. CD8(+) Lymphocytes Are Required for Maintaining Viral Suppression in SIV-Infected Macaques Treated with Short-Term Antiretroviral Therapy. Immunity 2016, 45, 656–668. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.B.; Walker, B.D. HIV-specific CD8(+) T cells and HIV eradication. J. Clin. Investig. 2016, 126, 455–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moranguinho, I.; Valente, S.T. Block-And-Lock: New Horizons for a Cure for HIV-1. Viruses 2020, 12, 1443. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Van Driessche, B.; Van Lint, C. HIV Latency: Should We Shock or Lock? Trends Immunol. 2017, 38, 217–228. [Google Scholar] [CrossRef]
- Mousseau, G.; Valente, S. Strategies to Block HIV Transcription: Focus on Small Molecule Tat Inhibitors. Biology 2012, 1, 668–697. [Google Scholar] [CrossRef] [Green Version]
- Mediouni, S.; Chinthalapudi, K.; Ekka, M.K.; Usui, I.; Jablonski, J.A.; Clementz, M.A.; Mousseau, G.; Nowak, J.; Macherla, V.R.; Beverage, J.N.; et al. Didehydro-Cortistatin A Inhibits HIV-1 by Specifically Binding to the Unstructured Basic Region of Tat. mBio 2019, 10, e02662-18. [Google Scholar] [CrossRef] [Green Version]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. mBio 2015, 6, e00465. [Google Scholar] [CrossRef] [Green Version]
- Elsheikh, M.M.; Tang, Y.; Li, D.; Jiang, G. Deep latency: A new insight into a functional HIV cure. EBioMedicine 2019, 45, 624–629. [Google Scholar] [CrossRef] [Green Version]
- Ait-Ammar, A.; Kula, A.; Darcis, G.; Verdikt, R.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Rohr, O.; Van Lint, C. Current Status of Latency Reversing Agents Facing the Heterogeneity of HIV-1 Cellular and Tissue Reservoirs. Front. Microbiol. 2019, 10, 3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donahue, D.A.; Wainberg, M.A. Cellular and molecular mechanisms involved in the establishment of HIV-1 latency. Retrovirology 2013, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Battistini, A.; Sgarbanti, M. HIV-1 latency: An update of molecular mechanisms and therapeutic strategies. Viruses 2014, 6, 1715–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Lint, C.; Bouchat, S.; Marcello, A. HIV-1 transcription and latency: An update. Retrovirology 2013, 10, 67. [Google Scholar] [CrossRef] [Green Version]
- Verdikt, R.; Hernalsteens, O.; Van Lint, C. Epigenetic Mechanisms of HIV-1 Persistence. Vaccines 2021, 9, 514. [Google Scholar] [CrossRef] [PubMed]
- Sloan, R.D.; Wainberg, M.A. The role of unintegrated DNA in HIV infection. Retrovirology 2011, 8, 52. [Google Scholar] [CrossRef] [Green Version]
- Kantor, B.; Ma, H.; Webster-Cyriaque, J.; Monahan, P.E.; Kafri, T. Epigenetic activation of unintegrated HIV-1 genomes by gut-associated short chain fatty acids and its implications for HIV infection. Proc. Natl. Acad. Sci. USA 2009, 106, 18786–18791. [Google Scholar] [CrossRef] [Green Version]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.C.; Martinez, J.P.; Zorita, E.; Meyerhans, A.; Filion, G.J. Position effects influence HIV latency reversal. Nat. Struct. Mol. Biol. 2017, 24, 47–54. [Google Scholar] [CrossRef]
- Colin, L.; Van Lint, C. Molecular control of HIV-1 postintegration latency: Implications for the development of new therapeutic strategies. Retrovirology 2009, 6, 111. [Google Scholar] [CrossRef] [Green Version]
- Verdin, E.; Paras, P., Jr.; Van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar] [CrossRef]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef] [Green Version]
- Bowman, G.D.; Poirier, M.G. Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev. 2015, 115, 2274–2295. [Google Scholar] [CrossRef] [Green Version]
- Ho, L.; Crabtree, G.R. Chromatin remodelling during development. Nature 2010, 463, 474–484. [Google Scholar] [CrossRef]
- Blazkova, J.; Trejbalova, K.; Gondois-Rey, F.; Halfon, P.; Philibert, P.; Guiguen, A.; Verdin, E.; Olive, D.; Van Lint, C.; Hejnar, J.; et al. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 2009, 5, e1000554. [Google Scholar] [CrossRef] [Green Version]
- Kauder, S.E.; Bosque, A.; Lindqvist, A.; Planelles, V.; Verdin, E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009, 5, e1000495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trejbalova, K.; Kovarova, D.; Blazkova, J.; Machala, L.; Jilich, D.; Weber, J.; Kucerova, D.; Vencalek, O.; Hirsch, I.; Hejnar, J. Development of 5’ LTR DNA methylation of latent HIV-1 provirus in cell line models and in long-term-infected individuals. Clin. Epigenetics 2016, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Blazkova, J.; Murray, D.; Justement, J.S.; Funk, E.K.; Nelson, A.K.; Moir, S.; Chun, T.W.; Fauci, A.S. Paucity of HIV DNA methylation in latently infected, resting CD4+ T cells from infected individuals receiving antiretroviral therapy. J. Virol. 2012, 86, 5390–5392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boltz, V.F.; Ceriani, C.; Rausch, J.W.; Shao, W.; Bale, M.J.; Keele, B.F.; Hoh, R.; Milush, J.M.; Deeks, S.G.; Maldarelli, F.; et al. CpG Methylation Profiles of HIV-1 Pro-Viral DNA in Individuals on ART. Viruses 2021, 13, 799. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.W.; Margolis, D.M. Chromatin Regulation and the Histone Code in HIV Latency. Yale J. Biol. Med. 2017, 90, 229–243. [Google Scholar] [PubMed]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Lusic, M.; Giacca, M. Regulation of HIV-1 latency by chromatin structure and nuclear architecture. J. Mol. Biol. 2015, 427, 688–694. [Google Scholar] [CrossRef]
- du Chene, I.; Basyuk, E.; Lin, Y.L.; Triboulet, R.; Knezevich, A.; Chable-Bessia, C.; Mettling, C.; Baillat, V.; Reynes, J.; Corbeau, P.; et al. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007, 26, 424–435. [Google Scholar] [CrossRef]
- Friedman, J.; Cho, W.K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef] [Green Version]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomont, N. HIV enters deep sleep in people who naturally control the virus. Nature 2020, 585, 190–191. [Google Scholar] [CrossRef]
- Jiang, C.; Lian, X.; Gao, C.; Sun, X.; Einkauf, K.B.; Chevalier, J.M.; Chen, S.M.Y.; Hua, S.; Rhee, B.; Chang, K.; et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature 2020, 585, 261–267. [Google Scholar] [CrossRef]
- Chavez, L.; Calvanese, V.; Verdin, E. HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells. PLoS Pathog. 2015, 11, e1004955. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.R.; Pollack, R.A.; Capoferri, A.; Ambinder, R.F.; Durand, C.M.; Siliciano, R.F. Rapamycin-mediated mTOR inhibition uncouples HIV-1 latency reversal from cytokine-associated toxicity. J. Clin. Investig. 2017, 127, 651–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, J.M.; Jurriaans, S.; van Praag, R.M.; Blaak, H.; van Rij, R.; Schellekens, P.T.; ten Berge, I.J.; Yong, S.L.; Fox, C.H.; Roos, M.T.; et al. Immuno-activation with anti-CD3 and recombinant human IL-2 in HIV-1-infected patients on potent antiretroviral therapy. AIDS 1999, 13, 2405–2410. [Google Scholar] [CrossRef] [PubMed]
- Dingwall, C.; Ernberg, I.; Gait, M.J.; Green, S.M.; Heaphy, S.; Karn, J.; Lowe, A.D.; Singh, M.; Skinner, M.A. HIV-1 tat protein stimulates transcription by binding to a U-rich bulge in the stem of the TAR RNA structure. EMBO J. 1990, 9, 4145–4153. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D.; Grdina, T.A.; Bogerd, H.P.; Cullen, B.R. Recruitment of cyclin T1/P-TEFb to an HIV type 1 long terminal repeat promoter proximal RNA target is both necessary and sufficient for full activation of transcription. Proc. Natl. Acad. Sci. USA 1999, 96, 7791–7796. [Google Scholar] [CrossRef] [Green Version]
- Bosque, A.; Planelles, V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 2009, 113, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, M. Molecular mechanisms for the regulation of HIV replication, persistence and latency. AIDS 1997, 11 (Suppl. A), S25–S33. [Google Scholar]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Ruiz-Jarabo, C.M.; Verdin, E.; Greene, W.C. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006, 25, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Sgarbanti, M.; Borsetti, A.; Moscufo, N.; Bellocchi, M.C.; Ridolfi, B.; Nappi, F.; Marsili, G.; Marziali, G.; Coccia, E.M.; Ensoli, B.; et al. Modulation of human immunodeficiency virus 1 replication by interferon regulatory factors. J. Exp. Med. 2002, 195, 1359–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munier, S.; Delcroix-Genete, D.; Carthagena, L.; Gumez, A.; Hazan, U. Characterization of two candidate genes, NCoA3 and IRF8, potentially involved in the control of HIV-1 latency. Retrovirology 2005, 2, 73. [Google Scholar] [CrossRef] [PubMed]
- Viglianti, G.A.; Planelles, V.; Hanley, T.M. Interactions with Commensal and Pathogenic Bacteria Induce HIV-1 Latency in Macrophages through Altered Transcription Factor Recruitment to the LTR. J. Virol. 2021, 95, e02141-20. [Google Scholar] [CrossRef]
- Pedro, K.D.; Agosto, L.M.; Sewell, J.A.; Eberenz, K.A.; He, X.; Fuxman Bass, J.I.; Henderson, A.J. A functional screen identifies transcriptional networks that regulate HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2012835118. [Google Scholar] [CrossRef]
- Krasnopolsky, S.; Kuzmina, A.; Taube, R. Genome-wide CRISPR knockout screen identifies ZNF304 as a silencer of HIV transcription that promotes viral latency. PLoS Pathog. 2020, 16, e1008834. [Google Scholar] [CrossRef]
- Li, Z.; Hajian, C.; Greene, W.C. Identification of unrecognized host factors promoting HIV-1 latency. PLoS Pathog. 2020, 16, e1009055. [Google Scholar] [CrossRef]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.F.; Mu, Y.; Greene, W.C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, S.; Chen, B.K.; Kaneshima, H.; Nolan, G.P. Host control of HIV-1 parasitism in T cells by the nuclear factor of activated T cells. Cell 1998, 95, 595–604. [Google Scholar] [CrossRef] [Green Version]
- Cron, R.Q.; Bartz, S.R.; Clausell, A.; Bort, S.J.; Klebanoff, S.J.; Lewis, D.B. NFAT1 enhances HIV-1 gene expression in primary human CD4 T cells. Clin. Immunol. 2000, 94, 179–191. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, C.; Rao, A. Nuclear factor of activated T cells (NFAT)-dependent transactivation regulated by the coactivators p300/CREB-binding protein (CBP). J. Exp. Med. 1998, 187, 2031–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, S.; Su, L.; Amano, M.; Timmerman, L.A.; Kaneshima, H.; Nolan, G.P. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity 1997, 6, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.K.; Bhattacharyya, D.; Lassen, K.G.; Ruelas, D.; Greene, W.C. Calcium/calcineurin synergizes with prostratin to promote NF-kappaB dependent activation of latent HIV. PLoS ONE 2013, 8, e77749. [Google Scholar] [CrossRef]
- Yang, X.; Chen, Y.; Gabuzda, D. ERK MAP kinase links cytokine signals to activation of latent HIV-1 infection by stimulating a cooperative interaction of AP-1 and NF-kappaB. J. Biol. Chem. 1999, 274, 27981–27988. [Google Scholar] [CrossRef] [Green Version]
- Sgarbanti, M.; Remoli, A.L.; Marsili, G.; Ridolfi, B.; Borsetti, A.; Perrotti, E.; Orsatti, R.; Ilari, R.; Sernicola, L.; Stellacci, E.; et al. IRF-1 is required for full NF-kappaB transcriptional activity at the human immunodeficiency virus type 1 long terminal repeat enhancer. J. Virol. 2008, 82, 3632–3641. [Google Scholar] [CrossRef] [Green Version]
- Remoli, A.L.; Marsili, G.; Battistini, A.; Sgarbanti, M. The development of immune-modulating compounds to disrupt HIV latency. Cytokine Growth Factor Rev. 2012, 23, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Karn, J. The molecular biology of HIV latency: Breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin HIV AIDS 2011, 6, 4–11. [Google Scholar] [CrossRef]
- Razooky, B.S.; Pai, A.; Aull, K.; Rouzine, I.M.; Weinberger, L.S. A hardwired HIV latency program. Cell 2015, 160, 990–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tantale, K.; Garcia-Oliver, E.; Robert, M.C.; L’Hostis, A.; Yang, Y.; Tsanov, N.; Topno, R.; Gostan, T.; Kozulic-Pirher, A.; Basu-Shrivastava, M.; et al. Stochastic pausing at latent HIV-1 promoters generates transcriptional bursting. Nat. Commun. 2021, 12, 4503. [Google Scholar] [CrossRef]
- Jadlowsky, J.K.; Wong, J.Y.; Graham, A.C.; Dobrowolski, C.; Devor, R.L.; Adams, M.D.; Fujinaga, K.; Karn, J. Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol. Cell Biol. 2014, 34, 1911–1928. [Google Scholar] [CrossRef] [Green Version]
- Palangat, M.; Meier, T.I.; Keene, R.G.; Landick, R. Transcriptional pausing at +62 of the HIV-1 nascent RNA modulates formation of the TAR RNA structure. Mol. Cell 1998, 1, 1033–1042. [Google Scholar] [CrossRef]
- Zhang, Z.; Klatt, A.; Gilmour, D.S.; Henderson, A.J. Negative elongation factor NELF represses human immunodeficiency virus transcription by pausing the RNA polymerase II complex. J. Biol. Chem. 2007, 282, 16981–16988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, Y.H.; Rana, T.M. DSIF and NELF interact with RNA polymerase II elongation complex and HIV-1 Tat stimulates P-TEFb-mediated phosphorylation of RNA polymerase II and DSIF during transcription elongation. J. Biol. Chem. 2001, 276, 12951–12958. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.; Garber, M.E.; Fang, S.M.; Fischer, W.H.; Jones, K.A. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef]
- Taube, R.; Peterlin, M. Lost in transcription: Molecular mechanisms that control HIV latency. Viruses 2013, 5, 902–927. [Google Scholar] [CrossRef] [Green Version]
- Budhiraja, S.; Famiglietti, M.; Bosque, A.; Planelles, V.; Rice, A.P. Cyclin T1 and CDK9 T-loop phosphorylation are downregulated during establishment of HIV-1 latency in primary resting memory CD4+ T cells. J. Virol. 2013, 87, 1211–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, V.T.; Kiss, T.; Michels, A.A.; Bensaude, O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 2001, 414, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhu, Q.; Luo, K.; Zhou, Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 2001, 414, 317–322. [Google Scholar] [CrossRef]
- Barboric, M.; Kohoutek, J.; Price, J.P.; Blazek, D.; Price, D.H.; Peterlin, B.M. Interplay between 7SK snRNA and oppositely charged regions in HEXIM1 direct the inhibition of P-TEFb. EMBO J. 2005, 24, 4291–4303. [Google Scholar] [CrossRef] [Green Version]
- Michels, A.A.; Fraldi, A.; Li, Q.; Adamson, T.E.; Bonnet, F.; Nguyen, V.T.; Sedore, S.C.; Price, J.P.; Price, D.H.; Lania, L.; et al. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 2004, 23, 2608–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.K.; Mbonye, U.; Hokello, J.; Karn, J. T-Cell Receptor Signaling Enhances Transcriptional Elongation from Latent HIV Proviruses by Activating P-TEFb through an ERK-Dependent Pathway. J. Mol. Biol. 2011, 410, 896–916. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287. [Google Scholar] [CrossRef]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 2010, 38, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.D.; Wu, J.; Shao, R.; Xue, Y.H. Mechanism and factors that control HIV-1 transcription and latency activation. J. Zhejiang Univ. Sci. B 2014, 15, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Ott, M.; Geyer, M.; Zhou, Q. The control of HIV transcription: Keeping RNA polymerase II on track. Cell Host Microbe 2011, 10, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Mbonye, U.; Karn, J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu. Rev. Virol. 2017, 4, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef] [Green Version]
- Grau-Exposito, J.; Luque-Ballesteros, L.; Navarro, J.; Curran, A.; Burgos, J.; Ribera, E.; Torrella, A.; Planas, B.; Badia, R.; Martin-Castillo, M.; et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PLoS Pathog. 2019, 15, e1007991. [Google Scholar] [CrossRef] [Green Version]
- Contreras, X.; Barboric, M.; Lenasi, T.; Peterlin, B.M. HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog. 2007, 3, 1459–1469. [Google Scholar] [CrossRef]
- Darcis, G.; Kula, A.; Bouchat, S.; Fujinaga, K.; Corazza, F.; Ait-Ammar, A.; Delacourt, N.; Melard, A.; Kabeya, K.; Vanhulle, C.; et al. An In-Depth Comparison of Latency-Reversing Agent Combinations in Various In Vitro and Ex Vivo HIV-1 Latency Models Identified Bryostatin-1+JQ1 and Ingenol-B+JQ1 to Potently Reactivate Viral Gene Expression. PLoS Pathog. 2015, 11, e1005063. [Google Scholar] [CrossRef]
- Acchioni, C.; Remoli, A.L.; Marsili, G.; Acchioni, M.; Nardolillo, I.; Orsatti, R.; Farcomeni, S.; Palermo, E.; Perrotti, E.; Barreca, M.L.; et al. Alternate NF-kappaB-Independent Signaling Reactivation of Latent HIV-1 Provirus. J. Virol 2019, 93, e00495-19. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.C.; Wang, J.M.; Kikkawa, U.; Mukai, H.; Shen, M.R.; Morita, I.; Chen, B.K.; Chang, W.C. Calcineurin-mediated dephosphorylation of c-Jun Ser-243 is required for c-Jun protein stability and cell transformation. Oncogene 2008, 27, 2422–2429. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.K.; Huang, C.C.; Chang, W.C.; Chen, Y.J.; Kikkawa, U.; Nakahama, K.; Morita, I.; Chang, W.C. PP2B-mediated dephosphorylation of c-Jun C terminus regulates phorbol ester-induced c-Jun/Sp1 interaction in A431 cells. Mol. Biol. Cell 2007, 18, 1118–1127. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- de Almagro, M.C.; Vucic, D. The inhibitor of apoptosis (IAP) proteins are critical regulators of signaling pathways and targets for anti-cancer therapy. Exp. Oncol. 2012, 34, 200–211. [Google Scholar]
- Pache, L.; Dutra, M.S.; Spivak, A.M.; Marlett, J.M.; Murry, J.P.; Hwang, Y.; Maestre, A.M.; Manganaro, L.; Vamos, M.; Teriete, P.; et al. BIRC2/cIAP1 Is a Negative Regulator of HIV-1 Transcription and Can Be Targeted by Smac Mimetics to Promote Reversal of Viral Latency. Cell Host Microbe 2015, 18, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Pache, L.; Marsden, M.D.; Teriete, P.; Portillo, A.J.; Heimann, D.; Kim, J.T.; Soliman, M.S.A.; Dimapasoc, M.; Carmona, C.; Celeridad, M.; et al. Pharmacological Activation of Non-canonical NF-kappaB Signaling Activates Latent HIV-1 Reservoirs In Vivo. Cell Rep. Med. 2020, 1, 100037. [Google Scholar] [CrossRef]
- Bobardt, M.; Kuo, J.; Chatterji, U.; Chanda, S.; Little, S.J.; Wiedemann, N.; Vuagniaux, G.; Gallay, P.A. The inhibitor apoptosis protein antagonist Debio 1143 Is an attractive HIV-1 latency reversal candidate. PLoS ONE 2019, 14, e0211746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; Brooks, A.D.; Spagnuolo, R.A.; Irlbeck, D.M.; Mattingly, C.; Ho, P.T.; Schoof, N.; Cammon, C.G.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-kappaB signalling in vivo. Nature 2020, 578, 160–165. [Google Scholar] [CrossRef]
- Campbell, G.R.; Spector, S.A. DIABLO/SMAC mimetics selectively kill HIV-1-infected resting memory CD4(+) T cells: A potential role in a cure strategy for HIV-1 infection. Autophagy 2019, 15, 744–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBrien, J.B.; Mavigner, M.; Franchitti, L.; Smith, S.A.; White, E.; Tharp, G.K.; Walum, H.; Busman-Sahay, K.; Aguilera-Sandoval, C.R.; Thayer, W.O.; et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8(+) cells. Nature 2020, 578, 154–159. [Google Scholar] [CrossRef]
- Garrido, C.; Abad-Fernandez, M.; Tuyishime, M.; Pollara, J.J.; Ferrari, G.; Soriano-Sarabia, N.; Margolis, D.M. Interleukin-15-Stimulated Natural Killer Cells Clear HIV-1-Infected Cells following Latency Reversal Ex Vivo. J. Virol. 2018, 92, e00235-18. [Google Scholar] [CrossRef] [Green Version]
- Archin, N.M.; Kirchherr, J.L.; Sung, J.A.; Clutton, G.; Sholtis, K.; Xu, Y.; Allard, B.; Stuelke, E.; Kashuba, A.D.; Kuruc, J.D.; et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J. Clin. Investig. 2017, 127, 3126–3135. [Google Scholar] [CrossRef] [Green Version]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Sogaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef] [Green Version]
- Lehrman, G.; Hogue, I.B.; Palmer, S.; Jennings, C.; Spina, C.A.; Wiegand, A.; Landay, A.L.; Coombs, R.W.; Richman, D.D.; Mellors, J.W.; et al. Depletion of latent HIV-1 infection in vivo: A proof-of-concept study. Lancet 2005, 366, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Siliciano, J.D.; Lai, J.; Callender, M.; Pitt, E.; Zhang, H.; Margolick, J.B.; Gallant, J.E.; Cofrancesco, J., Jr.; Moore, R.D.; Gange, S.J.; et al. Stability of the latent reservoir for HIV-1 in patients receiving valproic acid. J. Infect. Dis. 2007, 195, 833–836. [Google Scholar] [CrossRef]
- Singh, V.; Dashti, A.; Mavigner, M.; Chahroudi, A. Latency Reversal 2.0: Giving the Immune System a Seat at the Table. Curr. HIV/AIDS Rep. 2021, 18, 117–127. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Lewin, S.R. Shocking HIV out of hiding: Where are we with clinical trials of latency reversing agents? Curr. Opin. HIV AIDS 2016, 11, 394–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archin, N.M.; Bateson, R.; Tripathy, M.K.; Crooks, A.M.; Yang, K.H.; Dahl, N.P.; Kearney, M.F.; Anderson, E.M.; Coffin, J.M.; Strain, M.C.; et al. HIV-1 expression within resting CD4+ T cells after multiple doses of vorinostat. J. Infect. Dis. 2014, 210, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.S.; Richard, J.; Lichtfuss, M.; Smith, A.B., 3rd; Park, J.; Courter, J.R.; Melillo, B.N.; Sodroski, J.G.; Kaufmann, D.E.; Finzi, A.; et al. Antibody-Dependent Cellular Cytotoxicity against Reactivated HIV-1-Infected Cells. J. Virol. 2016, 90, 2021–2030. [Google Scholar] [CrossRef] [Green Version]
- Fidler, S.; Stohr, W.; Pace, M.; Dorrell, L.; Lever, A.; Pett, S.; Kinloch-de Loes, S.; Fox, J.; Clarke, A.; Nelson, M.; et al. Antiretroviral therapy alone versus antiretroviral therapy with a kick and kill approach, on measures of the HIV reservoir in participants with recent HIV infection (the RIVER trial): A phase 2, randomised trial. Lancet 2020, 395, 888–898. [Google Scholar] [CrossRef]
- Mothe, B.; Rosas-Umbert, M.; Coll, P.; Manzardo, C.; Puertas, M.C.; Moron-Lopez, S.; Llano, A.; Miranda, C.; Cedeno, S.; Lopez, M.; et al. HIVconsv Vaccines and Romidepsin in Early-Treated HIV-1-Infected Individuals: Safety, Immunogenicity and Effect on the Viral Reservoir (Study BCN02). Front. Immunol. 2020, 11, 823. [Google Scholar] [CrossRef]
- Herzner, A.M.; Hagmann, C.A.; Goldeck, M.; Wolter, S.; Kubler, K.; Wittmann, S.; Gramberg, T.; Andreeva, L.; Hopfner, K.P.; Mertens, C.; et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat. Immunol. 2015, 16, 1025–1033. [Google Scholar] [CrossRef] [Green Version]
- Gulen, M.F.; Koch, U.; Haag, S.M.; Schuler, F.; Apetoh, L.; Villunger, A.; Radtke, F.; Ablasser, A. Signalling strength determines proapoptotic functions of STING. Nat. Commun. 2017, 8, 427. [Google Scholar] [CrossRef]
- Zevini, A.; Olagnier, D.; Hiscott, J. Crosstalk between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Wang, Q.; Li, G.; Banga, R.; Ma, J.; Yu, H.; Yasui, F.; Zhang, Z.; Pantaleo, G.; Perreau, M.; et al. TLR3 agonist and CD40-targeting vaccination induces immune responses and reduces HIV-1 reservoirs. J. Clin. Investig. 2018, 128, 4387–4396. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.Y.; Osuna, C.E.; Hraber, P.T.; Hesselgesser, J.; Gerold, J.M.; Barnes, T.L.; Sanisetty, S.; Seaman, M.S.; Lewis, M.G.; Geleziunas, R.; et al. TLR7 agonists induce transient viremia and reduce the viral reservoir in SIV-infected rhesus macaques on antiretroviral therapy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, A.; Irrinki, A.; Kaur, J.; Cihlar, T.; Kukolj, G.; Sloan, D.D.; Murry, J.P. Toll-Like Receptor 7 Agonist GS-9620 Induces HIV Expression and HIV-Specific Immunity in Cells from HIV-Infected Individuals on Suppressive Antiretroviral Therapy. J. Virol. 2017, 91, e02166-16. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.B.; Mueller, S.; O’Connor, R.; Rimpel, K.; Sloan, D.D.; Karel, D.; Wong, H.C.; Jeng, E.K.; Thomas, A.S.; Whitney, J.B.; et al. A Subset of Latency-Reversing Agents Expose HIV-Infected Resting CD4+ T-Cells to Recognition by Cytotoxic T-Lymphocytes. PLoS Pathog. 2016, 12, e1005545. [Google Scholar] [CrossRef] [Green Version]
- Tomescu, C.; Chehimi, J.; Maino, V.C.; Montaner, L.J. NK cell lysis of HIV-1-infected autologous CD4 primary T cells: Requirement for IFN-mediated NK activation by plasmacytoid dendritic cells. J. Immunol. 2007, 179, 2097–2104. [Google Scholar] [CrossRef] [Green Version]
- Del Prete, G.Q.; Alvord, W.G.; Li, Y.; Deleage, C.; Nag, M.; Oswald, K.; Thomas, J.A.; Pyle, C.; Bosche, W.J.; Coalter, V.; et al. TLR7 agonist administration to SIV-infected macaques receiving early initiated cART does not induce plasma viremia. JCI Insight 2019, 4, e127717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekerman, E.; Hesselgesser, J.; Carr, B.; Nagel, M.; Hung, M.; Wang, A.; Stapleton, L.; von Gegerfelt, A.; Elyard, H.A.; Lifson, J.D.; et al. PD-1 Blockade and TLR7 Activation Lack Therapeutic Benefit in Chronic Simian Immunodeficiency Virus-Infected Macaques on Antiretroviral Therapy. Antimicrob. Agents Chemother. 2019, 63, e01163-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borducchi, E.N.; Cabral, C.; Stephenson, K.E.; Liu, J.; Abbink, P.; Ng’ang’a, D.; Nkolola, J.P.; Brinkman, A.L.; Peter, L.; Lee, B.C.; et al. Ad26/MVA therapeutic vaccination with TLR7 stimulation in SIV-infected rhesus monkeys. Nature 2016, 540, 284–287. [Google Scholar] [CrossRef]
- Hsu, D.C.; Schuetz, A.; Imerbsin, R.; Silsorn, D.; Pegu, A.; Inthawong, D.; Sopanaporn, J.; Visudhiphan, P.; Chuenarom, W.; Keawboon, B.; et al. TLR7 agonist, N6-LS and PGT121 delayed viral rebound in SHIV-infected macaques after antiretroviral therapy interruption. PLoS Pathog. 2021, 17, e1009339. [Google Scholar] [CrossRef]
- Riddler, S.A.; Para, M.; Benson, C.A.; Mills, A.; Ramgopal, M.; DeJesus, E.; Brinson, C.; Cyktor, J.; Jacobs, J.; Koontz, D.; et al. Vesatolimod, a Toll-like Receptor 7 Agonist, Induces Immune Activation in Virally Suppressed Adults Living With Human Immunodeficiency Virus-1. Clin. Infect. Dis. 2021, 72, e815–e824. [Google Scholar] [CrossRef]
- Meas, H.Z.; Haug, M.; Beckwith, M.S.; Louet, C.; Ryan, L.; Hu, Z.; Landskron, J.; Nordbo, S.A.; Tasken, K.; Yin, H.; et al. Sensing of HIV-1 by TLR8 activates human T cells and reverses latency. Nat. Commun. 2020, 11, 147. [Google Scholar] [CrossRef]
- Schlaepfer, E.; Audige, A.; Joller, H.; Speck, R.F. TLR7/8 triggering exerts opposing effects in acute versus latent HIV infection. J. Immunol. 2006, 176, 2888–2895. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, E.; Speck, R.F. TLR8 activates HIV from latently infected cells of myeloid-monocytic origin directly via the MAPK pathway and from latently infected CD4+ T cells indirectly via TNF-alpha. J. Immunol. 2011, 186, 4314–4324. [Google Scholar] [CrossRef]
- Scheller, C.; Ullrich, A.; McPherson, K.; Hefele, B.; Knoferle, J.; Lamla, S.; Olbrich, A.R.; Stocker, H.; Arasteh, K.; ter Meulen, V.; et al. CpG oligodeoxynucleotides activate HIV replication in latently infected human T cells. J. Biol. Chem. 2004, 279, 21897–21902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheller, C.; Ullrich, A.; Lamla, S.; Dittmer, U.; Rethwilm, A.; Koutsilieri, E. Dual activity of phosphorothioate CpG oligodeoxynucleotides on HIV: Reactivation of latent provirus and inhibition of productive infection in human T cells. Ann. N. Y. Acad. Sci. 2006, 1091, 540–547. [Google Scholar] [CrossRef]
- Vibholm, L.; Schleimann, M.H.; Hojen, J.F.; Benfield, T.; Offersen, R.; Rasmussen, K.; Olesen, R.; Dige, A.; Agnholt, J.; Grau, J.; et al. Short-Course Toll-Like Receptor 9 Agonist Treatment Impacts Innate Immunity and Plasma Viremia in Individuals With Human Immunodeficiency Virus Infection. Clin. Infect. Dis. 2017, 64, 1686–1695. [Google Scholar] [CrossRef]
- Vibholm, L.K.; Konrad, C.V.; Schleimann, M.H.; Frattari, G.; Winckelmann, A.; Klastrup, V.; Jensen, N.M.; Jensen, S.S.; Schmidt, M.; Wittig, B.; et al. Effects of 24-week Toll-like receptor 9 agonist treatment in HIV type 1+ individuals. AIDS 2019, 33, 1315–1325. [Google Scholar] [CrossRef]
- Winckelmann, A.A.; Munk-Petersen, L.V.; Rasmussen, T.A.; Melchjorsen, J.; Hjelholt, T.J.; Montefiori, D.; Ostergaard, L.; Sogaard, O.S.; Tolstrup, M. Administration of a Toll-like receptor 9 agonist decreases the proviral reservoir in virologically suppressed HIV-infected patients. PLoS ONE 2013, 8, e62074. [Google Scholar] [CrossRef]
- Li, P.; Kaiser, P.; Lampiris, H.W.; Kim, P.; Yukl, S.A.; Havlir, D.V.; Greene, W.C.; Wong, J.K. Stimulating the RIG-I pathway to kill cells in the latent HIV reservoir following viral reactivation. Nat. Med. 2016, 22, 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palermo, E.; Acchioni, C.; Di Carlo, D.; Zevini, A.; Muscolini, M.; Ferrari, M.; Castiello, L.; Virtuoso, S.; Borsetti, A.; Antonelli, G.; et al. Activation of Latent HIV-1 T Cell Reservoirs with a Combination of Innate Immune and Epigenetic Regulators. J. Virol. 2019, 93, e01194-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Vidal, E.; Castellvi, M.; Pujantell, M.; Badia, R.; Jou, A.; Gomez, L.; Puig, T.; Clotet, B.; Ballana, E.; Riveira-Munoz, E.; et al. Evaluation of the Innate Immune Modulator Acitretin as a Strategy To Clear the HIV Reservoir. Antimicrob. Agents Chemother. 2017, 61, e01368-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Kanuma, T.; Takahama, S.; Okamura, T.; Moriishi, E.; Ishii, K.J.; Terahara, K.; Yasutomi, Y. STING agonists activate latently infected cells and enhance SIV-specific responses ex vivo in naturally SIV controlled cynomolgus macaques. Sci. Rep. 2019, 9, 5917. [Google Scholar] [CrossRef]
- Gutjahr, A.; Papagno, L.; Nicoli, F.; Kanuma, T.; Kuse, N.; Cabral-Piccin, M.P.; Rochereau, N.; Gostick, E.; Lioux, T.; Perouzel, E.; et al. The STING ligand cGAMP potentiates the efficacy of vaccine-induced CD8+ T cells. JCI Insight 2019, 4, e125107. [Google Scholar] [CrossRef]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L.; et al. In Vivo Suppression of HIV Rebound by Didehydro-Cortistatin A, a "Block-and-Lock" Strategy for HIV-1 Treatment. Cell Rep. 2017, 21, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Mousseau, G.; Valente, S.T. Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat. Epigenet. Chromatin 2019, 12, 23. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Chen, X. Triptolide inhibits human immunodeficiency virus type 1 replication by promoting proteasomal degradation of Tat protein. Retrovirology 2014, 11, 88. [Google Scholar] [CrossRef]
- Vranckx, L.S.; Demeulemeester, J.; Saleh, S.; Boll, A.; Vansant, G.; Schrijvers, R.; Weydert, C.; Battivelli, E.; Verdin, E.; Cereseto, A.; et al. LEDGIN-mediated Inhibition of Integrase-LEDGF/p75 Interaction Reduces Reactivation of Residual Latent HIV. EBioMedicine 2016, 8, 248–264. [Google Scholar] [CrossRef] [Green Version]
- Debyser, Z.; Vansant, G.; Bruggemans, A.; Janssens, J.; Christ, F. Insight in HIV Integration Site Selection Provides a Block-and-Lock Strategy for a Functional Cure of HIV Infection. Viruses 2018, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Santoso, N.; Power, D.; Simpson, S.; Dieringer, M.; Miao, H.; Gurova, K.; Giam, C.Z.; Elledge, S.J.; Zhu, J. FACT Proteins, SUPT16H and SSRP1, Are Transcriptional Suppressors of HIV-1 and HTLV-1 That Facilitate Viral Latency. J. Biol. Chem. 2015, 290, 27297–27310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean, M.J.; Hayashi, T.; Huang, H.; Brennan, J.; Simpson, S.; Purmal, A.; Gurova, K.; Keefer, M.C.; Kobie, J.J.; Santoso, N.G.; et al. Curaxin CBL0100 Blocks HIV-1 Replication and Reactivation through Inhibition of Viral Transcriptional Elongation. Front. Microbiol. 2017, 8, 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlenstiel, C.; Mendez, C.; Lim, S.T.; Marks, K.; Turville, S.; Cooper, D.A.; Kelleher, A.D.; Suzuki, K. Novel RNA Duplex Locks HIV-1 in a Latent State via Chromatin-mediated Transcriptional Silencing. Mol. Ther. Nucleic Acids 2015, 4, e261. [Google Scholar] [CrossRef]
- Mendez, C.; Ledger, S.; Petoumenos, K.; Ahlenstiel, C.; Kelleher, A.D. RNA-induced epigenetic silencing inhibits HIV-1 reactivation from latency. Retrovirology 2018, 15, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, I.; Low, J.S.; Weston, S.; Weinberger, M.; Zhyvoloup, A.; Labokha, A.A.; Corazza, G.; Kitson, R.A.; Moody, C.J.; Marcello, A.; et al. Heat shock protein 90 controls HIV-1 reactivation from latency. Proc. Natl. Acad. Sci. USA 2014, 111, E1528–E1537. [Google Scholar] [CrossRef] [Green Version]
- Joshi, P.; Maidji, E.; Stoddart, C.A. Inhibition of Heat Shock Protein 90 Prevents HIV Rebound. J. Biol. Chem. 2016, 291, 10332–10346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavegnano, C.; Detorio, M.; Montero, C.; Bosque, A.; Planelles, V.; Schinazi, R.F. Ruxolitinib and tofacitinib are potent and selective inhibitors of HIV-1 replication and virus reactivation in vitro. Antimicrob. Agents Chemother. 2014, 58, 1977–1986. [Google Scholar] [CrossRef] [Green Version]
- Gavegnano, C.; Brehm, J.H.; Dupuy, F.P.; Talla, A.; Ribeiro, S.P.; Kulpa, D.A.; Cameron, C.; Santos, S.; Hurwitz, S.J.; Marconi, V.C.; et al. Novel mechanisms to inhibit HIV reservoir seeding using Jak inhibitors. PLoS Pathog. 2017, 13, e1006740. [Google Scholar] [CrossRef] [Green Version]
- Niu, Q.; Liu, Z.; Alamer, E.; Fan, X.; Chen, H.; Endsley, J.; Gelman, B.B.; Tian, B.; Kim, J.H.; Michael, N.L.; et al. Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J. Clin. Investig. 2019, 129, 3361–3373. [Google Scholar] [CrossRef]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef]
- Li, Z.; Mbonye, U.; Feng, Z.; Wang, X.; Gao, X.; Karn, J.; Zhou, Q. The KAT5-Acetyl-Histone4-Brd4 axis silences HIV-1 transcription and promotes viral latency. PLoS Pathog. 2018, 14, e1007012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Giacca, M. HIV Latency TORn Down. Cell Host Microbe 2016, 20, 700–702. [Google Scholar] [CrossRef] [Green Version]
- Vargas, B.; Giacobbi, N.S.; Sanyal, A.; Venkatachari, N.J.; Han, F.; Gupta, P.; Sluis-Cremer, N. Inhibitors of Signaling Pathways That Block Reversal of HIV-1 Latency. Antimicrob. Agents Chemother. 2019, 63, e01744-18. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.; Ruggiero, A.; Paxton, W.A.; Pollakis, G. Measuring the Success of HIV-1 Cure Strategies. Front. Cell Infect. Microbiol. 2020, 10, 134. [Google Scholar] [CrossRef]
- Ren, Y.; Huang, S.H.; Patel, S.; Alberto, W.D.C.; Magat, D.; Ahimovic, D.; Macedo, A.B.; Durga, R.; Chan, D.; Zale, E.; et al. BCL-2 antagonism sensitizes cytotoxic T cell-resistant HIV reservoirs to elimination ex vivo. J. Clin. Investig. 2020, 130, 2542–2559. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.W.; Sainski-Nguyen, A.M.; Natesampillai, S.; Aboulnasr, F.; Kaufmann, S.; Badley, A.D. Maintenance of the HIV Reservoir Is Antagonized by Selective BCL2 Inhibition. J. Virol. 2017, 91, e00012-17. [Google Scholar] [CrossRef] [Green Version]
- Cummins, N.W.; Sainski, A.M.; Dai, H.; Natesampillai, S.; Pang, Y.P.; Bren, G.D.; de Araujo Correia, M.C.M.; Sampath, R.; Rizza, S.A.; O’Brien, D.; et al. Prime, Shock, and Kill: Priming CD4 T Cells from HIV Patients with a BCL-2 Antagonist before HIV Reactivation Reduces HIV Reservoir Size. J. Virol. 2016, 90, 4032–4048. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarabia, I.; Bosque, A. HIV-1 Latency and Latency Reversal: Does Subtype Matter? Viruses 2019, 11, 1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Zhou, X.; Nakaya, M.; Jin, W.; Cheng, X.; Sun, S.C. T cell-intrinsic function of the noncanonical NF-kappaB pathway in the regulation of GM-CSF expression and experimental autoimmune encephalomyelitis pathogenesis. J. Immunol. 2014, 193, 422–430. [Google Scholar] [CrossRef]
- Grandi, N.; Tramontano, E. Human Endogenous Retroviruses Are Ancient Acquired Elements Still Shaping Innate Immune Responses. Front. Immunol. 2018, 9, 2039. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Huang, J.; Zhu, F. Human Endogenous Retroviral Envelope Protein Syncytin-1 and Inflammatory Abnormalities in Neuropsychological Diseases. Front. Psychiatry 2018, 9, 422. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the HIV-1 transcription elongation process in a generic CD4+T memory cell, latently infected. Upon transcription initiation mediated by transcription factors (TFs) binding the Long Terminal Repeats (LTR) enhancer, a proportion of the viral transcripts paused forming the TAR RNA structure. The elongation factor PTEF-b, made by CycT1 and CDK9, is hijacked in the 7SK snRNP/HEXIM1 inhibitory complex in resting CD4+ T memory cells. The treatment of these cells with Hexamethylenebisacetamide (HMBA) determines PTEF-b release from the inhibitory complex. PTEF-b can be bound to the Bromodomain-containing protein (BRD) 4, thus stimulating transcription elongation from cellular genes. As soon as the viral transactivator Tat accumulates, it displaces PTEF-b from BRD4 to delocalize it to the HIV-1 Trans-activation response element (TAR), thus strongly promoting HIV-1 transcription elongation and virus replication. Bryostatin-1 treatment increases the level of available CycT1 and CDK9 in resting memory CD4+ T cells, while the BRD4 inhibitor JQ1 prevents BRD4 from binding PTEF-b. Also indicated are the AFF1/4, ELL1/2, and AF9/ENL factors forming, together with PTEF-b and Tat, the multi-subunit complex named the “super elongation complex” (SEC). The negative elongation factor (NELF), and the 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) sensitivity inducing factor (DSIF), are phosphorylated by CDK9 determining NELF release and DSIF turned into a positive elongating factor. CDK9 also phosphorylates the largest subunit of RNA polymerase II (Pol II), thus allowing transcription elongation to proceed.
Figure 1.
Schematic representation of the HIV-1 transcription elongation process in a generic CD4+T memory cell, latently infected. Upon transcription initiation mediated by transcription factors (TFs) binding the Long Terminal Repeats (LTR) enhancer, a proportion of the viral transcripts paused forming the TAR RNA structure. The elongation factor PTEF-b, made by CycT1 and CDK9, is hijacked in the 7SK snRNP/HEXIM1 inhibitory complex in resting CD4+ T memory cells. The treatment of these cells with Hexamethylenebisacetamide (HMBA) determines PTEF-b release from the inhibitory complex. PTEF-b can be bound to the Bromodomain-containing protein (BRD) 4, thus stimulating transcription elongation from cellular genes. As soon as the viral transactivator Tat accumulates, it displaces PTEF-b from BRD4 to delocalize it to the HIV-1 Trans-activation response element (TAR), thus strongly promoting HIV-1 transcription elongation and virus replication. Bryostatin-1 treatment increases the level of available CycT1 and CDK9 in resting memory CD4+ T cells, while the BRD4 inhibitor JQ1 prevents BRD4 from binding PTEF-b. Also indicated are the AFF1/4, ELL1/2, and AF9/ENL factors forming, together with PTEF-b and Tat, the multi-subunit complex named the “super elongation complex” (SEC). The negative elongation factor (NELF), and the 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) sensitivity inducing factor (DSIF), are phosphorylated by CDK9 determining NELF release and DSIF turned into a positive elongating factor. CDK9 also phosphorylates the largest subunit of RNA polymerase II (Pol II), thus allowing transcription elongation to proceed.

Figure 2.
Schematic representation of the proposed molecular mechanisms acting in different subsets of CD4+ T central memory (CM) cells driving HIV-1 transcription initiation. (A) Suggested molecular mechanisms acting in CD4+ T central memory (CM) cells driving HIV-1 transcription initiation, following Bryostatin-1 plus Ionomycin or HMBA treatments. Bryostatin-1 drives protein kinase C (PKC) activation that, in turn, activates p38 kinase. Ionomycin and, to a lesser extent, Hexamethylenebisacetamide (HMBA), activate Calcineurin (CaN) phosphatase, that, together with p38 produce the accumulation of c-Jun transcription factor. Full c-Jun activation is obtained by its p38-mediated phosphorylation. Activated c-Jun binds the NF-κB inhibitory dimer p50/p50, bound to the long terminal repeat enhancer (LTR), determining initiation of HIV-1 transcription. The nucleosomes (Nuc 0) and Nuc1 -are also indicated. (B) Schematic representation of the latency reversing action of the combined treatments with Ingenol and Romidepsin, effective in almost all latently infected resting memory CD4+ T cell subtypes. The PKC agonist Ingenol activates PKC, which in turn stimulates the IκB Kinase (IKK) complex to phosphorylate IκB, determining its degradation. NF-κB is therefore free to accumulate in the nucleus and to bind the LTR enhancer. Full start of transcriptional activity is obtained by simultaneous treatment with the histone deacetylase inhibitor (HDI) Romidepsin, able to determine acetylation of nuc1 and of crucial NF-κB residues.
Figure 2.
Schematic representation of the proposed molecular mechanisms acting in different subsets of CD4+ T central memory (CM) cells driving HIV-1 transcription initiation. (A) Suggested molecular mechanisms acting in CD4+ T central memory (CM) cells driving HIV-1 transcription initiation, following Bryostatin-1 plus Ionomycin or HMBA treatments. Bryostatin-1 drives protein kinase C (PKC) activation that, in turn, activates p38 kinase. Ionomycin and, to a lesser extent, Hexamethylenebisacetamide (HMBA), activate Calcineurin (CaN) phosphatase, that, together with p38 produce the accumulation of c-Jun transcription factor. Full c-Jun activation is obtained by its p38-mediated phosphorylation. Activated c-Jun binds the NF-κB inhibitory dimer p50/p50, bound to the long terminal repeat enhancer (LTR), determining initiation of HIV-1 transcription. The nucleosomes (Nuc 0) and Nuc1 -are also indicated. (B) Schematic representation of the latency reversing action of the combined treatments with Ingenol and Romidepsin, effective in almost all latently infected resting memory CD4+ T cell subtypes. The PKC agonist Ingenol activates PKC, which in turn stimulates the IκB Kinase (IKK) complex to phosphorylate IκB, determining its degradation. NF-κB is therefore free to accumulate in the nucleus and to bind the LTR enhancer. Full start of transcriptional activity is obtained by simultaneous treatment with the histone deacetylase inhibitor (HDI) Romidepsin, able to determine acetylation of nuc1 and of crucial NF-κB residues.

Figure 3.
Schematic representation of LRAs mechanism of action. Histone Deacetylase Inhibitors (HDIs) induce histones hyperacetylation, which leads to a chromatin conformational change, activating transcription, thereby causing latency reversal; HDIs increase plasma viraemia in HIV-1 positive patients and, when administered together with HIV-specific CD8+ T-cells, induce selective killing of reactivated CD4+ T-Cells in vitro. TLRs agonists reactivate HIV-1 replication in a NF-κB, AP-1, and Type-I IFNs—dependent manner; TLRs stimulation also results in increased plasma viraemia in patients and in SIV-infected macaques, and triggers an HIV-specific adaptive response. Combination of TLRs agonists with therapeutic vaccines or broadly-neutralizing antibodies (bnAbs) protected SIV-infected monkeys and SHIV-infected macaques from viral rebound after ART cessation. RLRs agonist Acitretin stimulates latency reversal by activation of Histone Acetyl-Transferase p300 mediated by type-I IFN; STING agonists induce reactivation in a NF-κB dependent manner. Both RLRs and STING agonists induce selective apoptosis of reactivated cells. * Activated in apoptotic signaling.
Figure 3.
Schematic representation of LRAs mechanism of action. Histone Deacetylase Inhibitors (HDIs) induce histones hyperacetylation, which leads to a chromatin conformational change, activating transcription, thereby causing latency reversal; HDIs increase plasma viraemia in HIV-1 positive patients and, when administered together with HIV-specific CD8+ T-cells, induce selective killing of reactivated CD4+ T-Cells in vitro. TLRs agonists reactivate HIV-1 replication in a NF-κB, AP-1, and Type-I IFNs—dependent manner; TLRs stimulation also results in increased plasma viraemia in patients and in SIV-infected macaques, and triggers an HIV-specific adaptive response. Combination of TLRs agonists with therapeutic vaccines or broadly-neutralizing antibodies (bnAbs) protected SIV-infected monkeys and SHIV-infected macaques from viral rebound after ART cessation. RLRs agonist Acitretin stimulates latency reversal by activation of Histone Acetyl-Transferase p300 mediated by type-I IFN; STING agonists induce reactivation in a NF-κB dependent manner. Both RLRs and STING agonists induce selective apoptosis of reactivated cells. * Activated in apoptotic signaling.

Figure 4.
Schematic representation of two different possibilities to combine the “Shock and Kill” and the “Block and Lock” approaches to reach a functional cure. (A) The sequential application of the “Shock and Kill” followed by the “Block and lock” could lead to an initial reduction/elimination of poorly latent reservoirs, easy to be reactivated by LRAs, followed by the blocking and locking of potential residual replication by LPAs. (B) “Shoc-K and B-Lock”. All-in-one cocktail of LRAs + LPAs could result in a tailored reactivation and killing of poorly latent reservoir and, at the same time, the long term locking of residual deeply latent proviruses.
Figure 4.
Schematic representation of two different possibilities to combine the “Shock and Kill” and the “Block and Lock” approaches to reach a functional cure. (A) The sequential application of the “Shock and Kill” followed by the “Block and lock” could lead to an initial reduction/elimination of poorly latent reservoirs, easy to be reactivated by LRAs, followed by the blocking and locking of potential residual replication by LPAs. (B) “Shoc-K and B-Lock”. All-in-one cocktail of LRAs + LPAs could result in a tailored reactivation and killing of poorly latent reservoir and, at the same time, the long term locking of residual deeply latent proviruses.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Acchioni, C.; Palermo, E.; Sandini, S.; Acchioni, M.; Hiscott, J.; Sgarbanti, M. Fighting HIV-1 Persistence: At the Crossroads of “Shoc-K and B-Lock”. Pathogens 2021, 10, 1517. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10111517
AMA Style
Acchioni C, Palermo E, Sandini S, Acchioni M, Hiscott J, Sgarbanti M. Fighting HIV-1 Persistence: At the Crossroads of “Shoc-K and B-Lock”. Pathogens. 2021; 10(11):1517. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10111517
Chicago/Turabian StyleAcchioni, Chiara, Enrico Palermo, Silvia Sandini, Marta Acchioni, John Hiscott, and Marco Sgarbanti. 2021. "Fighting HIV-1 Persistence: At the Crossroads of “Shoc-K and B-Lock”" Pathogens 10, no. 11: 1517. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10111517
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.