
Bacteriophage-Host Association in the Phytoplasma Insect Vector Euscelidius variegatus

 , and
, and
Abstract
:1. Introduction
2. Results
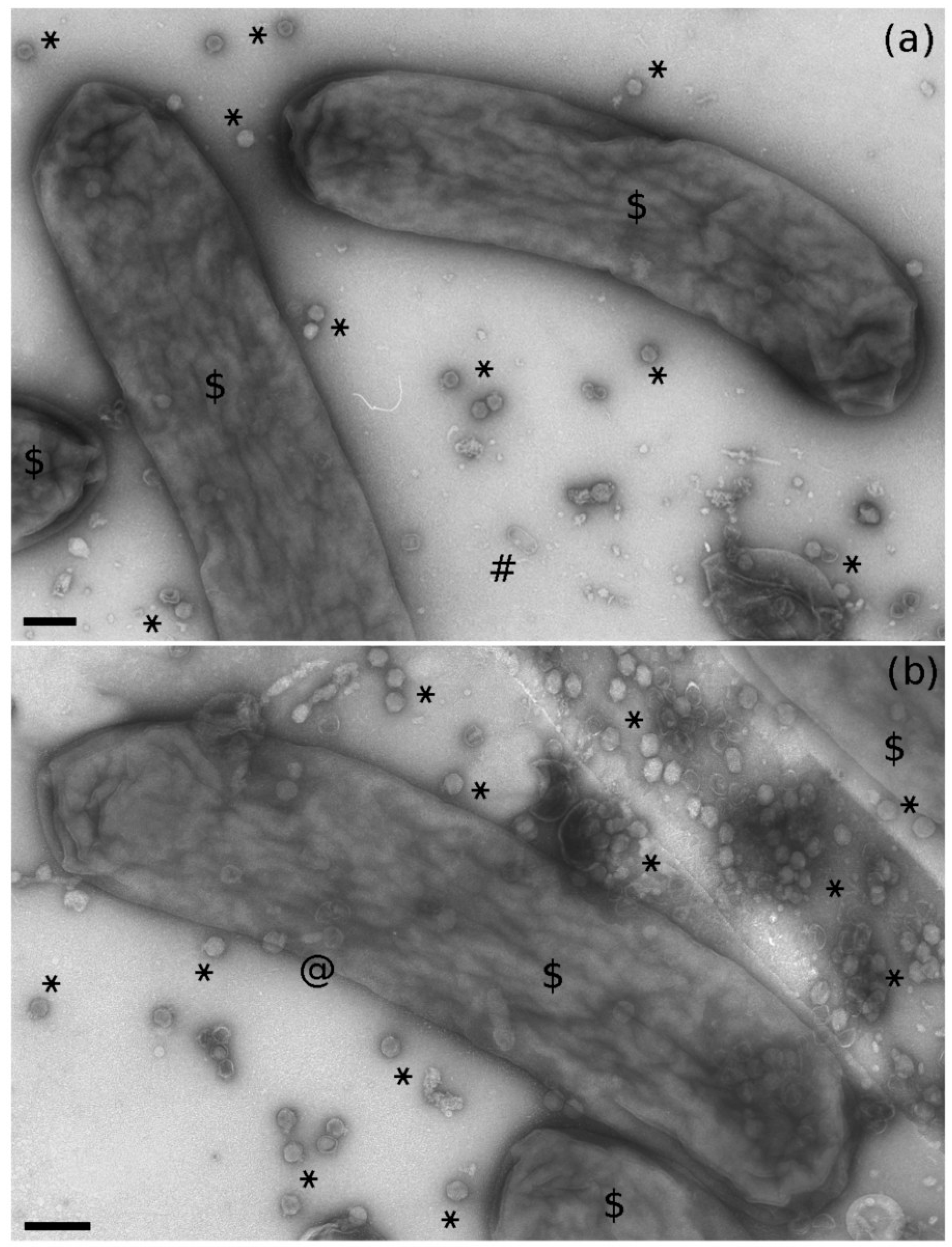
2.1. Bacteriophage-Like Particles in Euscelidius variegatus
2.2. Selection of Expressed Bacteriophage Sequences
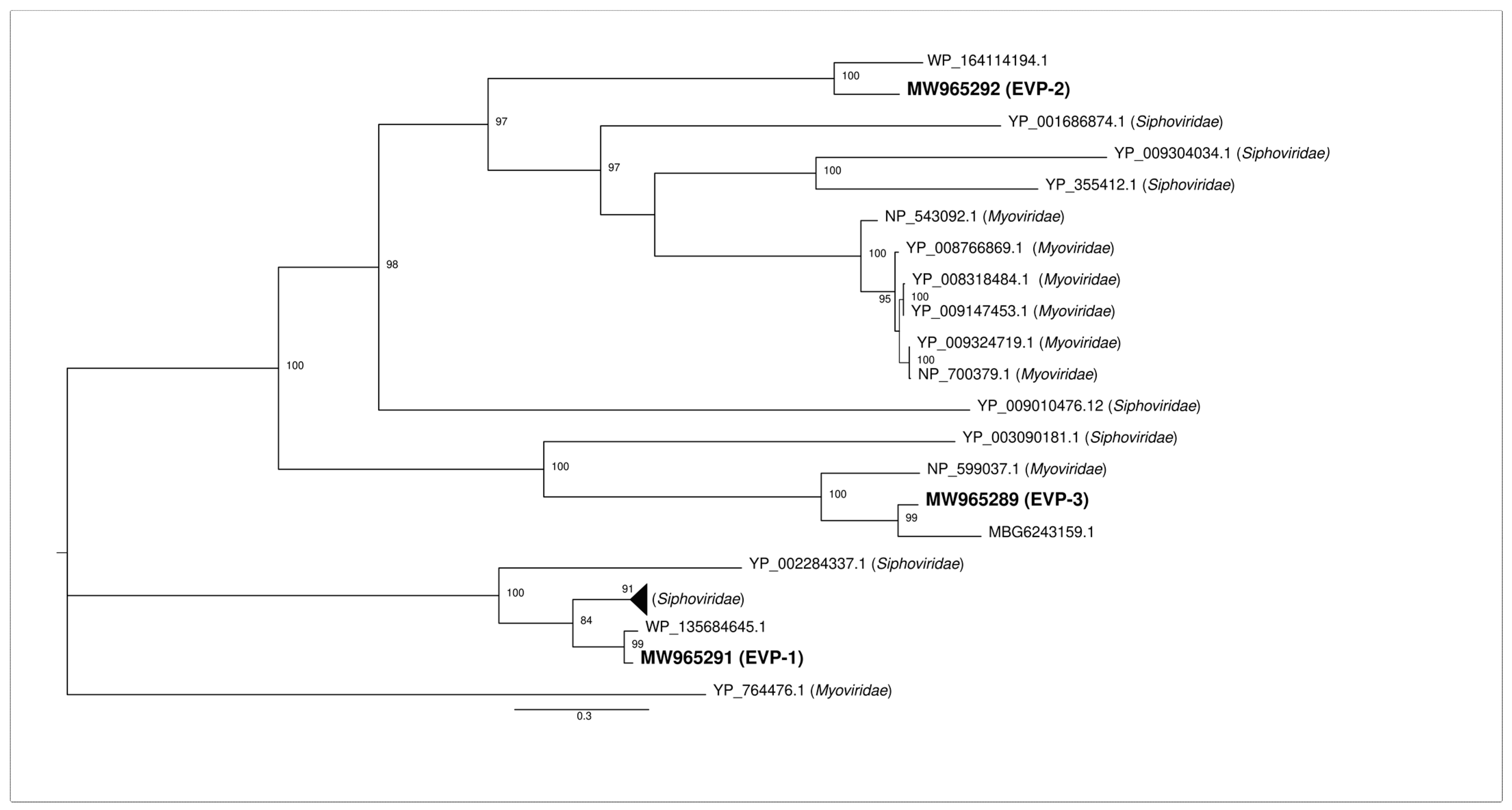
2.3. Phylogenetic Analysis of the Identified Major Capsid Proteins
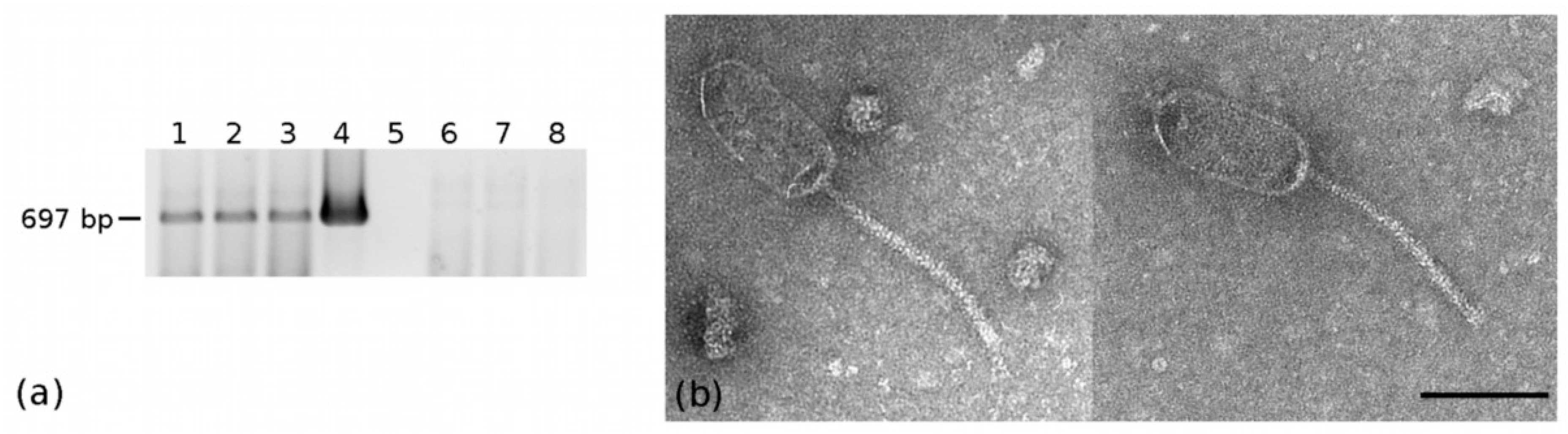
2.4. Detection and Prevalence of the Three Phages
2.5. Isolation of EVP-1 Bacterial Host
2.6. Multiple Phages in EVP-1 Host
3. Discussion
4. Materials and Methods
4.1. Insect Population
4.2. DNA and RNA Extraction
4.3. RNA-Seq and Bioinformatic Analysis
4.4. Accession Numbers
4.5. Phylogenetic Analysis
4.6. PCR and RT-PCR Amplifications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence | Target | Product Size (bp) | Annealing T (°C) | Citation |
|---|---|---|---|---|---|
| EvaTO_phage1f | CCGGTGGGTTCACTTTCC | MW965291 | 697 | 64 | This work |
| EvaTO_phage1r | CGTCCGCAGACCATTATCGG | ||||
| EvaTO_phage2f | CTTCTCTGGCTGGCCTACCC | MW965292 | 725 | 64 | This work |
| EvaTO_phage2r | GAGTATCGCCGGTCATCACG | ||||
| EvaTO_phage3f | AGGGTACTAGCCAGGACGAC | MW965289 | 524 | 64 | This work |
| EvaTO_phage3r | TGTGCCGCCATTTCGATAAG | ||||
| BEV3 | TTATGAGGTCCGCTTGCTCT | BEV 16S ribosomal DNA sequence | 1009 | 64 | [39] |
| BEV4 | CGATCCCTAGCTGGTCTGAG | ||||
| 27F | AGAGTTTGATCMTGGCTCAG | 16S ribosomal DNA sequence | 1507 | 58 | [76] |
| 1494R | CTACGGCTACCTTGTTACGA |
4.7. Bacterial Isolation
4.8. Transmission Electron Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Champomier Vergès, M.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Herrero Corral, G.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Gurung, K.; Wertheim, B.; Salles, J.F. The microbiome of pest insects: It is not just bacteria. Entomol. Exp. Appl. 2019, 167, 156–170. [Google Scholar] [CrossRef] [Green Version]
- Qadri, M.; Short, S.; Gast, K.; Hernandez, J.; Wong, A.C.N. Microbiome innovation in agriculture: Development of microbial based tools for insect pest management. Front. Sustain. Food Syst. 2020, 4, 547751. [Google Scholar] [CrossRef]
- Ishii, Y.; Matsuura, Y.; Kakizawa, S.; Nikoh, N.; Fukatsu, T. Diversity of bacterial endosymbionts associated with Macrosteles leafhoppers vectoring phytopathogenic phytoplasmas. Appl. Environ. Microb. 2013, 79, 5013–5022. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.F.F.; Willner, D.L.; Lim, Y.W.; Schmieder, R.; Chau, B.; Nilsson, C.; Anthony, S.; Ruan, Y.; Rohwer, F.; Breitbart, M. Broad surveys of DNA viral diversity obtained through viral metagenomics of mosquitoes. PLoS ONE 2011, 6, e20579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Leigh, B.A.; Bordenstein, S.R.; Brooks, A.W.; Mikaelyan, A.; Bordenstein, S.R. Finer-scale phylosymbiosis: Insights from insect viromes. mSystems 2018, 3, e00131-18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Z.; Shi, M.; Holmes, E.C. Using metagenomics to characterize an expanding virosphere. Cell 2018, 172, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- Varghese, F.S.; van Rij, R.P. Insect virus discovery by metagenomic and cell culture-based approaches. Methods Mol. Biol. 2018, 1746, 197–213. [Google Scholar] [CrossRef]
- Bonning, B.C. The Insect Virome: Opportunities and Challenges. Curr. Issues Mol. Biol. 2020, 34, 1–12. [Google Scholar] [CrossRef]
- Ottati, S.; Chiapello, M.; Galetto, L.; Bosco, D.; Marzachì, C.; Abbà, S. New viral sequences identified in the Flavescence Dorée phytoplasma vector Scaphoideus titanus. Viruses 2020, 12, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton, T.D.S.; Hill, C. Gut bacteriophage: Current understanding and challenges. Front. Endocrinol. 2019, 10, 784. [Google Scholar] [CrossRef] [PubMed]
- Guerin, E.; Hill, C. Shining light on human gut bacteriophages. Front. Cell. Infect. Microbiol. 2020, 10, 481. [Google Scholar] [CrossRef] [PubMed]
- Gurney, J.; Brown, S.P.; Kaltz, O.; Hochberg, M.E. Steering phages to combat bacterial pathogens. Trends Microbiol. 2020, 28, 85–94. [Google Scholar] [CrossRef]
- D’Accolti, M.; Soffritti, I.; Mazzacane, S.; Caselli, E. Bacteriophages as a potential 360-degree pathogen control strategy. Microorganisms 2021, 9, 261. [Google Scholar] [CrossRef]
- Obeng, N.; Pratama, A.A.; Elsas, J.D.V. The Significance of Mutualistic Phages for Bacterial Ecology and Evolution. Trends Microbiol. 2016, 24, 440–449. [Google Scholar] [CrossRef]
- Czajkowski, R.; Jackson, R.W.; Lindow, S.E. Editorial: Environmental bacteriophages: From biological control applications to directed bacterial evolution. Front. Microbiol. 2019, 10, 1830. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and their genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.J.; Williamson, D.L.; Oishi, K. SpV3 viruses of Drosophila spiroplasmas. Isr. J. Med. Sci. 1987, 23, 429–433. [Google Scholar]
- Van der Wilk, F.; Dullemans, A.M.; Verbeek, M.; van den Heuvel, J.F. Isolation and characterization of APSE-1, a bacteriophage infecting the secondary endosymbiont of Acyrthosiphon pisum. Virology 1999, 262, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Masui, S.; Kuroiwa, H.; Sasaki, T.; Inui, M.; Kuroiwa, T.; Ishikawa, H. Bacteriophage WO and virus-like particles in Wolbachia, an endosymbiont of arthropods. Biochem. Biophys. Res. Commun. 2001, 283, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Degnan, P.H.; Moran, N.A. Diverse phage-encoded toxins in a protective insect endosymbiont. Appl. Environ. Microb. 2008, 74, 6782–6791. [Google Scholar] [CrossRef] [Green Version]
- Degnan, P.H.; Yu, Y.; Sisneros, N.; Wing, R.A.; Moran, N.A. Hamiltonella defensa, genome evolution of protective bacterial endosymbiont from pathogenic ancestors. Proc. Natl. Acad. Sci. USA 2009, 106, 9063–9068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.H.; Xiao, J.H.; Huang, D.W. Distribution and evolution of the bacteriophage WO and its antagonism with Wolbachia. Front. Microbiol. 2020, 11, 595629. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.H.; Sun, B.F.; Xiong, T.L.; Wang, Y.K.; Murfin, K.E.; Xiao, J.H.; Huang, D.W. Bacteriophage WO can mediate horizontal gene transfer in endosymbiotic Wolbachia genomes. Front. Microbiol. 2016, 7, 1867. [Google Scholar] [CrossRef]
- LePage, D.P.; Metcalf, J.A.; Bordenstein, S.R.; On, J.; Perlmutter, J.I.; Shropshire, J.D.; Layton, E.M.; Funkhouser-Jones, L.J.; Beckmann, J.F.; Bordenstein, S.R. Prophage WO genes recapitulate and enhance Wolbachia-induced cytoplasmic incompatibility. Nature 2017, 543, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shropshire, J.D.; On, J.; Layton, E.M.; Zhou, H.; Bordenstein, S.R. One prophage WO gene rescues cytoplasmic incompatibility in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2018, 115, 4987–4991. [Google Scholar] [CrossRef] [Green Version]
- EFSA PLH Panel. Risk to plant health of Flavescence dorée for the EU territory. EFSA J. 2016, 14, e4603. [Google Scholar] [CrossRef]
- Tomkins, M.; Kliot, A.; Marée, A.F.M.; Hogenhout, S.A. A multi-layered mechanistic modelling approach to understand how effector genes extend beyond phytoplasma to modulate plant hosts, insect vectors and the environment. Curr. Opin. Plant Biol. 2018, 44, 39–48. [Google Scholar] [CrossRef]
- Weiss, B.; Aksoy, S. Microbiome influences on insect host vector competence. Trends Parasitol. 2011, 27, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, P.; Trivedi, C.; Grinyer, J.; Anderson, I.C.; Singh, B.K. Harnessing host-vector microbiome for sustainable plant disease management of phloem-limited bacteria. Front. Plant Sci. 2016, 7, 1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonella, E.; Tedeschi, R.; Crotti, E.; Alma, A. Multiple guests in a single host: Interactions across symbiotic and phytopathogenic bacteria in phloem-feeding vectors–a review. Entomol. Exp. Appl. 2019, 67, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.T.; Li, Y.; Li, T.P.; Liang, Y.; Hu, L.; Zhang, D.; Zhou, C.Y.; Yang, C.; Zhang, X.; Zha, S.S.; et al. Stable introduction of plant-virus-inhibiting Wolbachia into planthoppers for rice protection. Curr. Biol. 2020, 30, 4837–4845.e5. [Google Scholar] [CrossRef] [PubMed]
- Martinson, V.; Gawryluk, R.M.R.; Gowen, B.E.; Curtis, C.I.; Jaenike, J.; Perlman, S.J. Multiple origins of obligate nematode and insect symbionts by a clade of bacteria closely related to plant pathogens. Proc. Natl. Acad. Sci. USA 2020, 117, 31979–31986. [Google Scholar] [CrossRef] [PubMed]
- Abbà, S.; Galetto, L.; Vallino, M.; Rossi, M.; Turina, M.; Sicard, A.; Marzachì, C. Genome sequence, prevalence and quantification of the first iflavirus identified in a phytoplasma insect vector. Arch. Virol. 2017, 162, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Campbell, B.C.; Purcell, A.H. Phylogenetic affiliation of BEV, a bacterial parasite of the leafhopper Euscelidius variegatus, on the basis of 16S rDNA sequences. Curr. Microbiol. 1993, 26, 37–41. [Google Scholar] [CrossRef]
- Galetto, L.; Nardi, M.; Saracco, P.; Bressan, A.; Marzachì, C.; Bosco, D. Variation in vector competency depends on chrysanthemum yellows phytoplasma distribution within Euscelidius variegatus. Entomol. Exp. Appl. 2009, 131, 200–207. [Google Scholar] [CrossRef]
- Purcell, A.H.; Steiner, T.; Mégraud, F.; Bové, J. In vitro isolation of a transovarially transmitted bacterium from the leafhopper Euscelidius variegatus (Hemiptera: Cicadellidae). J. Invertebr. Pathol. 1986, 48, 66–73. [Google Scholar] [CrossRef]
- Cheung, W.W.K.; Purcell, A.H. Ultrastructure of the digestive system of the leafhopper Euscelidius variegatus Kirshbaum (Homoptera: Cicadellidae), with and without congenital bacterial infections. Int. J. Insect Morphol. Embryol. 1993, 22, 49–61. [Google Scholar] [CrossRef]
- Degnan, P.H.; Bittleston, L.S.; Hansen, A.K.; Sabree, Z.L.; Moran, N.A.; Almeida, R.P.P. Origin and examination of a leafhopper facultative endosymbiont. Curr. Microbiol. 2011, 62, 1565–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitbart, M.; Salamon, P.; Andresen, B.; Mahaffy, J.M.; Segall, A.M.; Mead, D.; Azam, F.; Rohwer, F. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. USA 2002, 99, 14250–14255. [Google Scholar] [CrossRef] [Green Version]
- Paez-Espino, D.; Eloe-Fadrosh, E.; Pavlopoulos, G.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Manrique, P.; Bolduc, B.; Walk, S.T.; van der Oost, J.; de Vos, W.M.; Young, M.J. Healthy human gut phageome. Proc. Natl. Acad. Sci. USA 2016, 113, 10400–10405. [Google Scholar] [CrossRef] [Green Version]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef] [Green Version]
- Yutin, N.; Makarova, K.S.; Gussow, A.B.; Krupovic, M.; Segall, A.; Edwards, R.A.; Koonin, E.V. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol. 2018, 3, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Guerin, E.; Shkoporov, A.; Stockdale, S.R.; Clooney, A.G.; Ryan, F.J.; Sutton, T.D.S.; Draper, L.A.; Gonzalez-Tortuero, E.; Ross, R.P.; Hill, C. Biology and taxonomy of crAss-like bacteriophages, the most abundant virus in the human gut. Cell Host Microbe 2018, 24, 653–664.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Chen, C.; Shen, W.; Huang, G.; Le, S.; Lu, S.; Li, M.; Zhao, Y.; Wang, J.; Rao, X.; et al. Global Transcriptomic Analysis of Interactions between Pseudomonas aeruginosa and Bacteriophage PaP3. Sci. Rep. 2016, 6, 19237. [Google Scholar] [CrossRef] [Green Version]
- Mojardín, L.; Salas, M. Global Transcriptional Analysis of Virus-Host Interactions between Phageφ29 and Bacillus subtilis. J. Virol. 2016, 90, 9293–9304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacher, J.C.; Flint, A.; Butcher, J.; Blasdel, B.; Reynolds, H.M.; Lavigne, R.; Stintzi, A.; Szymanski, C.M. Transcriptomic analysis of the campylobacter Jejuni response to T4-like phage NCTC 12673 infection. Viruses 2018, 10, 332. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, S.R.; Janowski, A.B.; Zhao, G.; Barouch, D.; Wang, D. Hyperexpansion of RNA Bacteriophage Diversity. PLoS Biol. 2016, 14, e1002409. [Google Scholar] [CrossRef] [PubMed]
- Starr, E.; Nuccio, E.E.; Pett-Ridge, J.; Banfield, J.F.; Firestone, M.K. Metatranscriptomic reconstruction reveals RNA viruses with the potential to shape carbon cycling in soil. Proc. Natl. Acad. Sci. USA 2019, 116, 25900–25908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquiod, S.; Nunes, I.; Brejnrod, A.; Hansen, M.A.; Holm, P.E.; Johansen, A.; Brandt, K.K.; Priemé, A.; Sørensen, A.J. Long-term soil metal exposure impaired temporal variation in microbial metatranscriptomes and enriched active phages. Microbiome 2018, 6, 223. [Google Scholar] [CrossRef]
- Sieradzki, E.; Ignacio-Espinoza, J.C.; Needham, D.; Fichot, E.B.; Fuhrman, J.A. Dynamic marine viral infections and major contribution to photosynthetic processes shown by spatiotemporal picoplankton metatranscriptomes. Nat. Commun. 2019, 10, 1169. [Google Scholar] [CrossRef]
- Galetto, L.; Abbà, S.; Rossi, M.; Vallino, M.; Pesando, M.; Arricau-Bouvery, N.; Dubrana, M.P.; Chitarra, W.; Pegoraro, M.; Bosco, D.; et al. Two phytoplasmas elicit different responses in the insect vector Euscelidius variegatus Kirschbaum. Infect. Immun. 2018, 86, e00042-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, R.A.; McNair, K.; Faust, K.; Raes, J.; Dutilh, B.E. Computational approaches to predict bacteriophage–host relationships. FEMS Microbiol. Rev. 2016, 40, 258–272. [Google Scholar] [CrossRef] [Green Version]
- Boeckaerts, D.; Stock, M.; Criel, B.; Gerstmans, H.; De Baets, B.; Briers, Y. Predicting bacteriophage hosts based on sequences of annotated receptor-binding proteins. Sci. Rep. 2021, 11, 1467. [Google Scholar] [CrossRef]
- Gonella, E.; Crotti, E.; Mandrioli, M.; Daffonchio, D.; Alma, A. Asaia symbionts interfere with infection by “flavescence dorée” phytoplasma in leafhoppers. J. Pest Sci. 2018, 91, 1033–1046. [Google Scholar] [CrossRef]
- Tvedte, E.S.; Walden, K.K.O.; McElroy, K.E.; Werren, J.H.; Forbes, A.A.; Hood, G.R.; Logsdon, J.M.; Feder, J.L.; Robertson, H.M. Genome of the parasitoid wasp Diachasma alloeum, an emerging model for ecological speciation and transitions to asexual reproduction. Genome Biol. Evol. 2019, 11, 2767–2773. [Google Scholar] [CrossRef]
- Argov, T.; Sapir, S.R.; Pasechnek, A.; Azulay, G.; Stadnyuk, O.; Rabinovich, L.; Sigal, N.; Borovok, I.; Herskovits, A.A. Coordination of cohabiting phage elements supports bacteria–phage cooperation. Nat. Commun. 2019, 10, 5288. [Google Scholar] [CrossRef] [Green Version]
- Williamson, K.E.; Schnitker, J.B.; Radosevich, M.; Smith, D.W.; Wommack, K.E. Cultivation-based assessment of lysogeny among soil bacteria. Microb. Ecol. 2008, 56, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Burns, N.; Chloe, E.; James, C.E.; Harrison, E. Polylysogeny magnifies competitiveness of a bacterial pathogen in vivo. Evol. Appl. 2015, 8, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [Green Version]
- Refardt, D. Within-host competition determines reproductive success of temperate bacteriophages. ISME J. 2011, 5, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Marzachì, C.; Veratti, F.; Bosco, D. Direct PCR detection of phytoplasmas in experimentally infected insects. Ann. Appl. Biol. 1998, 133, 45–54. [Google Scholar] [CrossRef]
- Ottati, S.; Persico, A.; Rossi, M.; Bosco, D.; Vallino, M.; Abbà, S.; Molinatto, G.; Palmano, S.; Balestrini, R.; Galetto, L.; et al. Biological characterization of Euscelidius variegatus iflavirus 1. J. Invertebr. Pathol. 2020, 173, 107370. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. The CIPRES science gateway: Enabling high-impact science for phylogenetics researchers with limited resources. In Proceedings of the 1st Conference of the Extreme Science and Engineering Discovery Environment: Bridging from the eXtreme to the campus and beyond (XSEDE ‘12), Chicago, IL, USA, 16–19 July 2012; Association for Computing Machinery: New York, NY, USA, 2012. Article 39. pp. 1–8. [Google Scholar] [CrossRef]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley & Sons: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]





| Transcripts IDs and Predicted ORFs | Length (bp) | Range | RPK | Hit Description [Organism] | % Identities | E-Value | % Query Coverage |
|---|---|---|---|---|---|---|---|
| MW965290 | 5544 | 111.7 | |||||
| ORF1 | 398 | 1–398 | WP_195316289.1 phage tail tape measure protein [Serratia marcescens] | 70 | 2 × 10−51 | 97 | |
| ORF2 # | 357 | 398–754 | WP_039351538.1 phage tail protein [Pectobacterium fontis] | 90 | 1 × 10−75 | 97 | |
| ORF3 # | 753 | 804–1556 | MBG6243408.1 phage minor tail protein L [Candidatus Symbiopectobacterium sp. Dall1.0] | 96 | 0.0 | 100 | |
| ORF4 # | 576 | 1713–2288 | MBG6243407.1 peptidase P60 [Candidatus Symbiopectobacterium sp. Dall1.0] | 95 | 8 × 10−165 | 94 | |
| ORF5 # | 606 | 2272–2877 | WP_104212022.1 tail assembly protein [Pectobacterium brasiliense] | 81 | 2 × 10−85 | 100 | |
| ORF6 | 2611 | 2934–5544 | WP_104212026.1 phage tail protein [Pectobacterium brasiliense] | 91 | 2 × 10−70 | 100 | |
| MW965281 | 2188 | 112 | |||||
| ORF1 | 318 | 255–572 | WP_021179416 fimbrial protein TcfA [Serratia fonticola] | 62 | 4 × 10−29 | 90 | |
| ORF2 # | 252 | 557–808 | WP_146751463.1 ANR family transcriptional regulator [Enterobacter cloacae complex] | 56 | 5 × 10−7 | 60 | |
| ORF3 # | 1089 | 859–1947 | WP_187497555.1 phage tail protein [Pantoea Psp39-30] | 43 | 2 × 10−82 | 98 | |
| MW965289 | 1777 | 128.3 | |||||
| ORF1 | 99 | 1–99 | EFC4054519.1 HK97 family phage prohead protease [Escherichia coli] | 81 | 2 × 10−9 | 100 | |
| ORF2 # | 1227 | 109–1335 | MBG6243159.1 phage major capsid protein [Candidatus Symbiopectobacterium sp. Dall1.0] | 85 | 0.0 | 99 | |
| ORF3 # | 300 | 1426–1725 | WP_044208854.1Phage gp6-like head-tail connector family protein [Pectobacterium odoriferum] | 87 | 3 × 10−57 | 100 | |
| MW965291 | 6115 | 213.4 | |||||
| ORF1 | 580 | 1–580 | WP_108703399 Terminase small subunit [Enterobacter hormaechei] | 96 | 2 × 10−86 | 96 | |
| ORF2 # | 1659 | 583–2241 | WP_108703400 terminase large subunit [Citrobacter europaeus] | 98 | 0.0 | 100 | |
| ORF3 # | 1935 | 2324–4258 | WP_135684645.1 phage major capsid protein [Klebsiella pneumoniae] | 95 | 0.0 | 99 | |
| ORF4 # | 168 | 4297–4464 | WP_181941880.1 hypothetical protein [Klebsiella pneumoniae] | 89 | 8 × 10−28 | 100 | |
| ORF5 # | 1359 | 4464–5822 | NIC64170.1 phage portal protein [Klebsiella pneumoniae] | 94 | 0.0 | 100 | |
| ORF6 | 297 | 5819–6115 | RTO54147.1 phage gp6-like head-tail connector protein, partial [Enterobacter hormaechei] | 80 | 8 × 10−47 | 100 | |
| MW965287 | 1419 | 71.2 | |||||
| ORF1 | 482 | 1–482 | MBD2797976.1 HK97 family phage prohead protease [Xenorhabdus sp. 18] | 72 | 6 × 10−74 | 96 | |
| ORF2 | 953 | 467–1419 | QBY47020.1 phage portal protein [Arsenophonus nasoniae] | 92 | 0.0 | 100 | |
| MW965282 | 770 | 61.0 | WP_187497555.1 putative phage tail protein [Plautia stali symbiont] | 99 | 1 × 10−158 | 84 | |
| MW965283 | 3268 | 114.1 | WP_113869621.1 phage tail tape measure protein [Brenneria salicis] | 82 | 0.0 | 82 | |
| MW965286 * | 251 | 47.8 | SPW64604.1 putative head-tail adaptor [Escherichia coli] | 98 | 1 × 10−20 | 73 | |
| MW965288 | 768 | 50.8 | MBJ9599707.1 phage portal protein [Citrobacter werkmanii] | 99 | 4 × 10−179 | 94 | |
| MW965292 # | 1852 | 123.1 | WP_164114194.1 phage major capsid protein [Serratia marcescens] | 63 | 0.0 | 77 | |
| MW965285 | 299 | 23.4 | WP_010281992.1 portal protein [Pectobacterium brasiliense] | 95 | 8 × 10−55 | 92 | |
| MW965284 * | 345 | 46.4 | SUH06759.1 portal protein [Salmonella enterica subsp. enterica] | 87 | 1 × 10−27 | 66 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallino, M.; Rossi, M.; Ottati, S.; Martino, G.; Galetto, L.; Marzachì, C.; Abbà, S. Bacteriophage-Host Association in the Phytoplasma Insect Vector Euscelidius variegatus. Pathogens 2021, 10, 612. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10050612
Vallino M, Rossi M, Ottati S, Martino G, Galetto L, Marzachì C, Abbà S. Bacteriophage-Host Association in the Phytoplasma Insect Vector Euscelidius variegatus. Pathogens. 2021; 10(5):612. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10050612
Chicago/Turabian StyleVallino, Marta, Marika Rossi, Sara Ottati, Gabriele Martino, Luciana Galetto, Cristina Marzachì, and Simona Abbà. 2021. "Bacteriophage-Host Association in the Phytoplasma Insect Vector Euscelidius variegatus" Pathogens 10, no. 5: 612. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10050612