
Transmission Dynamics of Bovine Viral Diarrhea Virus in Hokkaido, Japan by Phylogenetic and Epidemiological Network Approaches


Abstract
:1. Introduction
2. Results
2.1. Phylogenetic Analysis
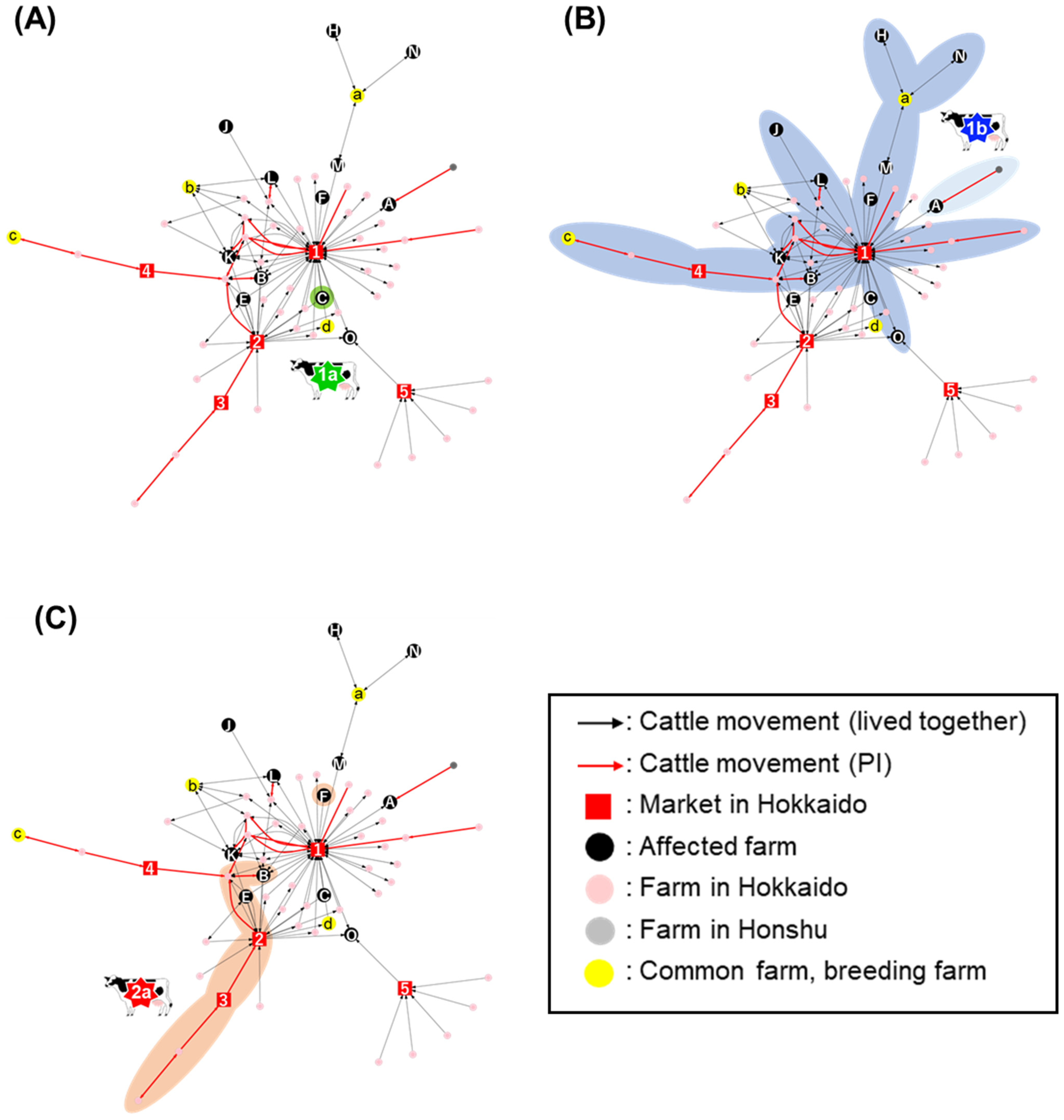
2.2. Network Analysis of Cattle Movement
2.3. Centrality Analysis in the Network
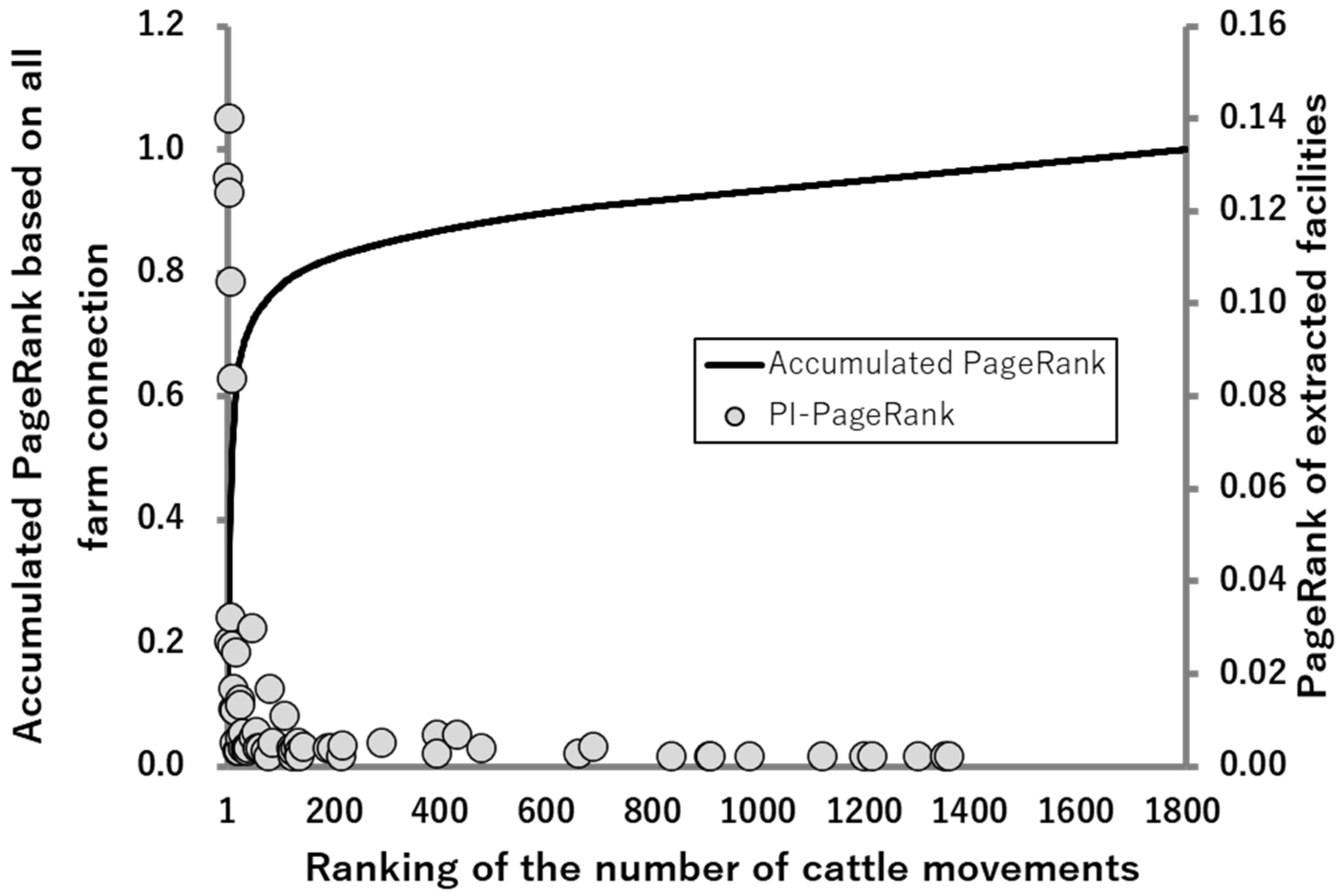
2.3.1. PageRank Analysis against the PI-Network (PI-PageRank)
2.3.2. Association between PI PageRank and the PageRank Based on All Farm Connections
3. Discussion
4. Materials and Methods
4.1. Characteristics of the Farms and Animals in This Study
4.2. Phylogenetic Analysis
4.2.1. Viruses and Cells
4.2.2. RT-PCR and Sequencing
4.2.3. Phylogenetic Tree
4.3. Epidemiological Analysis
4.3.1. Network Analysis
4.3.2. PageRank Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Committee on Taxonomy of Viruses. Available online: https://talk.ictvonline.org/taxonomy (accessed on 6 July 2021).
- Tautz, N.; Tews, B.A.; Meyers, G. The Molecular biology of pestiviruses. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2015; Volume 93, pp. 47–160. ISBN 978-0-12-802179-8. [Google Scholar]
- Mirosław, P.; Polak, M.P. Variability of E2 protein-coding sequences of bovine viral diarrhea virus in Polish cattle. Virus Genes 2020, 56, 515–521. [Google Scholar] [CrossRef]
- Hoppe, I.B.A.L.; de Souza-Pollo, A.; de Medeiros, A.S.R.; Samara, S.I.; Carvalho, A.A.B. HoBi-like pestivirus infection in an outbreak of bovine respiratory disease. Res. Vet. Sci. 2019, 126, 184–191. [Google Scholar] [CrossRef]
- Lanyon, S.R.; Hill, F.I.; Reichel, M.P.; Brownlie, J. Bovine viral diarrhoea: Pathogenesis and diagnosis. Vet. J. 2014, 199, 201–209. [Google Scholar] [CrossRef]
- Meyling, A.; Houe, H.; Jensen, A. Epidemiology of bovine virus diarrhoea virus. Rev. Sci. Technol. Int. Off. Epizoot. 1990, 9, 75–93. [Google Scholar] [CrossRef]
- Brownlie, J.; Hooper, L.; Thompson, I.; Collins, M. Maternal recognition of foetal infection with bovine virus diarrhoea virus (BVDV)—the bovine pestivirus. Clin. Diagn. Virol. 1998, 10, 141–150. [Google Scholar] [CrossRef]
- Baker, J.C. The Clinical manifestations of bovine viral diarrhea infection. Vet. Clin. N. Am. Food Anim. Pract. 1995, 11, 425–445. [Google Scholar] [CrossRef]
- Ministry of Agriculture, Forestry and Fisheries. Guidelines for Prevention and Control of Bovine Viral Diarrhea and Mucosal Disease. Available online: https://www.maff.go.jp/j/syouan/douei/pdf/bvd_md_gl.pdf (accessed on 30 March 2021). (In Japanese)
- Kadohira, M.; Tajima, M. A case control study of bovine viral diarrhea virus (BVDV) persistent infection (PI) in Betsukai, Hokkaido, Japan. J. Vet. Med. Sci. 2010, 72, 635–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hokkaido Government Livestock Promotion Division, Production Promotion Bureau. Outbreaks of Infectious Diseases in Livestock. Available online: http://www.pref.hokkaido.lg.jp/ns/tss/kachikueisei/densenseisippei.htm (accessed on 30 March 2021). (In Japanese).
- Løken, T.; Nyberg, O. Eradication of BVDV in cattle: The Norwegian project. Vet. Rec. 2013, 172, 661. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, K.; Alenius, S. BVDV control and eradication in Europe—An update. Jpn. J. Vet. Res. 2012, 60, S31–S39. [Google Scholar] [PubMed]
- Isoda, N.; Asano, A.; Ichijo, M.; Ohno, H.; Sato, K.; Okamoto, H.; Nakao, S.; Kato, H.; Saito, K.; Ito, N.; et al. Assessment of the cost effectiveness of compulsory testing of introduced animals and bulk tank milk testing for bovine viral diarrhea in Japan. J. Vet. Med. Sci. 2019, 81, 577–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomann, B.; Tschopp, A.; Magouras, I.; Meylan, M.; Schüpbach-Regula, G.; Häsler, B. Economic evaluation of the eradication program for bovine viral diarrhea in the Swiss dairy sector. Prev. Vet. Med. 2017, 145, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, R.E.; Thomas, C.J.; El-Attar, L.M.; Gunn, G.; Brownlie, J. A Phylogenetic Analysis of bovine viral diarrhoea virus (BVDV) isolates from six different regions of the UK and links to animal movement data. Vet. Res. 2013, 44, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekiguchi, S.; Presi, P.; Omori, R.; Staerk, K.; Schuppers, M.; Isoda, N.; Yoshikawa, Y.; Umemura, T.; Nakayama, H.; Fujii, Y.; et al. Evaluation of bovine viral diarrhoea virus control strategies in dairy herds in Hokkaido, Japan, using stochastic modelling. Transbound. Emerg. Dis. 2018, 65, e135–e144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fèvre, E.M.; Bronsvoort, B.M.d.C.; Hamilton, K.A.; Cleaveland, S. Animal movements and the spread of infectious diseases. Trends Microbiol. 2006, 14, 125–131. [Google Scholar] [CrossRef] [PubMed]
- de Menezes, T.C.; Luna, I.; de Miranda, S.H.G. Network analysis of cattle movement in Mato Grosso Do Sul (Brazil) and implications for foot-and-mouth disease. Front. Vet. Sci. 2020, 7, 219. [Google Scholar] [CrossRef] [PubMed]
- Beaunée, G.; Vergu, E.; Ezanno, P. Modelling of paratuberculosis spread between dairy cattle farms at a regional scale. Vet. Res. 2015, 46, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanderWaal, K.; Perez, A.; Torremorrell, M.; Morrison, R.M.; Craft, M. Role of animal movement and indirect contact among farms in transmission of porcine epidemic diarrhea virus. Epidemics 2018, 24, 67–75. [Google Scholar] [CrossRef]
- Notsu, K.; Wiratsudakul, A.; Mitoma, S.; Daous, H.E.; Kaneko, C.; El-Khaiat, H.M.; Norimine, J.; Sekiguchi, S. Quantitative risk assessment for the introduction of bovine leukemia virus-infected cattle using a cattle movement network analysis. Pathogens 2020, 9, 903. [Google Scholar] [CrossRef]
- Abe, Y.; Tamura, T.; Torii, S.; Wakamori, S.; Nagai, M.; Mitsuhashi, K.; Mine, J.; Fujimoto, Y.; Nagashima, N.; Yoshino, F.; et al. Genetic and antigenic characterization of bovine viral diarrhea viruses isolated from cattle in Hokkaido, Japan. J. Vet. Med. Sci. 2016, 78, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Nagai, M.; Hayashi, M.; Itou, M.; Fukutomi, T.; Akashi, H.; Kida, H.; Sakoda, Y. Identification of new genetic subtypes of bovine viral diarrhea virus genotype 1 isolated in Japan. Virus Genes 2008, 36, 135–139. [Google Scholar] [CrossRef]
- Matsuno, K.; Sakoda, Y.; Kameyama, K.; Tamai, K.; Ito, A.; Kida, H. Genetic and pathobiological characterization of bovine viral diarrhea viruses recently isolated from cattle in Japan. J. Vet. Med. Sci. 2007, 69, 515–520. [Google Scholar] [CrossRef] [Green Version]
- Tinsley, M.; Lewis, F.I.; Brülisauer, F. Network modeling of BVD transmission. Vet. Res. 2012, 43, 11. [Google Scholar] [CrossRef] [Green Version]
- Alban, L.; Stryhn, H.; Kjeldsen, A.M.; Ersbøll, A.K.; Skjøth, F.; Christensen, J.; Bitsch, V.; Chriél, M.; Strøger, U. Estimating transfer of bovine virus-diarrhoea virus in Danish cattle by use of register data. Prev. Vet. Med. 2001, 52, 133–146. [Google Scholar] [CrossRef]
- Schärrer, S.; Widgren, S.; Schwermer, H.; Lindberg, A.; Vidondo, B.; Zinsstag, J.; Reist, M. Evaluation of farm-level parameters derived from animal movements for use in risk-based surveillance programmes of cattle in Switzerland. BMC Vet. Res. 2015, 11, 149. [Google Scholar] [CrossRef] [Green Version]
- Akagami, M.; Seki, S.; Kashima, Y.; Yamashita, K.; Oya, S.; Fujii, Y.; Takayasu, M.; Yaguchi, Y.; Suzuki, A.; Ono, Y.; et al. Risk factors associated with the within-farm transmission of bovine viral diarrhea virus and the incidence of persistently infected cattle on dairy farms from Ibaraki prefecture of Japan. Res. Vet. Sci. 2020, 129, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Han, J.-H.; Holter, J.; Moffat, J.; Weston, J.F.; Heuer, C.; Gates, M.C. Using bayesian network modelling to untangle farm management risk factors for bovine viral diarrhoea virus infection. Prev. Vet. Med. 2018, 161, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Littlejohns, I. Complications to the Study of the Relationship between Bovine Lymphocyte Antigens and Mucosal Disease; Deptartment of Veterinary Microbiology and Pathology, Washington State University: Washington, DC, USA, 1985. [Google Scholar]
- McGowan, M.; Kirkland, P.; Richards, S.; Littlejohns, I. Increased reproductive losses in cattle infected with bovine pestivirus around the time of insemination. Vet. Rec. 1993, 133, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Reardon, F.; Graham, D.A.; Clegg, T.A.; Tratalos, J.A.; O’Sullivan, P.; More, S.J. Quantifying the role of trojan dams in the between-herd spread of bovine viral diarrhoea virus (BVDv) in Ireland. Prev. Vet. Med. 2018, 152, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Isoda, K. Assessment of Various Testing Methods for Bovine Viral Diarrhea; Tokyo Metropolitan Livestock Hygiene Service Center: Tokyo, Japan, 2015. (In Japanese) [Google Scholar]
- Vilček, Š.; Herring, A.J.; Herring, J.A.; Nettleton, P.F.; Lowings, J.P.; Paton, D.J. Pestiviruses Isolated from Pigs, Cattle and Sheep Can Be Allocated into at Least Three Genogroups Using Polymerase Chain Reaction and Restriction Endonuclease Analysis. Arch. Virol. 1994, 136, 309–323. [Google Scholar] [CrossRef]
- Kameyama, K.; Sakoda, Y.; Tamai, K.; Nagai, M.; Akashi, H.; Kida, H. Genetic recombination at different points in the Npro-coding region of bovine viral diarrhea viruses and the potentials to change their antigenicities and pathogenicities. Virus Res. 2006, 116, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Kameyama, K.; Sakoda, Y.; Tamai, K.; Igarashi, H.; Tajima, M.; Mochizuki, T.; Namba, Y.; Kida, H. Development of an immunochromatographic test kit for rapid detection of bovine viral diarrhea virus antigen. J. Virol. Methods 2006, 138, 140–146. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozasa, T.; Abe, Y.; Mitsuhashi, K.; Tamura, T.; Aoki, H.; Ishimaru, M.; Nakamura, S.; Okamatsu, M.; Kida, H.; Sakoda, Y. Analysis of a pair of END+ and END− viruses derived from the same bovine viral diarrhea virus stock reveals the amino acid determinants in Npro responsible for inhibition of type I interferon production. J. Vet. Med. Sci. 2015, 77, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Csárdi, G.; Nepusz, T. The igraph software package for complex network research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Brin, S.; Page, L. The anatomy of a large-scale hypertextual web search engine. Comput. Netw. ISDN Syst. 1998, 30, 107–117. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Number of Cattle | Number of PI Cattle | Isolates of Each Genotype | |||
|---|---|---|---|---|---|
| 1a | 1b | 2a | |||
| A | 608 | 4 | 0 | 3 | 1 |
| B | 605 | 3 | 0 | 2 | 1 |
| C | 1693 | 2 | 2 | 0 | 0 |
| D | 118 | 6 | 0 | 6 | 0 |
| E | 613 | 1 | 0 | 1 | 0 |
| F | 30 | 1 | 0 | 0 | 1 |
| G | 110 | 4 | 0 | 4 | 0 |
| H | 83 | 1 | 0 | 1 | 0 |
| I | 107 | 1 | 0 | 1 | 0 |
| J | 128 | 4 | 0 | 4 | 0 |
| K | 4120 | 1 | 0 | 1 | 0 |
| L | 78 | 2 | 0 | 2 | 0 |
| M | 295 | 6 | 0 | 6 | 0 |
| N | 84 | 2 | 0 | 2 | 0 |
| O | 723 | 3 | 0 | 3 | 0 |
| Total | 9395 | 41 | 2 | 36 | 3 |
| Ranking in the PI Network | PI-PageRank | Facility Status |
|---|---|---|
| 1 | 0.140 | Farm K |
| 2 | 0.128 | Market 1 |
| 3 | 0.124 | Farm in Hokkaido |
| 4 | 0.105 | Common farm a |
| 5 | 0.084 | Farm A |
| 6 | 0.032 | Market 2 |
| 7 | 0.030 | Breeding farm b |
| 8 | 0.027 | Farm O |
| 9 | 0.026 | Farm B |
| 10 | 0.025 | Farm in Hokkaido |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirose, S.; Notsu, K.; Ito, S.; Sakoda, Y.; Isoda, N. Transmission Dynamics of Bovine Viral Diarrhea Virus in Hokkaido, Japan by Phylogenetic and Epidemiological Network Approaches. Pathogens 2021, 10, 922. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10080922
Hirose S, Notsu K, Ito S, Sakoda Y, Isoda N. Transmission Dynamics of Bovine Viral Diarrhea Virus in Hokkaido, Japan by Phylogenetic and Epidemiological Network Approaches. Pathogens. 2021; 10(8):922. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10080922
Chicago/Turabian StyleHirose, Shizuka, Kosuke Notsu, Satoshi Ito, Yoshihiro Sakoda, and Norikazu Isoda. 2021. "Transmission Dynamics of Bovine Viral Diarrhea Virus in Hokkaido, Japan by Phylogenetic and Epidemiological Network Approaches" Pathogens 10, no. 8: 922. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10080922