
Comparative Analysis of Structural Features in SLiMs from Eukaryotes, Bacteria, and Viruses with Importance for Host-Pathogen Interactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. The Majority of Instances in the ELM Database Bind Ligands and Are from Human
2.2. Accessibility and Lack of Secondary Structure Influence SLiM Functionality More than Disorder
2.3. SLiMs from Viruses Are Less Disordered
2.4. Most SLiMs Lack Secondary Structure
2.5. Disordered or Flexible?
2.5.1. SLiMs Are Found in Flexible Regions
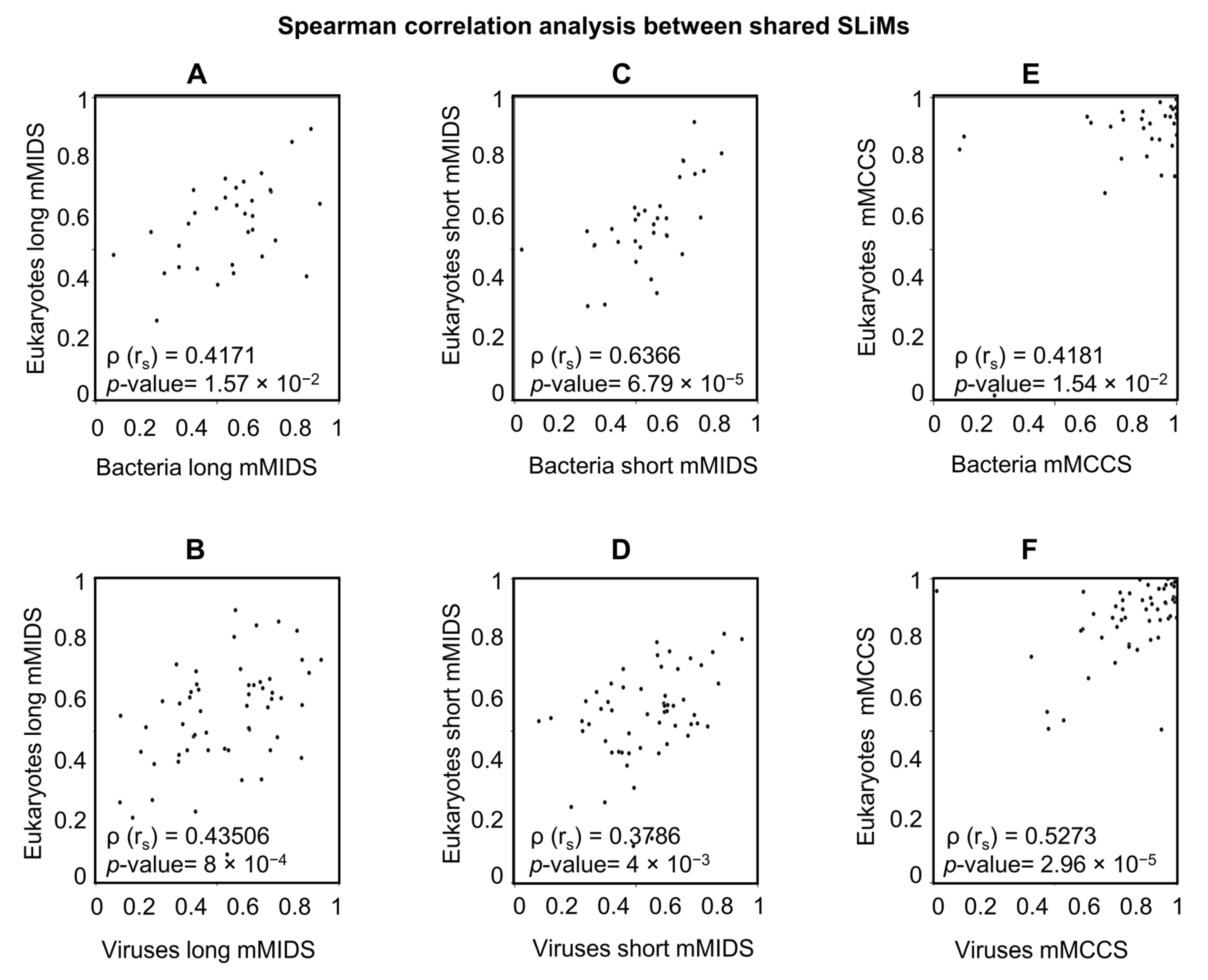
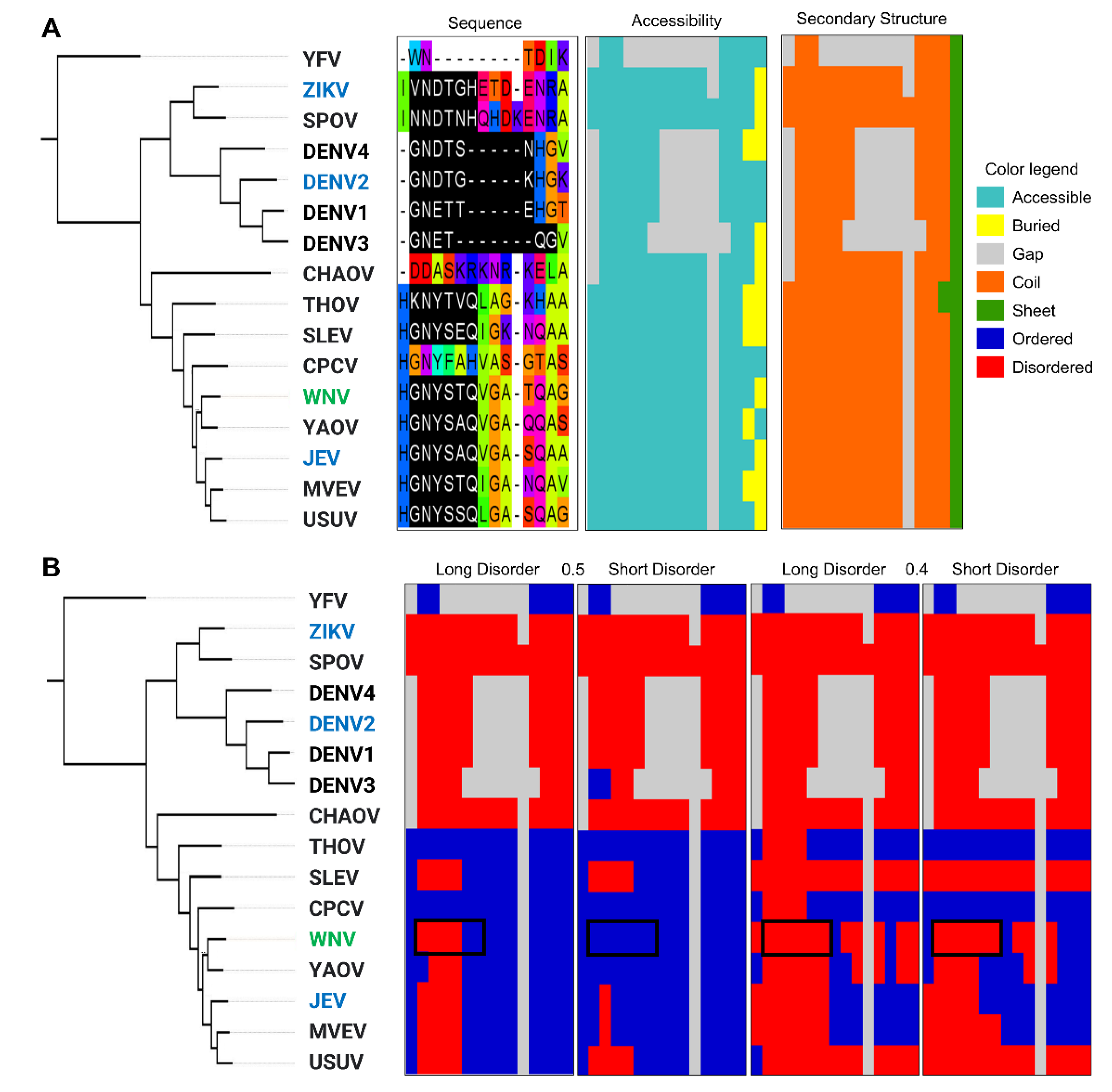
2.5.2. A Comparison of Viral and Bacterial Motifs with Their Corresponding Eukaryotic Motifs
2.5.3. To Fold or Not to Fold: A Tale of Two Motifs
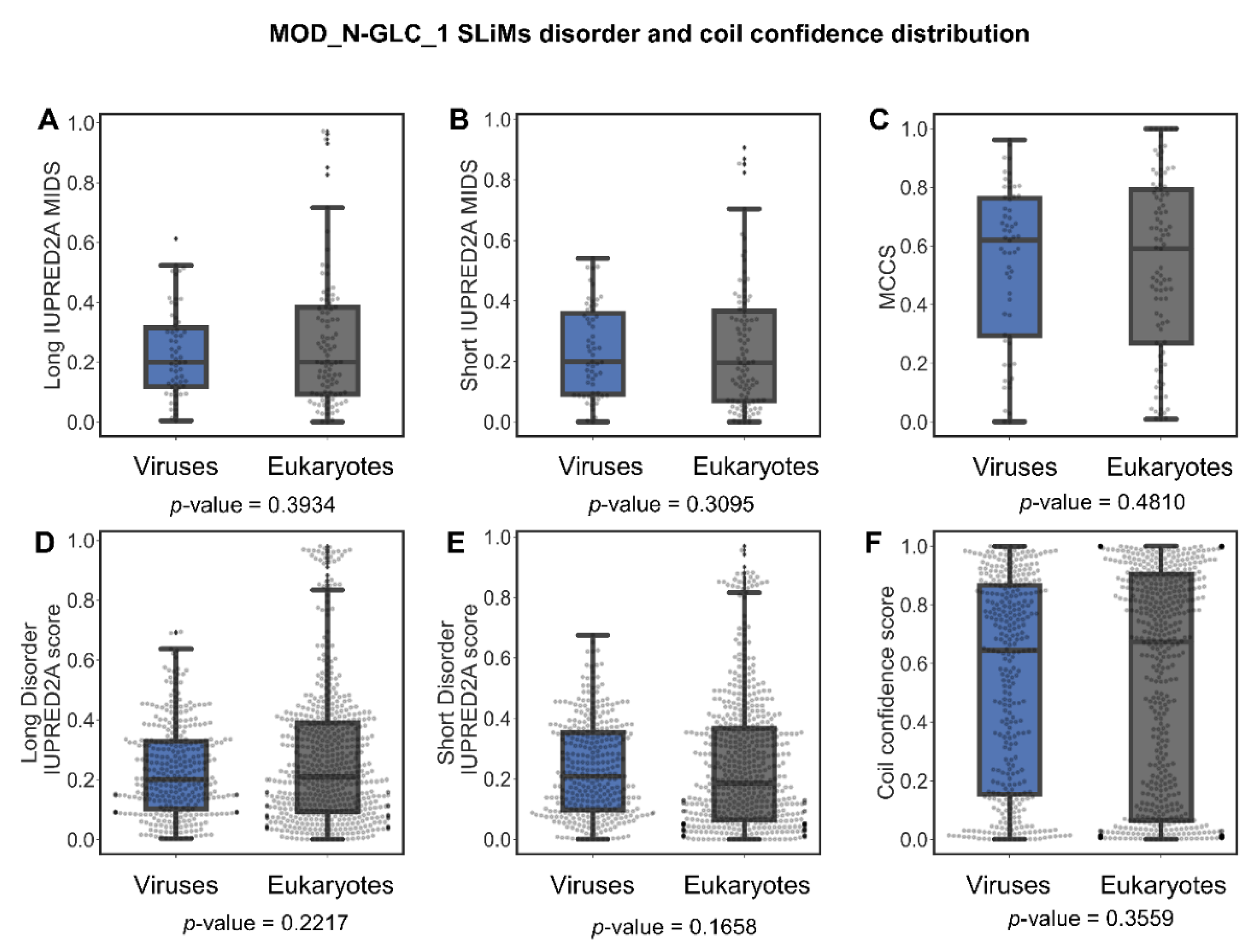
Are MOD_N-GLC_1 Instances Indeed Predominantly Ordered in Viruses or Is This Perhaps Due to Insufficient Data?
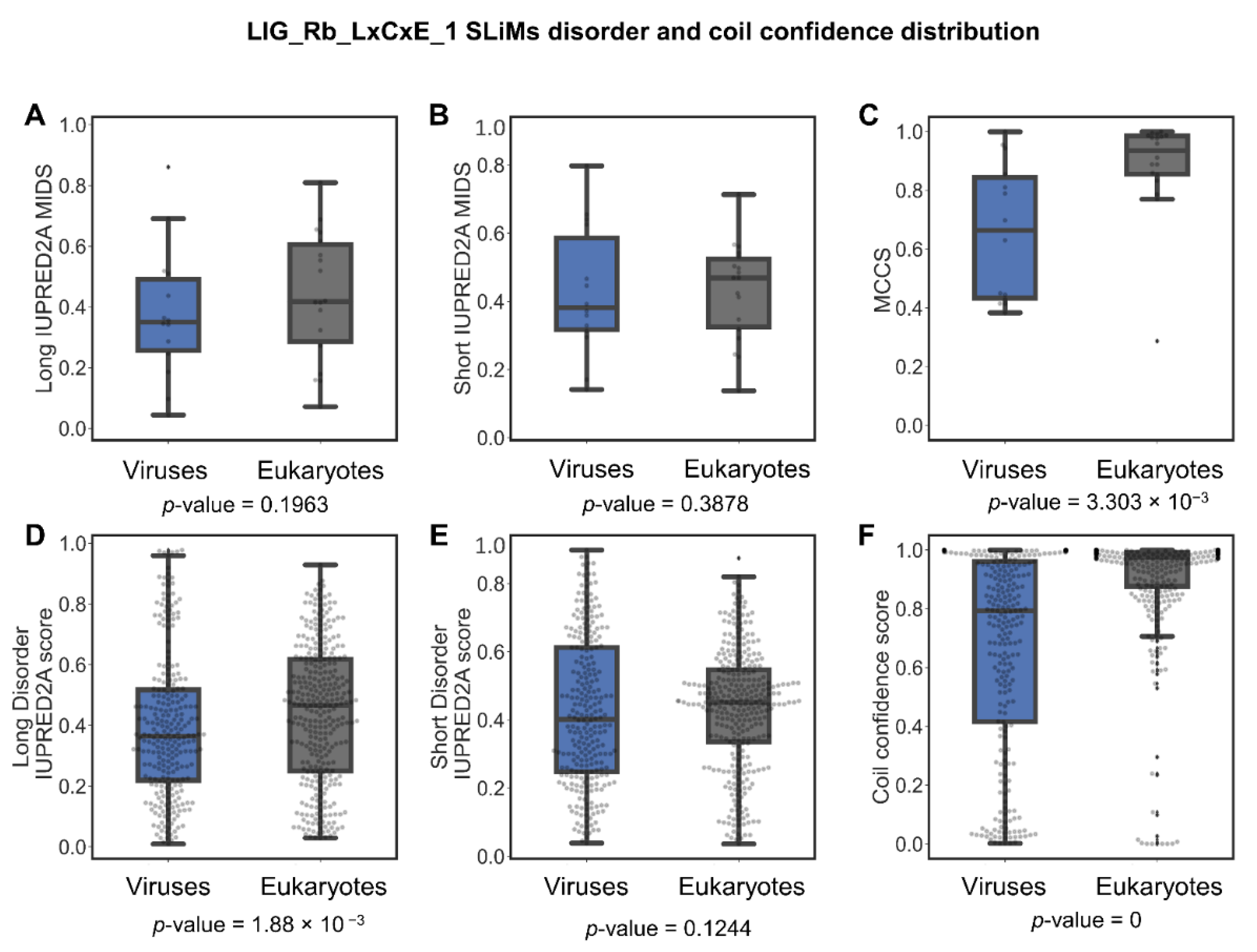
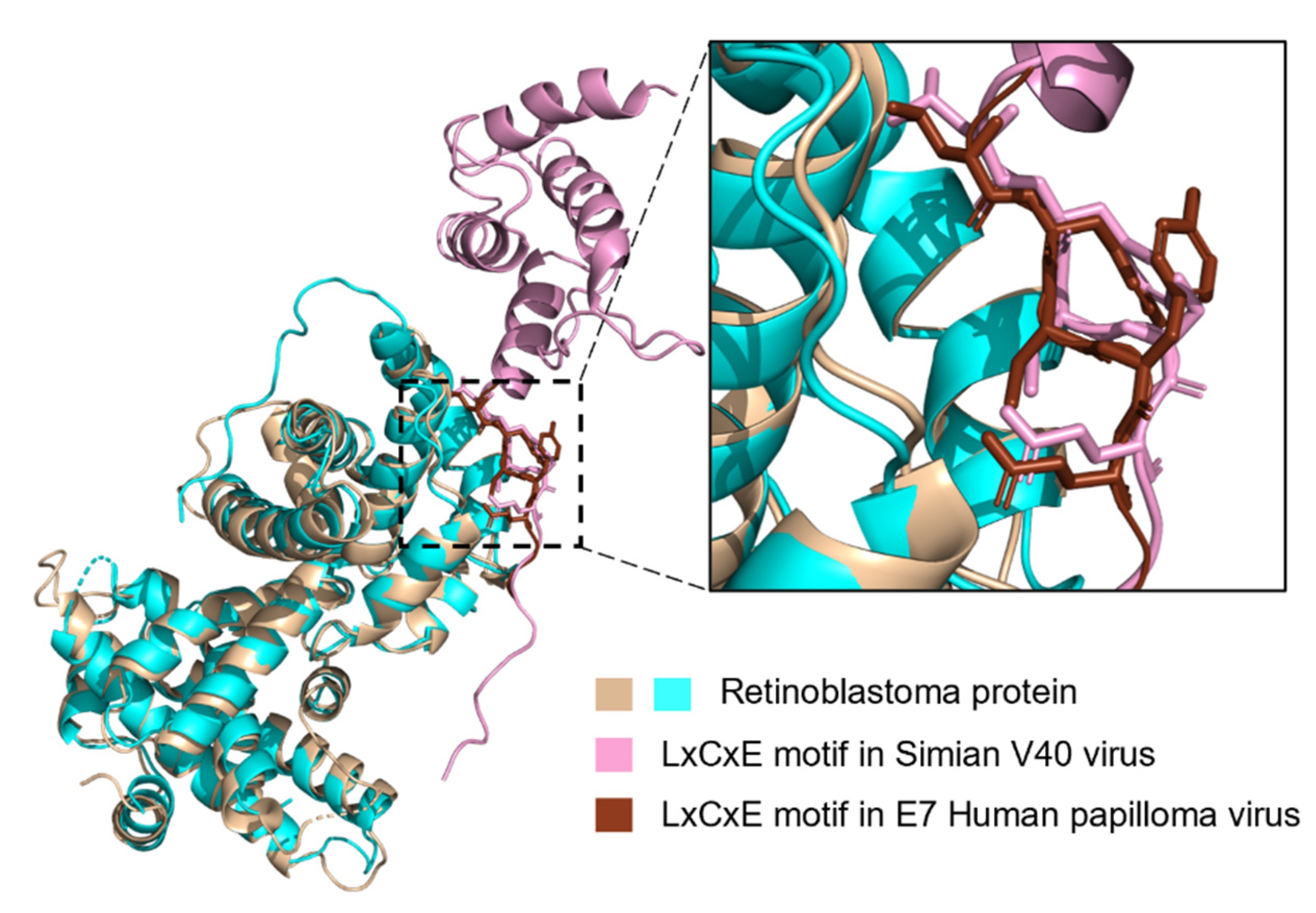
LIG_Rb_LxCxE_1 Is Less Disordered in Viruses
3. Conclusions
4. Methods
4.1. The ELM Dataset
4.2. Sequence-Based Structural Predictions
4.2.1. Intrinsic Disorder Prediction
4.2.2. Relative Solvent Accessibility and Secondary Structure Predictions
4.3. Phylogenetic Tree Analysis
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PPI | Protein-protein interactions |
| ELM | Eukaryotic Linear Motifs |
| SLiMs | Short Linear Motifs |
| IDR | Intrinsically Disordered protein Region |
| IDP | Intrinsically Disordered Protein |
| MIDS | Mean IUPRED2A Disorder Score |
| MCCS | Mean Coil Confidence Score |
| mMIDS | mean MIDS |
| mMCCS | mean MCCS |
| LIG | Ligand binding motifs |
| MOD | Post-translational modification motifs |
| TRG | Targeting motifs |
| DOC | Docking motifs |
| CLV | Cleavage sites motifs |
| DEG | Degradation motifs |
| WNV | West Nile Virus |
| Rb | Retinoblastoma protein |
| DSSP | Dictionary of Secondary Structure of Proteins |
| PDB | Protein Data Bank |
References
- Rao, V.S.; Srinivas, K.; Sujini, G.N.; Kumar, G.N.S. Protein-protein interaction detection: Methods and analysis. Int. J. Proteom. 2014, 2014, 147648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, P.; Gingras, A.C. History of protein–protein interactions: From egg-white to complex networks. Proteomics 2012, 12, 1478–1498. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets: The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett. 2015, 589, 2498–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davey, N.E.; van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol. Biosyst. 2012, 8, 268–281. [Google Scholar] [CrossRef]
- Guven-Maiorov, E.; Tsai, C.J.; Nussinov, R. Pathogen mimicry of host protein-protein interfaces modulates immunity. Semin. Cell Dev. Biol. 2016, 58, 136–145. [Google Scholar] [CrossRef]
- Sámano-Sánchez, H.; Gibson, T.J. Mimicry of short linear motifs by bacterial pathogens: A drugging opportunity. Trends Biochem. Sci. 2020, 45, 526–544. [Google Scholar] [CrossRef]
- Davey, N.E.; Travé, G.; Gibson, T.J. How viruses hijack cell regulation. Trends Biochem. Sci. 2011, 36, 159–169. [Google Scholar] [CrossRef]
- Hraber, P.; O’Maille, P.E.; Silberfarb, A.; Davis-Anderson, K.; Generous, N.; McMahon, B.H.; Fair, J.M. Resources to discover and use short linear motifs in viral proteins. Trends Biotechnol. 2020, 38, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Fuxreiter, M.; Tompa, P.; Simon, I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics 2007, 23, 950–956. [Google Scholar] [CrossRef]
- Davey, N.E.; Cyert, M.S.; Moses, A.M. Short linear motifs—Ex nihilo evolution of protein regulation. Cell Commun. Signal. 2015, 13, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Intrinsically disordered proteins and their “mysterious” (meta)physics. Front. Phys. 2019, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Babu, M.M.; Kriwacki, R.W.; Pappu, R.V. Versatility from protein disorder. Science 2012, 337, 1460–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, R.J.; Paulsen, K.; Aguilar Gomez, C.M.; Pérez-Bercoff, Å. Computational prediction of disordered protein motifs using SLiMSuite. Methods Mol. Biol. 2020, 2141, 37–72. [Google Scholar] [CrossRef] [PubMed]
- Gould, C.M.; Diella, F.; Via, A.; Puntervoll, P.; Gemünd, C.; Chabanis-Davidson, S.; Michael, S.; Sayadi, A.; Bryne, J.C.; Chica, C.; et al. ELM: The status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res. 2010, 38, D167–D180. [Google Scholar] [CrossRef]
- Gibson, T.J.; Dinkel, H.; van Roey, K.; Diella, F. Experimental detection of short regulatory motifs in eukaryotic proteins: Tips for good practice as well as for bad. Cell Commun. Signal. 2015, 13, 42. [Google Scholar] [CrossRef] [Green Version]
- Dinkel, H.; Sticht, H. A computational strategy for the prediction of functional linear peptide motifs in proteins. Bioinformatics 2007, 23, 3297–3303. [Google Scholar] [CrossRef]
- Via, A.; Gould, C.M.; Gemünd, C.; Gibson, T.J.; Helmer-Citterich, M. A structure filter for the eukaryotic linear motif resource. BMC Bioinform. 2009, 10, 351. [Google Scholar] [CrossRef]
- Davey, N.E.; Shields, D.C.; Edwards, R.J. Masking residues using context-specific evolutionary conservation significantly improves short linear motif discovery. Bioinformatics 2009, 25, 443–450. [Google Scholar] [CrossRef]
- Elkhaligy, H.; Balbin, C.A.; Gonzalez, J.L.; Liberatore, T.; Siltberg-Liberles, J. Dynamic, but Not necessarily disordered, human-virus interactions mediated through SLiMs in viral proteins. Viruses 2021, 13, 2369. [Google Scholar] [CrossRef]
- Pushker, R.; Mooney, C.; Davey, N.E.; Jacqué, J.-M.; Shields, D.C. Marked variability in the extent of protein disorder within and between viral families. PLoS ONE 2013, 8, e60724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastano, K.; Erdős, G.; Mier, P.; Alanis-Lobato, G.; Promponas, V.J.; Dosztányi, Z.; Andrade-Navarro, M.A. Evolutionary study of disorder in protein sequences. Biomolecules 2020, 10, 1413. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: Comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci. 2015, 72, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gouw, M.; Michael, S.; Sámano-Sánchez, H.; Pancsa, R.; Glavina, J.; Diakogianni, A.; Valverde, J.A.; Bukirova, D.; Čalyševa, J.; et al. ELM—The eukaryotic linear motif resource in 2020. Nucleic Acids Res. 2020, 48, D296–D306. [Google Scholar] [CrossRef] [Green Version]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Sharma, R.; Raduly, Z.; Miskei, M.; Fuxreiter, M. Fuzzy complexes: Specific binding without complete folding. FEBS Lett. 2015, 589, 2533–2542. [Google Scholar] [CrossRef] [Green Version]
- Fuxreiter, M. Fuzziness: Linking regulation to protein dynamics. Mol. Biosyst. 2011, 8, 168–177. [Google Scholar] [CrossRef]
- Lis, H.; Sharon, N. Protein glycosylation: Structural and functional aspects. Eur. J. Biochem. 1993, 218, 1–27. [Google Scholar] [CrossRef]
- Vigerust, D.J.; Shepherd, V.L. Virus glycosylation: Role in virulence and immune interactions. Trends Microbiol. 2007, 15, 211–218. [Google Scholar] [CrossRef]
- Bosques, C.J.; Tschampel, S.M.; Woods, R.J.; Imperiali, B. Effects of glycosylation on peptide conformation: A synergistic experimental and computational study. J. Am. Chem. Soc. 2004, 126, 8421–8425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitling, J.; Aebi, M. N-linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Canada, C.; Kelleher, D.J.; Gilmore, R. Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 2009, 136, 272–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, S.; Chaudhary, B.P.; Zoetewey, D. Structural insight into the mechanism of N-linked glycosylation by oligosaccharyltransferase. Biomolecules 2020, 10, 624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelleher, D.J.; Karaoglu, D.; Mandon, E.C.; Gilmore, R. Oligosaccharyltransferase isoforms that contain different catalytic STT3 subunits have distinct enzymatic properties. Mol. Cell 2003, 12, 101–111. [Google Scholar] [CrossRef]
- Nybakken, G.E.; Nelson, C.A.; Chen, B.R.; Diamond, M.S.; Fremont, D.H. Crystal structure of the west nile virus envelope glycoprotein. J. Virol. 2006, 80, 11467. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Cherrier, M.V.; Kaufmann, B.; Nybakken, G.E.; Lok, S.M.; Warren, J.T.; Chen, B.R.; Nelson, C.A.; Kostyuchenko, V.A.; Holdaway, H.A.; Chipman, P.R.; et al. Structural basis for the preferential recognition of immature flaviviruses by a fusion-loop antibody. EMBO J. 2009, 28, 3269. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, T.; Romero, P.R.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in the protein data bank. J. Biomol. Struct. Dyn. 2007, 24, 325–341. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Xue, B.; Van, Y.Y.; Ulrich, E.L.; Markley, J.L.; Dunker, A.K.; Uversky, V.N. Utilization of protein intrinsic disorder knowledge in structural proteomics. Biochim. Biophys. Acta 2013, 1834, 487. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Stec, B.; Godzik, A. Between order and disorder in protein structures—Analysis of “dual personality” fragments in proteins. Structure 2007, 15, 1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, S.L.; Pierson, T.C.; Sanchez, M.D.; Ahmed, A.A.; Murtadha, M.M.; Doms, R.W. N-linked glycosylation of west nile virus envelope proteins influences particle assembly and infectivity. J. Virol. 2005, 79, 13262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondotte, J.A.; Lozach, P.-Y.; Amara, A.; Gamarnik, A.V. Essential role of dengue virus envelope protein N glycosylation at asparagine-67 during viral propagation. J. Virol. 2007, 81, 7136–7148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbaugh, D.L.; Baric, R.S.; Lazear, H.M. Envelope protein glycosylation mediates zika virus pathogenesis. J. Virol. 2019, 93, e00113-19. [Google Scholar] [CrossRef] [Green Version]
- Moudy, R.M.; Payne, A.F.; Dodson, B.L.; Kramer, L.D. Requirement of glycosylation of west nile virus envelope protein for infection of, but not spread within, culex quinquefasciatus mosquito vectors. Am. J. Trop. Med. Hyg. 2011, 85, 374. [Google Scholar] [CrossRef] [Green Version]
- Fall, G.; di Paola, N.; Faye, M.; Dia, M.; Freire, C.C.M.; Loucoubar, C.; Zanotto, P.M.A.; Faye, O.; Sall, A.A. Biological and phylogenetic characteristics of West African lineages of West Nile virus. PLoS Negl. Trop. Dis. 2017, 11, e0006078. [Google Scholar] [CrossRef]
- Wang, P.; Hu, K.; Luo, S.; Zhang, M.; Deng, X.; Li, C.; Jin, W.; Hu, B.; He, S.; Li, M.; et al. DC-SIGN as an attachment factor mediates Japanese encephalitis virus infection of human dendritic cells via interaction with a single high-mannose residue of viral E glycoprotein. Virology 2016, 488, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, A.; Gavin, M.R.; Luo, R.X.; Dean, D.C. Role of the LXCXE binding site in Rb function. Mol. Cell. Biol. 2000, 20, 6799–6805. [Google Scholar] [CrossRef] [Green Version]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671. [Google Scholar] [CrossRef]
- Fischer, M.; Müller, G.A. Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 638–662. [Google Scholar] [CrossRef] [Green Version]
- Palopoli, N.; Foutel, N.S.G.; Gibson, T.J.; Chemes, L.B. Short linear motif core and flanking regions modulate retinoblastoma protein binding affinity and specificity. Protein Eng. Des. Sel. 2018, 31, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Felsani, A.; Mileo, A.M.; Paggi, M.G. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene 2006, 25, 5277–5285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narisawa-Saito, M.; Kiyono, T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: Roles of E6 and E7 proteins. Cancer Sci. 2007, 98, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Helt, A.M.; Galloway, D.A. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 2003, 24, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, V.; Reiss, K.; Khalili, K.; de Falco, G.; Giordano, A. Role of the interaction between large T antigen and Rb family members in the oncogenicity of JC virus. Oncogene 2006, 25, 5294–5301. [Google Scholar] [CrossRef]
- Fan, Y.; Sanyal, S.; Bruzzone, R. Breaking bad: How viruses subvert the cell cycle. Front. Cell. Infect. Microbiol. 2018, 8, 396. [Google Scholar] [CrossRef]
- Chemes, L.B.; Sánchez, I.E.; de Prat-Gay, G. Kinetic recognition of the retinoblastoma tumor suppressor by a specific protein target. J. Mol. Biol. 2011, 412, 267–284. [Google Scholar] [CrossRef]
- Kim, H.Y.; Ahn, B.Y.; Cho, Y. Structural basis for the inactivation of retinoblastoma tumor suppressor by SV40 large T antigen. EMBO J. 2001, 20, 295. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.O.; Russo, A.A.; Pavletich, N.P. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 1998, 391, 859–865. [Google Scholar] [CrossRef]
- Davey, N.E.; Seo, M.-H.; Yadav, V.K.; Jeon, J.; Nim, S.; Krystkowiak, I.; Blikstad, C.; Dong, D.; Markova, N.; Kim, P.M.; et al. Discovery of short linear motif-mediated interactions through phage display of intrinsically disordered regions of the human proteome. FEBS J. 2017, 284, 485–498. [Google Scholar] [CrossRef]
- Wadie, B.; Kleshchevnikov, V.; Sandaltzopoulou, E.; Benz, C.; Correspondence, E.P.; Petsalaki, E. Use of viral motif mimicry improves the proteome-wide discovery of human linear motifs. Cell Rep. 2022, 39, 110764. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Sønderby, C.K.; Sommer, M.O.A.; Winther, O.; Nielsen, M.; Petersen, B.; et al. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins Struct. Funct. Bioinform. 2019, 87, 520–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; von Den Driesch, L.; Galiez, C.; Martin, M.J.; Soding, J.; Steinegger, M. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 2017, 45, D170–D176. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkhaligy, H.; Balbin, C.A.; Siltberg-Liberles, J. Comparative Analysis of Structural Features in SLiMs from Eukaryotes, Bacteria, and Viruses with Importance for Host-Pathogen Interactions. Pathogens 2022, 11, 583. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11050583
Elkhaligy H, Balbin CA, Siltberg-Liberles J. Comparative Analysis of Structural Features in SLiMs from Eukaryotes, Bacteria, and Viruses with Importance for Host-Pathogen Interactions. Pathogens. 2022; 11(5):583. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11050583
Chicago/Turabian StyleElkhaligy, Heidy, Christian A. Balbin, and Jessica Siltberg-Liberles. 2022. "Comparative Analysis of Structural Features in SLiMs from Eukaryotes, Bacteria, and Viruses with Importance for Host-Pathogen Interactions" Pathogens 11, no. 5: 583. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11050583