
Proteomic Characterization of the Oral Pathogen Filifactor alocis Reveals Key Inter-Protein Interactions of Its RTX Toxin: FtxA

 , and
, and 
Abstract
:1. Introduction
2. Results
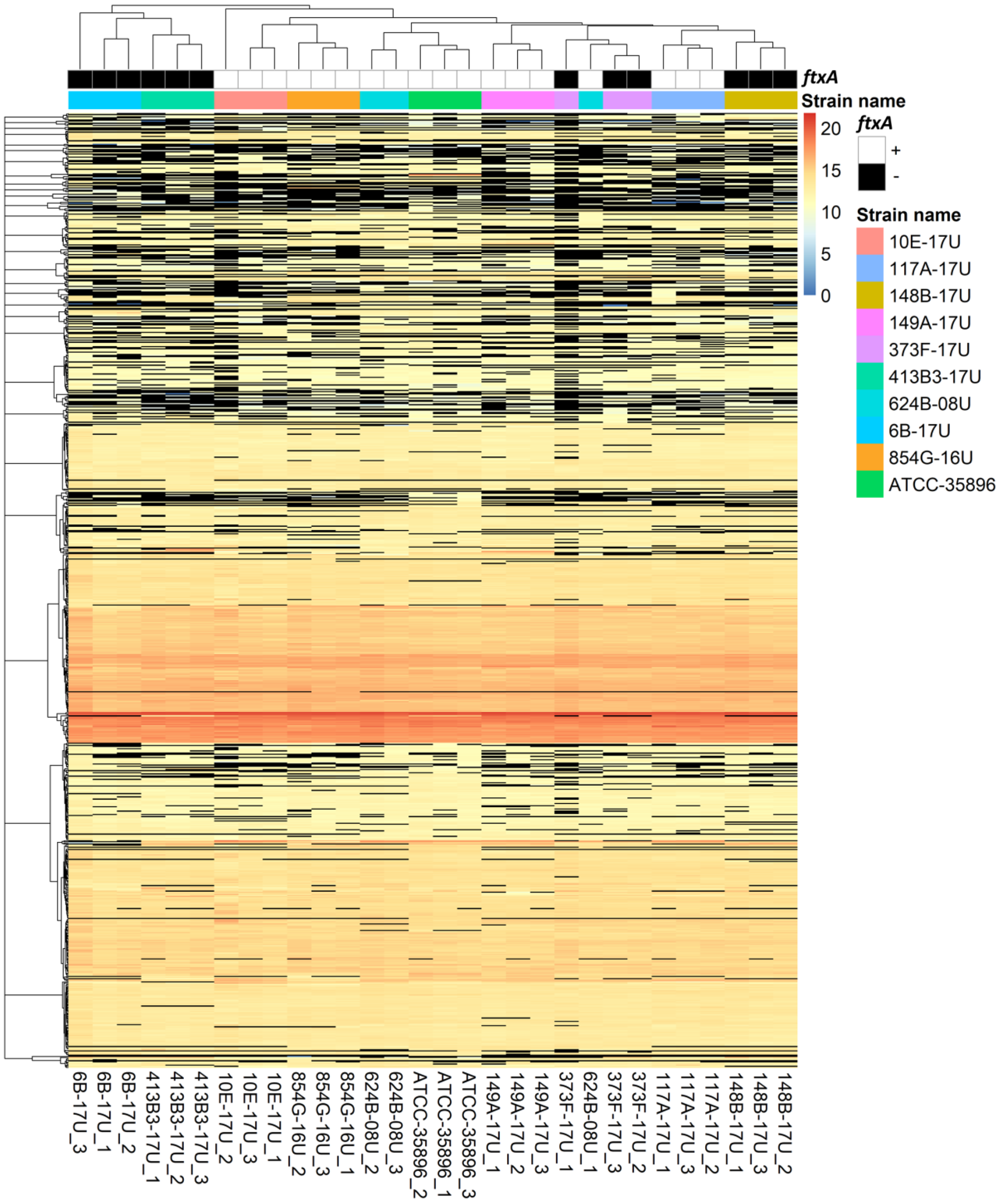
2.1. Proteome Profiles of Ten F. alocis Strains
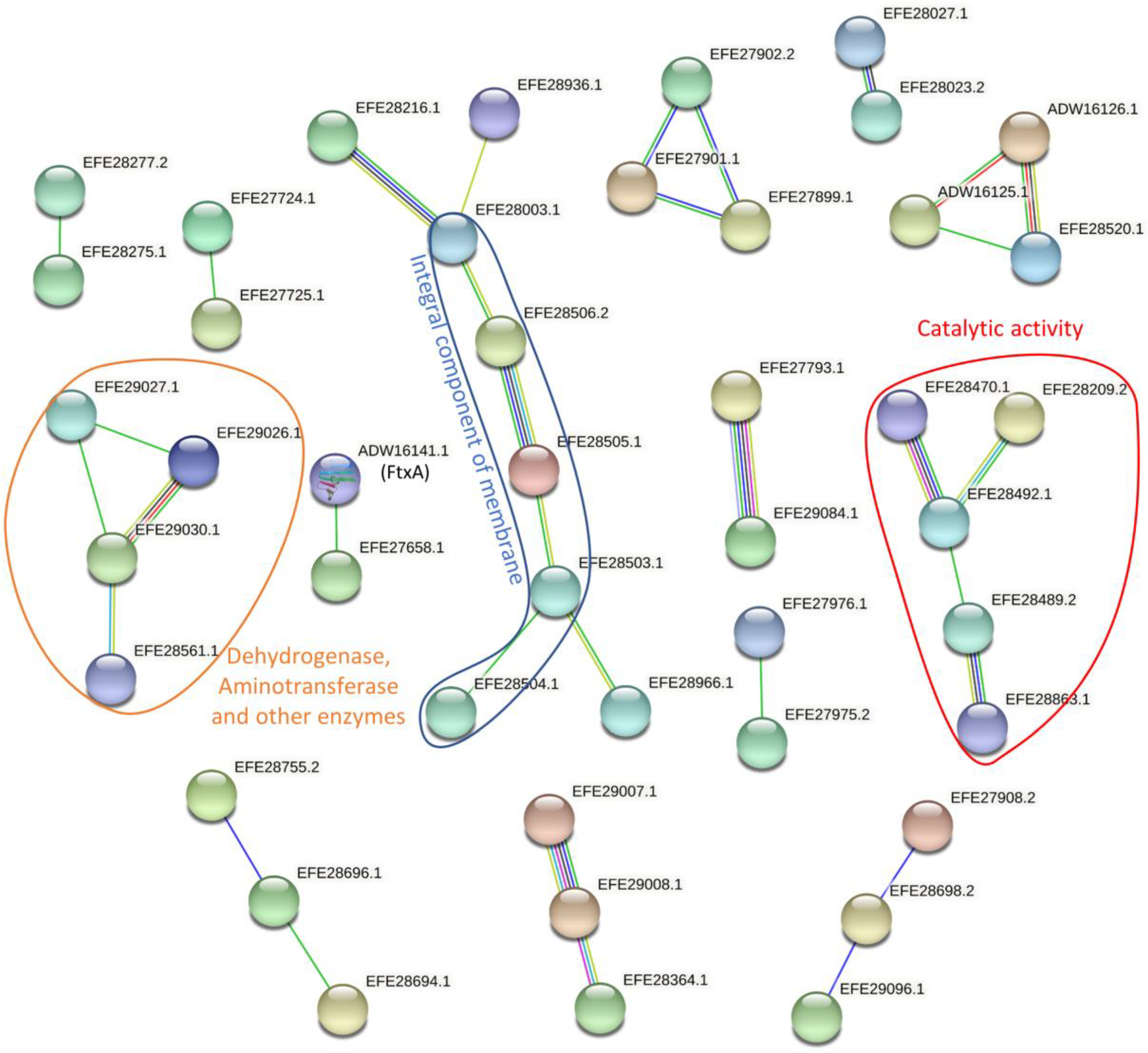
2.2. Differential Expression of Proteins between FtxA-Positive and -Negative Strains and Their Predicted Functional Protein Association Networks
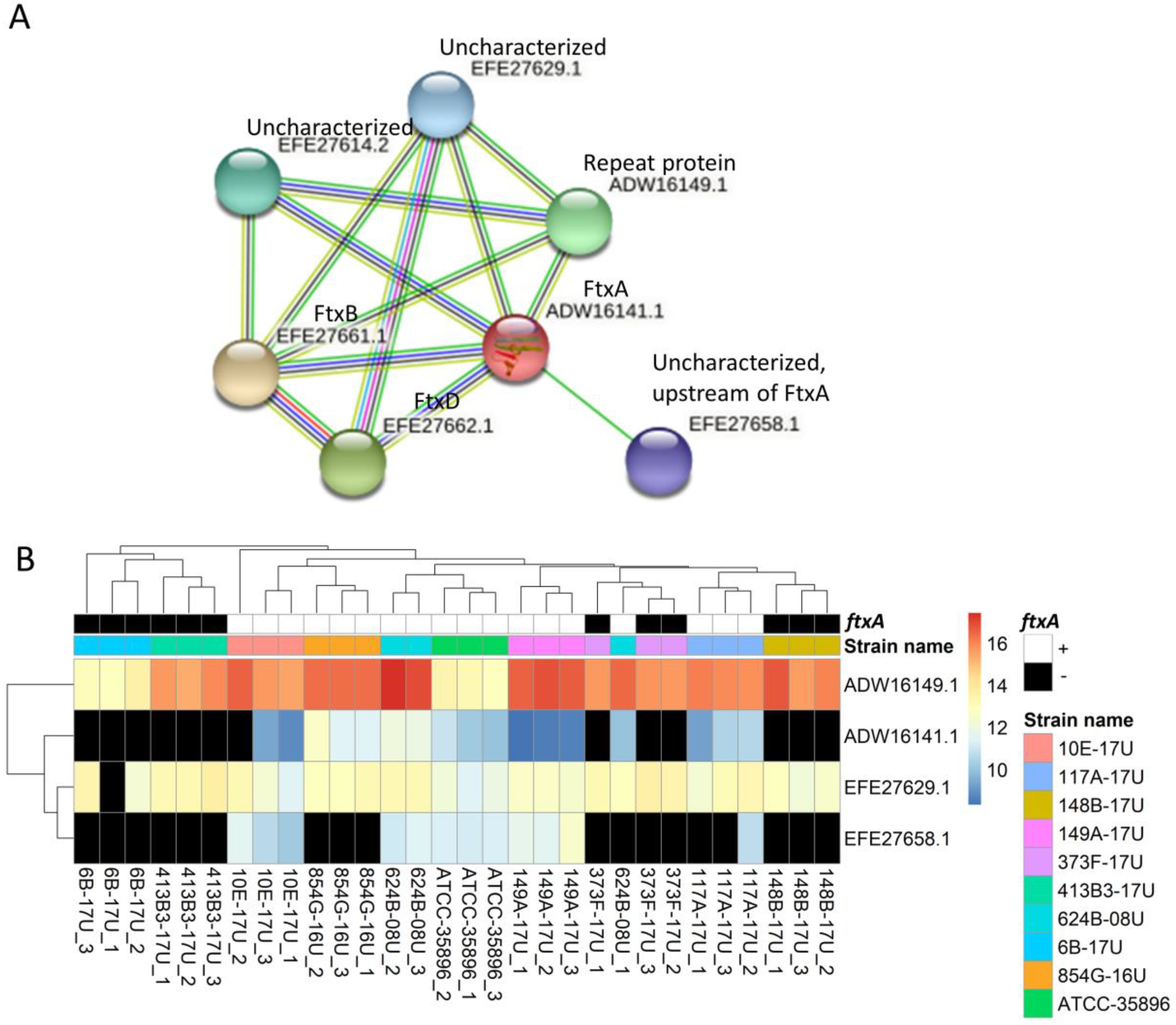
2.3. Predicted Functional Protein Association Network for FtxA
3. Discussion
4. Materials and Methods
4.1. F. alocis Strains and Growth Conditions
4.2. Bacterial Protein Extraction
4.3. LC-MS/MS Analysis
4.4. Label-Free Quantification
4.5. Data Clustering and Heat Maps for Regulated Proteins
4.6. Functional Analysis for Regulated Proteins
4.7. Predicted Interaction for FtxA
4.8. Image Processing
4.9. Ethical Considerations
4.10. Data Availability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aja, E.; Mangar, M.; Fletcher, H.M.; Mishra, A. Filifactor alocis: Recent Insights and Advances. J. Dent. Res. 2021, 100, 790–797. [Google Scholar] [CrossRef]
- Aruni, W.; Chioma, O.; Fletcher, H.M. Filifactor alocis: The Newly Discovered Kid on the Block with Special Talents. J. Dent. Res. 2014, 93, 725–732. [Google Scholar] [CrossRef] [Green Version]
- Gomes, B.; Louzada, L.M.; Almeida-Gomes, R.F.; Pinheiro, E.T.; Sousa, E.L.R.; Jacinto, R.C.; Arruda-Vasconcelos, R. Investigation of Filifactor alocis in primary and in secondary endodontic infections: A molecular study. Arch. Oral. Biol. 2020, 118, 104826. [Google Scholar] [CrossRef]
- Greenwood, D.; Afacan, B.; Emingil, G.; Bostanci, N.; Belibasakis, G.N. Salivary Microbiome Shifts in Response to Periodontal Treatment Outcome. Proteom. Clin. Appl. 2020, 14, e2000011. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Martin, I.; Doolittle-Hall, J.; Teles, R.P.; Patel, M.; Belibasakis, G.N.; Hammerle, C.H.F.; Jung, R.E.; Teles, F.R.F. Exploring the microbiome of healthy and diseased peri-implant sites using Illumina sequencing. J. Clin. Periodontol. 2017, 44, 1274–1284. [Google Scholar] [CrossRef]
- Schlafer, S.; Riep, B.; Griffen, A.L.; Petrich, A.; Hubner, J.; Berning, M.; Friedmann, A.; Gobel, U.B.; Moter, A. Filifactor alocis--involvement in periodontal biofilms. BMC Microbiol. 2010, 10, 66. [Google Scholar] [CrossRef] [Green Version]
- Zehnder, M.; Rechenberg, D.K.; Thurnheer, T.; Luthi-Schaller, H.; Belibasakis, G.N. FISHing for gutta-percha-adhered biofilms in purulent post-treatment apical periodontitis. Mol. Oral. Microbiol. 2017, 32, 226–235. [Google Scholar] [CrossRef]
- Armstrong, C.L.; Klaes, C.K.; Vashishta, A.; Lamont, R.J.; Uriarte, S.M. Filifactor alocis manipulates human neutrophils affecting their ability to release neutrophil extracellular traps induced by PMA. Innate Immun. 2018, 24, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Edmisson, J.S.; Tian, S.; Armstrong, C.L.; Vashishta, A.; Klaes, C.K.; Miralda, I.; Jimenez-Flores, E.; Le, J.; Wang, Q.; Lamont, R.J.; et al. Filifactor alocis modulates human neutrophil antimicrobial functional responses. Cell. Microbiol. 2018, 20, e12829. [Google Scholar] [CrossRef]
- Miralda, I.; Vashishta, A.; Uriarte, S.M. Neutrophil Interaction with Emerging Oral Pathogens: A Novel View of the Disease Paradigm. Adv. Exp. Med. Biol. 2019, 1197, 165–178. [Google Scholar] [CrossRef]
- Uriarte, S.M.; Edmisson, J.S.; Jimenez-Flores, E. Human neutrophils and oral microbiota: A constant tug-of-war between a harmonious and a discordant coexistence. Immunol. Rev. 2016, 273, 282–298. [Google Scholar] [CrossRef] [PubMed]
- Vashishta, A.; Jimenez-Flores, E.; Klaes, C.K.; Tian, S.; Miralda, I.; Lamont, R.J.; Uriarte, S.M. Putative Periodontal Pathogens, Filifactor Alocis and Peptoanaerobacter Stomatis, Induce Differential Cytokine and Chemokine Production by Human Neutrophils. Pathogens 2019, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, A.V.B.; Nokhbehsaim, M.; Damanaki, A.; Eick, S.; Kirschneck, C.; Schroder, A.; Jantsch, J.; Deschner, J. Filifactor alocis and Tumor Necrosis Factor-Alpha Stimulate Synthesis of Visfatin by Human Macrophages. Int. J. Mol. Sci. 2021, 22, 1235. [Google Scholar] [CrossRef]
- Oscarsson, J.; Claesson, R.; Bao, K.; Brundin, M.; Belibasakis, G.N. Phylogenetic Analysis of Filifactor alocis Strains Isolated from Several Oral Infections Identified a Novel RTX Toxin, FtxA. Toxins 2020, 12, 687. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lim, Y.; An, S.J.; Choi, B.K. Characterization and immunostimulatory activity of extracellular vesicles from Filifactor alocis. Mol. Oral. Microbiol. 2020, 35, 1–9. [Google Scholar] [CrossRef]
- Song, M.K.; Kim, H.Y.; Choi, B.K.; Kim, H.H. Filifactor alocis-derived extracellular vesicles inhibit osteogenesis through TLR2 signaling. Mol. Oral. Microbiol. 2020, 35, 202–210. [Google Scholar] [CrossRef]
- Aruni, A.W.; Roy, F.; Sandberg, L.; Fletcher, H.M. Proteome variation among Filifactor alocis strains. Proteomics 2012, 12, 3343–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, J.N.; Sebo, P. Bacterial RTX toxins and host immunity. Curr. Opin. Infect. Dis. 2021, 34, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Linhartova, I.; Bumba, L.; Masin, J.; Basler, M.; Osicka, R.; Kamanova, J.; Prochazkova, K.; Adkins, I.; Hejnova-Holubova, J.; Sadilkova, L.; et al. RTX proteins: A highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 2010, 34, 1076–1112. [Google Scholar] [CrossRef] [Green Version]
- Ristow, L.C.; Welch, R.A. RTX Toxins Ambush Immunity’s First Cellular Responders. Toxins 2019, 11, 720. [Google Scholar] [CrossRef] [Green Version]
- Haubek, D.; Poulsen, K.; Kilian, M. Microevolution and patterns of dissemination of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans. Infect. Immun. 2007, 75, 3080–3088. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.C.; Ho, Y.P.; Chou, Y.S.; Ho, K.Y.; Wu, Y.M.; Lin, Y.C. Aggregatibacter (Actinobacillus) actimycetemcomitans leukotoxin and human periodontitis-A historic review with emphasis on JP2. Kaohsiung J. Med. Sci. 2018, 34, 186–193. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, R.; Card, R.; Nunes, C.; AbuOun, M.; Bagnall, M.C.; Nunez, J.; Mendonca, N.; Anjum, M.F.; da Silva, G.J. Virulence Characterization of Salmonella enterica by a New Microarray: Detection and Evaluation of the Cytolethal Distending Toxin Gene Activity in the Unusual Host S. Typhimurium. PLoS ONE 2015, 10, e0135010. [Google Scholar] [CrossRef]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [Green Version]
- Greene, N.P.; Kaplan, E.; Crow, A.; Koronakis, V. Antibiotic Resistance Mediated by the MacB ABC Transporter Family: A Structural and Functional Perspective. Front. Microbiol. 2018, 9, 950. [Google Scholar] [CrossRef] [Green Version]
- Ebbes, M.; Bleymuller, W.M.; Cernescu, M.; Nolker, R.; Brutschy, B.; Niemann, H.H. Fold and function of the InlB B-repeat. J. Biol. Chem. 2011, 286, 15496–15506. [Google Scholar] [CrossRef] [Green Version]
- Cato, E.P.; Moore, L.V.; Moore, W.E.C. Fusobacterium alocis sp. nov. and Fusobacterium sulci sp. nov. from the human gingival sulcus. Int. J. Syst. Bacteriol. 1985, 35, 475–477. [Google Scholar] [CrossRef] [Green Version]
- Jalava, J.; Eerola, E. Phylogenetic analysis of Fusobacterium alocis and Fusobacterium sulci based on 16S rRNA gene sequences: Proposal of Filifactor alocis (Cato, Moore and Moore) comb. nov. and Eubacterium sulci (Cato, Moore and Moore) comb. nov. Int. J. Syst. Bacteriol. 1999, 49, 1375–1379. [Google Scholar] [CrossRef] [Green Version]
- Bao, K.; Bostanci, N.; Thurnheer, T.; Belibasakis, G.N. Proteomic shifts in multi-species oral biofilms caused by Anaeroglobus geminatus. Sci. Rep. 2017, 7, 4409. [Google Scholar] [CrossRef] [Green Version]
- Käll, L.; Storey, J.D.; MacCoss, M.J.; Noble, W.S. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 2008, 7, 29–34. [Google Scholar] [CrossRef]
- Turker, C.; Akal, F.; Schlapbach, R. Life sciences data and application integration with B-fabric. J. Integr. Bioinform. 2011, 8, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Strain Name | ATCC 35896 | 854G-16U | 117A-17U | 149A-17U | 624B-08U | 10E-17U | 373F-17U | 6B-17U | 413B3-17U | 148B-17U |
|---|---|---|---|---|---|---|---|---|---|---|
| No. of proteins | 802 | 755 | 762 | 761 | 785 | 744 | 746 | 762 | 746 | 762 |
| No. | First Accession | Unique Peptides | log2FC FtxA+/FtxA− | t-Test | Description |
|---|---|---|---|---|---|
| 1 | EFE27669.1 | 3 | −6.98 | 5 × 10−13 | hypothetical protein HMPREF0389_01588 |
| 2 | EFE28470.1 | 4 | −4.23 | 7 × 10−3 | DNA polymerase III, alpha subunit |
| 3 | EFE28966.1 | 9 | −2.57 | 3 × 10−2 | ABC transporter, substrate-binding protein, family 3 |
| 4 | EFE28389.1 | 12 | −2.28 | 3 × 10−2 | UDP-N-acetylmuramoyl-tripeptide--D-alanyl-D-alanine ligase |
| 5 | EFE29056.2 | 4 | −1.78 | 3 × 10−2 | hypothetical protein HMPREF0389_00978 |
| 6 | EFE29171.2 | 4 | −1.65 | 4 × 10−2 | HIRAN domain protein |
| 7 | EFE29153.1 | 9 | −1.58 | 3 × 10−2 | transcriptional regulator, Spx/MgsR family |
| 8 | WP_083799680.1 | 2 | −1.43 | 6 × 10−3 | aromatic acid exporter family protein |
| 9 | EFE28489.2 | 13 | −1.31 | 9 × 10−4 | GTP-binding protein TypA |
| 10 | EFE28520.1 | 2 | −1.30 | 2 × 10−2 | Sua5/YciO/YrdC/YwlC family protein |
| 11 | EFE27675.1 | 10 | −1.18 | 3 × 10−3 | RelA/SpoT domain protein |
| 12 | EFE28371.1 | 4 | −1.11 | 3 × 10−3 | aminotransferase, class V |
| 13 | EFE28992.1 | 6 | −1.03 | 4 × 10−4 | B3/4 domain protein |
| 14 | EFE28003.1 | 3 | −1.02 | 3 × 10−4 | comEA protein |
| 15 | EFE28083.2 | 27 | 1.02 | 6 × 10−3 | LPXTG-motif cell-wall anchor domain protein |
| 16 | EFE28698.2 | 6 | 1.06 | 4 × 10−2 | hypothetical protein HMPREF0389_00616 |
| 17 | EFE28364.1 | 25 | 1.07 | 7 × 10−8 | endopeptidase La |
| 18 | EFE27626.1 | 13 | 1.08 | 4 × 10−2 | signal recognition particle-docking protein FtsY |
| 19 | EFE28863.1 | 7 | 1.11 | 8 × 10−3 | ABC transporter, ATP-binding protein |
| 20 | EFE27917.1 | 7 | 1.28 | 1 × 10−2 | CRISPR-associated protein, Csd1 family |
| 21 | EFE28620.1 | 7 | 1.68 | 5 × 10−3 | transcriptional regulator, effector binding domain protein |
| 22 | EFE27793.1 | 61 | 1.70 | 6 × 10−5 | LPXTG-motif cell-wall anchor domain protein |
| 23 | EFE28561.1 | 4 | 1.77 | 3 × 10−2 | aspartate–ammonia ligase |
| 24 | EFE28977.1 | 2 | 1.81 | 3 × 10−2 | cobalt transport protein |
| 25 | EFE29084.1 | 30 | 2.18 | 3 × 10−3 | LPXTG-motif cell-wall anchor domain protein |
| 26 | EFE27653.2 | 3 | 2.40 | 3 × 10−3 | hypothetical protein HMPREF0389_01572 |
| 27 | EFE27796.2 | 3 | 2.40 | 3 × 10−2 | hypothetical protein HMPREF0389_01427 |
| 28 | EFE29085.1 | 11 | 2.41 | 2 × 10−2 | purine nucleoside phosphorylase I, inosine and guanosine-specific |
| 29 | EFE28936.1 | 20 | 2.80 | 9 × 10−4 | pyridoxal 5’-phosphate synthase, synthase subunit Pdx1 |
| 30 | EFE28939.1 | 10 | 3.60 | 4 × 10−2 | thermonuclease |
| 31 | EFE29027.1 | 28 | 5.05 | 3 × 10−7 | TRAP transporter solute receptor, DctP family |
| 32 | EFE29030.1 | 27 | 5.44 | 8 × 10−6 | aminotransferase, class I/II |
| 33 | EFE29026.1 | 22 | 8.84 | 2 × 10−6 | 4-phosphoerythronate dehydrogenase |
| 34 | EFE29077.1 | 2 | 12.39 | 4 × 10−3 | response regulator receiver domain protein |
| No. | First Accession | Unique Peptides | Discovered in FtxA+ or FtxA− Strains | Description |
|---|---|---|---|---|
| 1 | ADW16126.1 | 2 | FtxA+ | protein-(glutamine-N5) methyltransferase, release factor-specific |
| 2 | ADW16141.1 | 9 | FtxA+ | type I secretion target GGXGXDXXX repeat (2 copies) |
| 3 | ADW16178.1 | 2 | FtxA+ | N-acetylmuramoyl-L-alanine amidase |
| 4 | EFE27600.1 | 2 | FtxA+ | hypothetical protein HMPREF0389_01518 |
| 5 | EFE27658.1 | 4 | FtxA+ | hypothetical protein HMPREF0389_01577 |
| 6 | EFE27681.2 | 53 | FtxA+ | poly(R)-hydroxyalkanoic acid synthase, class III, PhaE subunit |
| 7 | EFE27724.1 | 2 | FtxA+ | amino acid carrier protein |
| 8 | EFE27725.1 | 26 | FtxA+ | peptidase, M20/M25/M40 family |
| 9 | EFE27785.1 | 2 | FtxA+ | DEAD/DEAH box helicase |
| 10 | EFE27899.1 | 2 | FtxA+ | hypothetical protein HMPREF0389_01151 |
| 11 | EFE27901.1 | 6 | FtxA+ | hypothetical protein HMPREF0389_01153 |
| 12 | EFE27902.2 | 5 | FtxA+ | hypothetical protein HMPREF0389_01154 |
| 13 | EFE27946.1 | 2 | FtxA+ | peptidase family T4 |
| 14 | EFE27975.2 | 13 | FtxA+ | hypothetical protein HMPREF0389_01227 |
| 15 | EFE27976.1 | 2 | FtxA+ | hypothetical protein HMPREF0389_01228 |
| 16 | EFE27979.1 | 2 | FtxA+ | IclR helix-turn-helix domain protein |
| 17 | EFE28064.1 | 2 | FtxA+ | hypothetical protein HMPREF0389_01317 |
| 18 | EFE28209.2 | 4 | FtxA+ | MutS2 family protein |
| 19 | EFE28216.1 | 2 | FtxA+ | DNA protecting protein DprA |
| 20 | EFE28277.2 | 9 | FtxA+ | hypothetical protein HMPREF0389_00192 |
| 21 | EFE28328.2 | 3 | FtxA+ | addiction module antitoxin, RelB/DinJ family |
| 22 | EFE28411.1 | 2 | FtxA+ | hypothetical protein HMPREF0389_00326 |
| 23 | EFE28456.1 | 2 | FtxA+ | ACT domain protein |
| 24 | EFE28503.1 | 19 | FtxA+ | prepilin-type cleavage/methylation N-terminal domain protein |
| 25 | EFE28504.1 | 7 | FtxA+ | hypothetical protein HMPREF0389_00419 |
| 26 | EFE28505.1 | 18 | FtxA+ | prepilin-type cleavage/methylation N-terminal domain protein |
| 27 | EFE28506.2 | 8 | FtxA+ | bacterial type II secretion system domain protein F |
| 28 | EFE28572.1 | 3 | FtxA+ | transcriptional regulator, Fur family |
| 29 | EFE28693.1 | 7 | FtxA+ | hypothetical protein HMPREF0389_00610 |
| 30 | EFE28694.1 | 4 | FtxA+ | hypothetical protein HMPREF0389_00612 |
| 31 | EFE28696.1 | 3 | FtxA+ | hypothetical protein HMPREF0389_00614 |
| 32 | EFE28755.2 | 3 | FtxA+ | hypothetical protein HMPREF0389_00675 |
| 33 | EFE28756.1 | 11 | FtxA+ | alcohol dehydrogenase, iron-dependent |
| 34 | EFE28838.1 | 2 | FtxA+ | DNA-binding helix-turn-helix protein |
| 35 | EFE29023.1 | 4 | FtxA+ | hypothetical protein HMPREF0389_00945 |
| 36 | EFE29055.1 | 2 | FtxA+ | DNA-binding helix-turn-helix protein |
| 37 | EFE29096.1 | 2 | FtxA+ | hypothetical protein HMPREF0389_01018 |
| 38 | EFE29109.1 | 3 | FtxA+ | hypothetical protein HMPREF0389_01031 |
| 39 | EFE29118.2 | 4 | FtxA+ | type I restriction modification DNA specificity domain protein |
| 40 | EFE29197.1 | 2 | FtxA+ | hypothetical protein HMPREF0389_01122 |
| 41 | WP_041250771.1 | 7 | FtxA+ | acyl carrier protein |
| 42 | EFE28023.2 | 3 | FtxA− | iron permease FTR1 family |
| 43 | EFE29170.2 | 2 | FtxA− | hypothetical protein HMPREF0389_01093 |
| 44 | EFE28866.1 | 2 | FtxA− | dihydroorotate dehydrogenase, electron transfer subunit |
| 45 | EFE28492.1 | 4 | FtxA− | excinuclease ABC, B subunit |
| 46 | ADW16125.1 | 2 | FtxA− | hypothetical protein HMPREF0389_01679 |
| 47 | EFE27676.1 | 2 | FtxA− | VanW-like protein |
| 48 | EFE27908.2 | 2 | FtxA− | hypothetical protein HMPREF0389_01160 |
| No. | First Accession | Unique Peptides | log2FC FtxA+/FtxA− | Description |
|---|---|---|---|---|
| 1 | EFE27582.2 | 2 | 1.32 | hypothetical protein HMPREF0389_01655 |
| 2 | EFE27612.1 | 4 | 1.40 | phospholipase D domain protein |
| 3 | EFE29008.1 | 8 | 1.88 | 3-oxoacyl-[acyl-carrier-protein] reductase |
| 4 | EFE27694.1 | 2 | 2.31 | adenosylmethionine-8-amino-7-oxononanoate transaminase |
| 5 | EFE29007.1 | 5 | 3.73 | beta-ketoacyl-acyl-carrier-protein synthase II |
| 6 | EFE28275.1 | 9 | 3.75 | DNA topoisomerase |
| 7 | EFE28848.1 | 12 | 4.87 | hypothetical protein HMPREF0389_00770 |
| 8 | EFE27992.2 | 2 | 5.71 | EDD domain protein, DegV family |
| 9 | EFE28027.1 | 2 | −3.13 | efflux ABC transporter, permease protein |
| 10 | EFE27594.2 | 2 | −3.05 | hypothetical protein HMPREF0389_01512 |
| 11 | EFE28953.1 | 2 | −2.47 | hypothetical protein HMPREF0389_00875 |
| 12 | EFE28704.2 | 2 | −2.23 | hypothetical protein HMPREF0389_00623 |
| Accession | Protein Domain (Pfam) * |
|---|---|
| ADW16141.1 | RTX calcium-binding nonapeptide repeat (4 copies); Hemolysin-type calcium binding protein related domain; RTX C-terminal domain |
| ADW16149.1 | Copper amine oxidase N-terminal domain; Listeria–Bacteroides repeat domain (List_Bact_rpt); Divergent InlB B-repeat domain; Family of unknown function (DUF6273) |
| EFE27629.1 | Outer membrane efflux protein; SMODS- and SLOG-associating 2TM vector domain 1; Hydrogenase/urease nickel incorporation, metallochaperone |
| EFE27658.1 | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, K.; Claesson, R.; Gehrig, P.; Grossmann, J.; Oscarsson, J.; Belibasakis, G.N. Proteomic Characterization of the Oral Pathogen Filifactor alocis Reveals Key Inter-Protein Interactions of Its RTX Toxin: FtxA. Pathogens 2022, 11, 590. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11050590
Bao K, Claesson R, Gehrig P, Grossmann J, Oscarsson J, Belibasakis GN. Proteomic Characterization of the Oral Pathogen Filifactor alocis Reveals Key Inter-Protein Interactions of Its RTX Toxin: FtxA. Pathogens. 2022; 11(5):590. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11050590
Chicago/Turabian StyleBao, Kai, Rolf Claesson, Peter Gehrig, Jonas Grossmann, Jan Oscarsson, and Georgios N. Belibasakis. 2022. "Proteomic Characterization of the Oral Pathogen Filifactor alocis Reveals Key Inter-Protein Interactions of Its RTX Toxin: FtxA" Pathogens 11, no. 5: 590. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11050590