
Flavivirus NS1 Triggers Tissue-Specific Disassembly of Intercellular Junctions Leading to Barrier Dysfunction and Vascular Leak in a GSK-3β-Dependent Manner

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
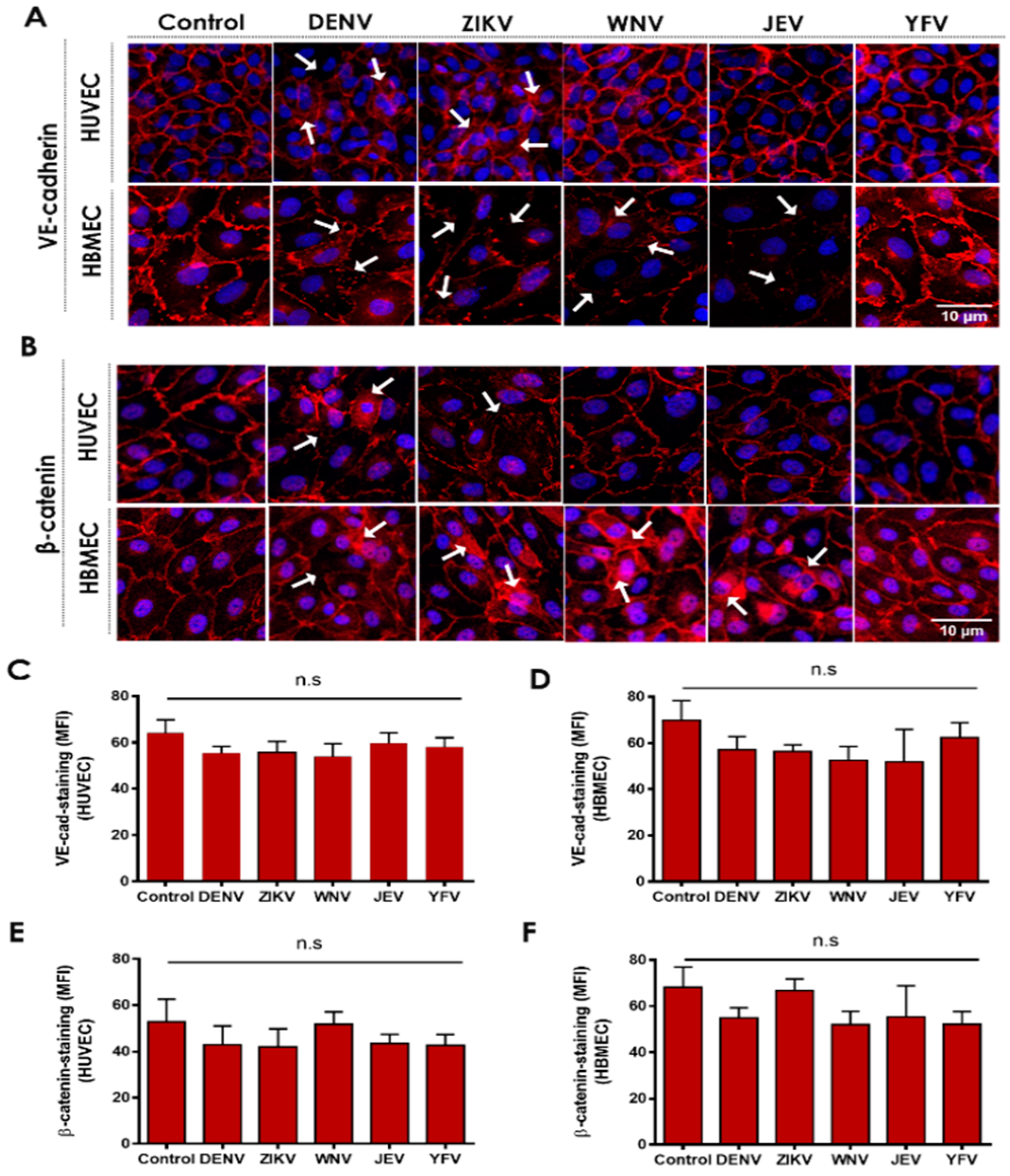
2.1. Flavivirus NS1 Proteins Alter the Localization of the AJ Proteins VE-Cadherin and β-Catenin in Human Endothelial Cells from the Brain and Umbilical Vein
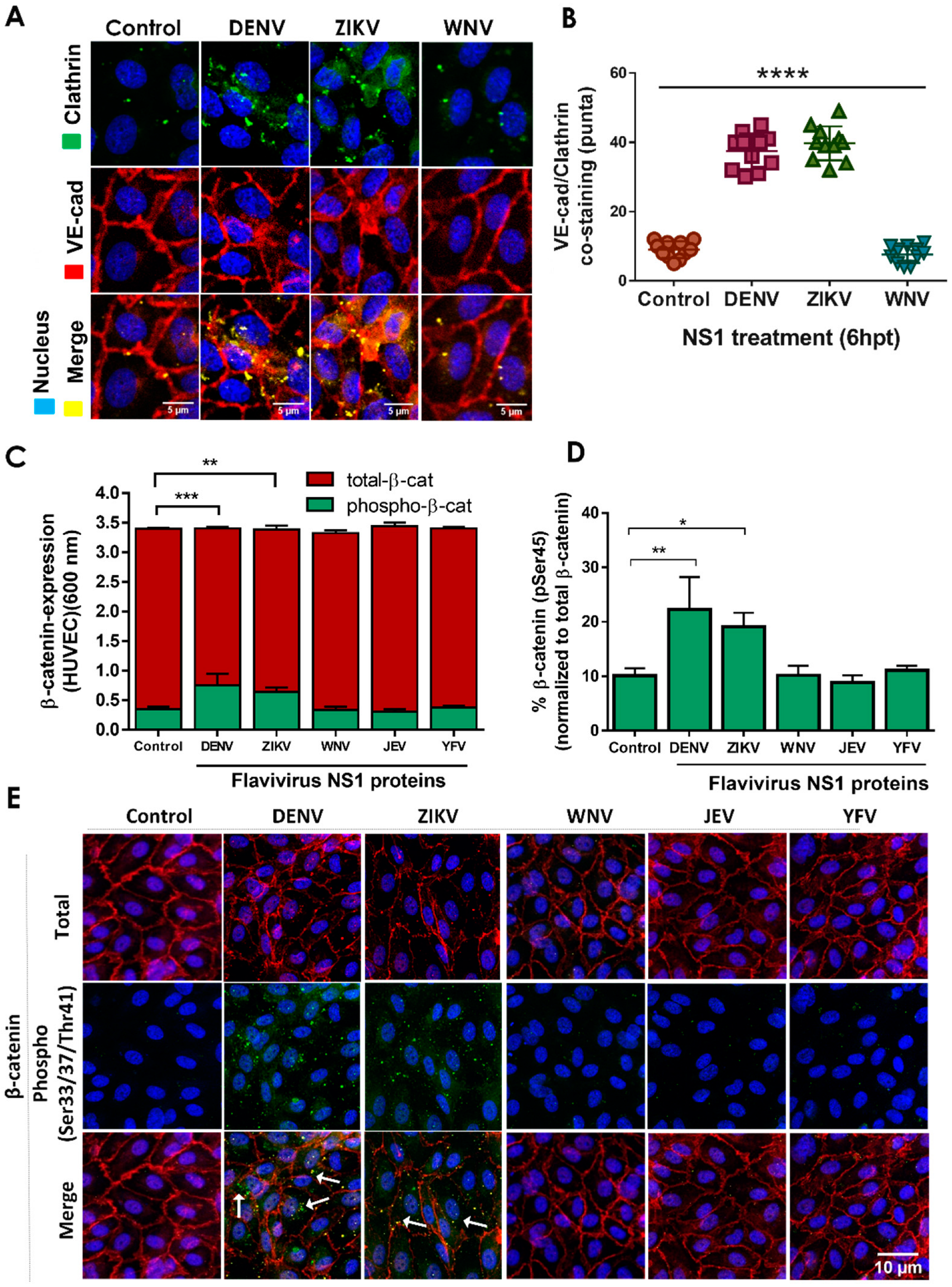
2.2. NS1 Triggers Endocytosis of VE-Cadherin and Phosphorylation of β-Catenin in Human Endothelial Cells
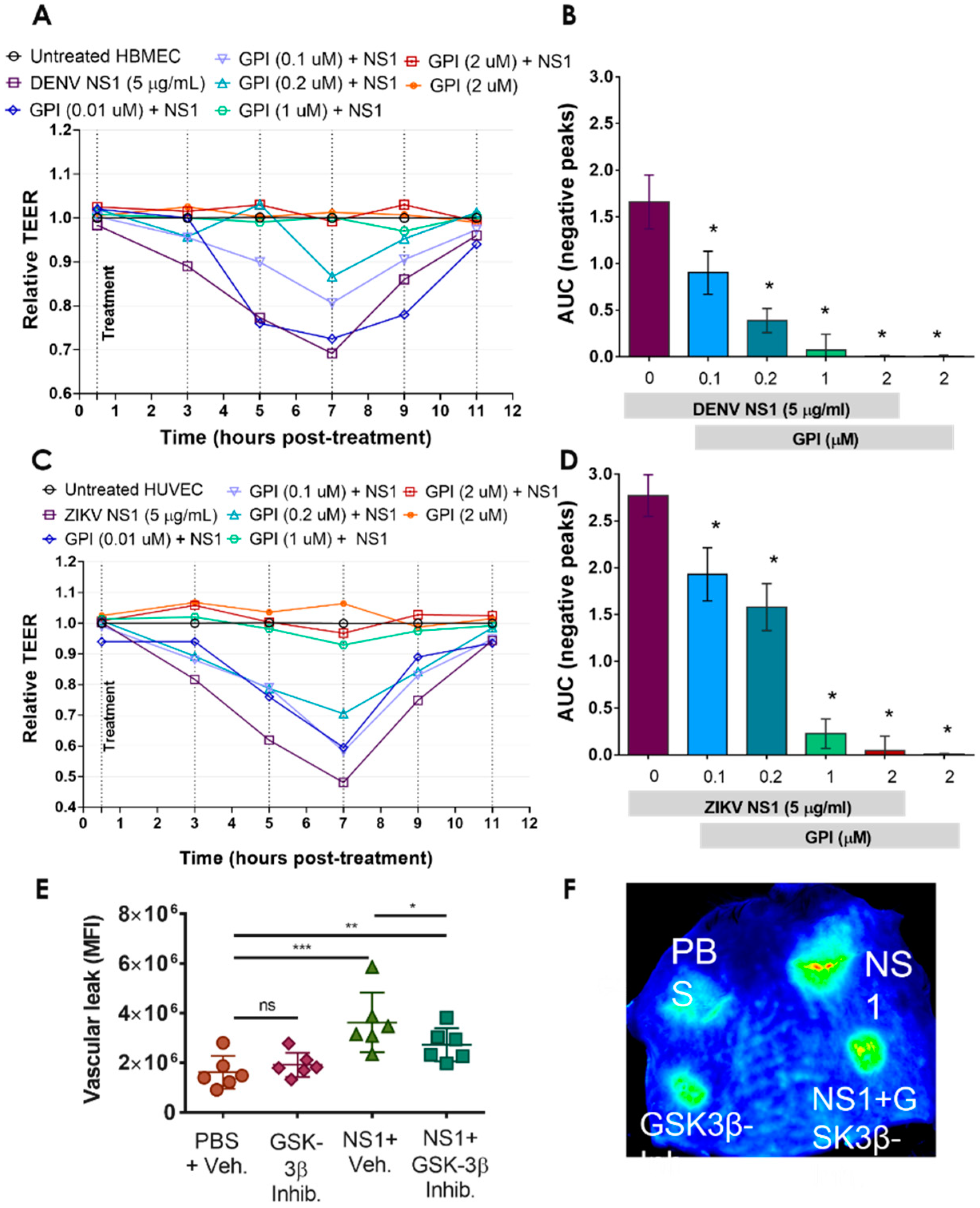
2.3. GSK-3β Is Required for NS1-Mediated Endothelial Hyperpermeability and Vascular Leak
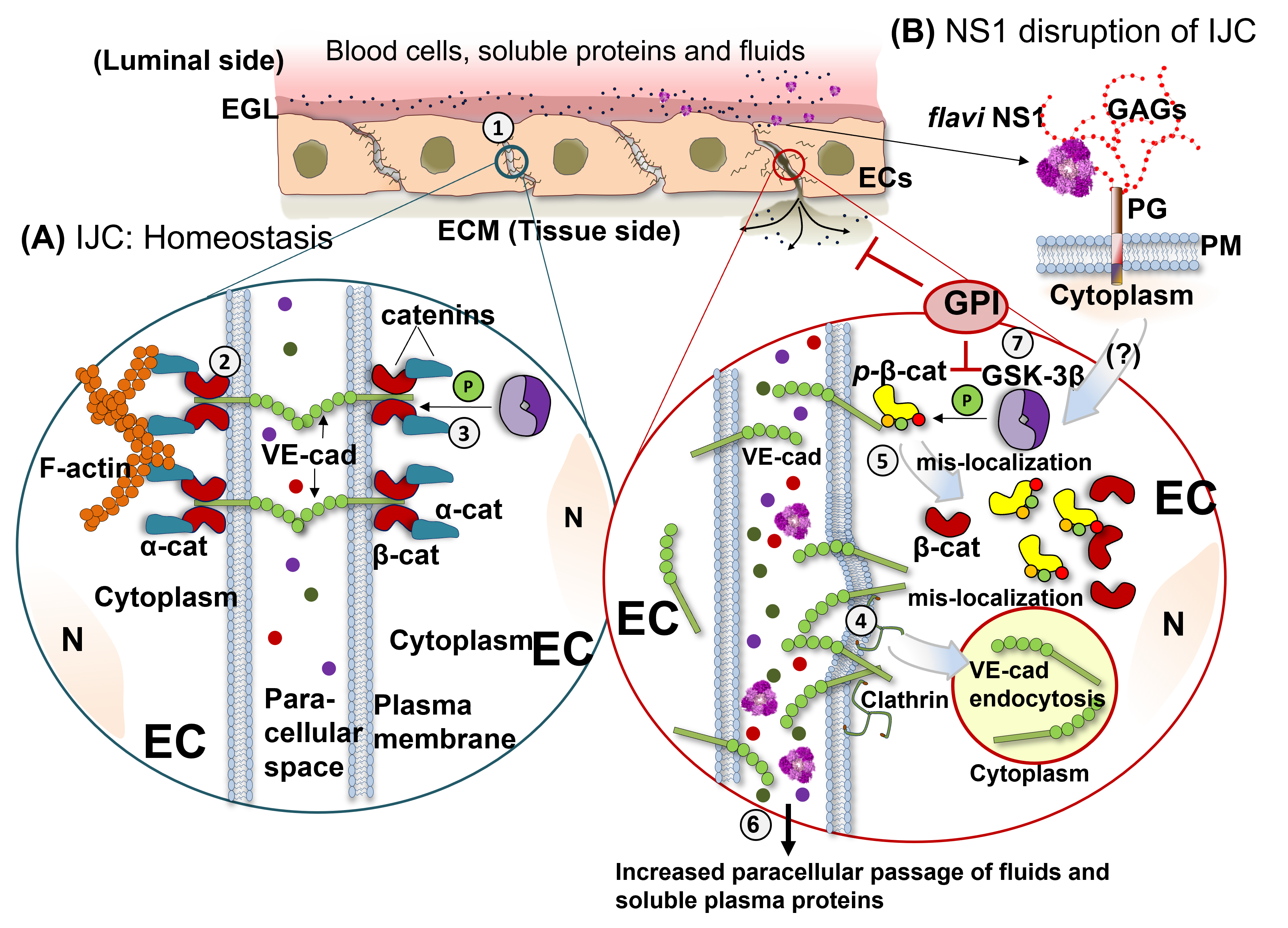
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Mice
4.3. Cell Culture
4.4. Recombinant NS1 Proteins
4.5. Monoclonal Antibodies, and Inhibitors
4.6. Trans-Endothelial Electrical Resistance (TEER)
4.7. Fluorescence Microscopy
4.8. Western Blot Analyses
4.9. ELISA
4.10. Localized Vascular Leak Murine Model Assay
4.11. Data Analyses and Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.A.; Solomon, T. Pathogenic flaviviruses. Lancet 2008, 371, 500–509. [Google Scholar] [CrossRef]
- Fernandez-Garcia, M.D.; Mazzon, M.; Jacobs, M.; Amara, A. Pathogenesis of flavivirus infections: Using and abusing the host cell. Cell Host Microbe 2009, 5, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Barrows, N.J.; Campos, R.K.; Liao, K.-C.; Prasanth, K.R.; Soto-Acosta, R.; Yeh, S.-C.; Schott-Lerner, G.; Pompon, J.; Sessions, O.M.; Bradrick, S.S.; et al. Biochemistry and Molecular Biology of Flaviviruses. Chem. Rev. 2018, 118, 4448–4482. [Google Scholar] [CrossRef]
- Akey, D.L.; Brown, W.C.; Jose, J.; Kuhn, R.J.; Smith, J.L. Structure-guided insights on the role of NS1 in flavivirus infection. Bioessays 2015, 37, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Muller, D.A.; Young, P.R. The flavivirus NS1 protein: Molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antivir. Res. 2013, 98, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Glasner, D.R.; Puerta-Guardo, H.; Beatty, P.R.; Harris, E. The Good, the Bad, and the Shocking: The Multiple Roles of Dengue Virus Nonstructural Protein 1 in Protection and Pathogenesis. Annu. Rev. Virol. 2018, 5, 227–253. [Google Scholar] [CrossRef]
- Puerta-Guardo, H.; Glasner, D.R.; Harris, E. Dengue Virus NS1 Disrupts the Endothelial Glycocalyx, Leading to Hyperpermeability. PLoS Pathog. 2016, 12, e1005738. [Google Scholar] [CrossRef] [Green Version]
- Glasner, D.R.; Ratnasiri, K.; Puerta-Guardo, H.; Espinosa, D.A.; Beatty, P.R.; Harris, E. Dengue virus NS1 cytokine-independent vascular leak is dependent on endothelial glycocalyx components. PLoS Pathog. 2017, 13, e1006673. [Google Scholar] [CrossRef]
- Chen, H.R.; Chao, C.H.; Liu, C.C.; Ho, T.S.; Tsai, H.P.; Perng, G.C.; Lin, Y.S.; Wang, J.R.; Yeh, T.M. Macrophage migration inhibitory factor is critical for dengue NS1-induced endothelial glycocalyx degradation and hyperpermeability. PLoS Pathog. 2018, 14, e1007033. [Google Scholar] [CrossRef]
- Beatty, P.R.; Puerta-Guardo, H.; Killingbeck, S.S.; Glasner, D.R.; Hopkins, K.; Harris, E. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci. Transl. Med. 2015, 7, 304ra141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alphonsus, C.S.; Rodseth, R.N. The endothelial glycocalyx: A review of the vascular barrier. Anaesthesia 2014, 69, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Maksimenko, A.V.; Turashev, A.D. Endothelial glycocalyx of blood circulation. II. Biological functions, state at norm and pathology, bioengineering application. Russ. J. Bioorganic Chem. 2014, 40, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Koo, A.; Dewey, C.F., Jr.; García-Cardeña, G. Hemodynamic shear stress characteristic of atherosclerosis-resistant regions promotes glycocalyx formation in cultured endothelial cells. Am. J. Physiol. Physiol. 2013, 304, C137–C146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suwarto, S.; Sasmono, R.T.; Sinto, R.; Ibrahim, E.; Suryamin, M. Association of Endothelial Glycocalyx and Tight and Adherens Junctions With Severity of Plasma Leakage in Dengue Infection. J. Infect. Dis. 2017, 215, 992–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, T.H.; Alonso, S.; Ng, L.F.; Thein, T.L.; Pang, J.; Leo, Y.S.; Lye, D.C.; Yeo, T.W. Author Correction: Increased Serum Hyaluronic Acid and Heparan Sulfate in Dengue Fever: Association with Plasma Leakage and Disease Severity. Sci. Rep. 2021, 11, 13346. [Google Scholar] [CrossRef]
- Lam, P.K.; McBride, A.; Le, D.H.T.; Huynh, T.T.; Vink, H.; Wills, B.; Yacoub, S. Visual and Biochemical Evidence of Glycocalyx Disruption in Human Dengue Infection, and Association With Plasma Leakage Severity. Front. Med. (Lausanne) 2020, 7, 545813. [Google Scholar] [CrossRef]
- Lin, C.Y.; Kolliopoulos, C.; Huang, C.H.; Tenhunen, J.; Heldin, C.H.; Chen, Y.H.; Heldin, P. High levels of serum hyaluronan is an early predictor of dengue warning signs and perturbs vascular integrity. EBioMedicine 2019, 48, 425–441. [Google Scholar] [CrossRef] [Green Version]
- Puerta-Guardo, H.; Glasner, D.R.; Espinosa, D.A.; Biering, S.B.; Patana, M.; Ratnasiri, K.; Wang, C.; Beatty, P.R.; Harris, E. Flavivirus NS1 Triggers Tissue-Specific Vascular Endothelial Dysfunction Reflecting Disease Tropism. Cell Rep. 2019, 26, 1598–1613.e1598. [Google Scholar] [CrossRef] [Green Version]
- Modhiran, N.; Watterson, D.; Muller, D.A.; Panetta, A.K.; Sester, D.P.; Liu, L.; Hume, D.A.; Stacey, K.J.; Young, P.R. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 2015, 7, 304ra142. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-R.; Chuang, Y.-C.; Lin, Y.-S.; Liu, H.-S.; Liu, C.-C.; Perng, G.-C.; Yeh, T.-M. Dengue Virus Nonstructural Protein 1 Induces Vascular Leakage through Macrophage Migration Inhibitory Factor and Autophagy. PLoS Negl. Trop. Dis. 2016, 10, e0004828. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Puerta-Guardo, H.; Biering, S.B.; Glasner, D.R.; Tran, E.B.; Patana, M.; Gomberg, T.A.; Malvar, C.; Lo, N.T.N.; Espinosa, D.A.; et al. Endocytosis of flavivirus NS1 is required for NS1-mediated endothelial hyperpermeability and is abolished by a single N-glycosylation site mutation. PLOS Pathog. 2019, 15, e1007938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puerta-Guardo, H.; Tabata, T.; Petitt, M.; Dimitrova, M.; Glasner, D.R.; Pereira, L.; Harris, E. Zika Virus Nonstructural Protein 1 Disrupts Glycosaminoglycans and Causes Permeability in Developing Human Placentas. J. Infect. Dis. 2020, 221, 313–324. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F.; Molendini, C.; Baluk, P.; McDonald, D.M. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 2009, 335, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzoni, G.; Dejana, E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef] [Green Version]
- Dejana, E.; Orsenigo, F. Endothelial adherens junctions at a glance. J. Cell Sci. 2013, 126, 2545–2549. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.I.; Naydenov, N.G. Chapter Two—Dynamics and Regulation of Epithelial Adherens Junctions: Recent Discoveries and Controversies. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 303, pp. 27–99. [Google Scholar]
- Dejana, E.; Vestweber, D. The role of VE-cadherin in vascular morphogenesis and permeability control. Prog. Mol. Biol. Transl. Sci. 2013, 116, 119–144. [Google Scholar] [CrossRef]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.H.; Zheng, G. E-cadherin/β-catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011, 567305. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Sawhney, M.; DattaGupta, S.; Shukla, N.K.; Srivastava, A.; Walfish, P.G.; Ralhan, R. Clinical significance of altered expression of β-catenin and E-cadherin in oral dysplasia and cancer: Potential link with ALCAM expression. PLoS ONE 2013, 8, e67361. [Google Scholar] [CrossRef]
- Mehta, S.; Nijhuis, A.; Kumagai, T.; Lindsay, J.; Silver, A. Defects in the adherens junction complex (E-cadherin/β-catenin) in inflammatory bowel disease. Cell Tissue Res. 2015, 360, 749–760. [Google Scholar] [CrossRef]
- Harris, T.J.C.; Tepass, U. Adherens junctions: From molecules to morphogenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, R.L.; Gottardi, C.J. Phospho-regulation of Beta-catenin adhesion and signaling functions. Physiology 2007, 22, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [Green Version]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Kato, Y.; Zhang, Z.; Do, V.M.; Yankner, B.A.; He, X. beta-Trcp couples beta-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc. Natl. Acad. Sci. USA 1999, 96, 6273–6278. [Google Scholar] [CrossRef] [Green Version]
- Latres, E.; Chiaur, D.S.; Pagano, M. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene 1999, 18, 849–854. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.; Li, G.; Shen, M.; Yu, Z.; Ge, W.; Lao, Z.; Fan, Y.; Chen, K.; Ding, Z.; Wang, W.; et al. DENV NS1 and MMP-9 cooperate to induce vascular leakage by altering endothelial cell adhesion and tight junction. PLoS Pathog. 2021, 17, e1008603. [Google Scholar] [CrossRef]
- Barbachano-Guerrero, A.; Endy, T.P.; King, C.A. Dengue virus non-structural protein 1 activates the p38 MAPK pathway to decrease barrier integrity in primary human endothelial cells. J. Gen. Virol. 2020, 101, 484–496. [Google Scholar] [CrossRef]
- Plotkin, B.; Kaidanovich, O.; Talior, I.; Eldar-Finkelman, H. Insulin Mimetic Action of Synthetic Phosphorylated Peptide Inhibitors of Glycogen Synthase Kinase-3. J. Pharmacol. Exp. Ther. 2003, 305, 974. [Google Scholar] [CrossRef]
- Turner, J.R. Molecular basis of epithelial barrier regulation: From basic mechanisms to clinical application. Am. J. Pathol. 2006, 169, 1901–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L. Tight junctions on the move: Molecular mechanisms for epithelial barrier regulation. Ann. N. Y. Acad. Sci. 2012, 1258, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerutti, C.; Ridley, A.J. Endothelial cell-cell adhesion and signaling. Exp. Cell Res. 2017, 358, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Sawada, N. Tight junction-related human diseases. Pathol. Int. 2013, 63, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.A.; Uppada, S.; Achkar, I.W.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A Functional Crosstalk. Front. Physiol. 2019, 9, 1942. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, M.B.; Dhawan, P.; Baumert, T.F. Tight junction proteins in gastrointestinal and liver disease. Gut 2019, 68, 547. [Google Scholar] [CrossRef]
- Bednarczyk, J.; Lukasiuk, K. Tight junctions in neurological diseases. Acta Neurobiol. Exp. (Wars) 2011, 71, 393–408. [Google Scholar]
- Utech, M.; Mennigen, R.; Bruewer, M. Endocytosis and Recycling of Tight Junction Proteins in Inflammation. J. Biomed. Biotechnol. 2010, 2010, 484987. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.I.; Nusrat, A.; Parkos, C.A. Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol. Biol. Cell 2004, 15, 176–188. [Google Scholar] [CrossRef] [Green Version]
- Stamatovic, S.M.; Johnson, A.M.; Sladojevic, N.; Keep, R.F.; Andjelkovic, A.V. Endocytosis of tight junction proteins and the regulation of degradation and recycling. Ann. N. Y. Acad. Sci. 2017, 1397, 54–65. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Anderson, J.M. Phosphorylation of tight junction transmembrane proteins: Many sites, much to do. Tissue Barriers 2018, 6, e1382671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, D.; Xie, W.; Liu, M. Alteration of cell junctions during viral infection. Thorac. Cancer 2020, 11, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Malavige, G.N.; Ogg, G.S. Pathogenesis of vascular leak in dengue virus infection. Immunology 2017, 151, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Puerta-Guardo Henry, B.S.; Harris, E.; Pavia-Ruz, N.; Vázquez-Prokopec, G.; Ayora-Talavera, G.; Manrique-Saide, P. Dengue Immunopathogenesis: A Crosstalk between Host and Viral Factors Leading to Disease: Part I—Dengue Virus Tropism, Host Innate Immune Responses, and Subversion of Antiviral Responses. In Dengue Fever in a One Health Perspective; IntechOpen Book Series; IntechOpen, Ed.; Intechopen: London, UK, 2020; pp. 1–34. [Google Scholar] [CrossRef]
- Puerta-Guardo Henry, B.S.; Harris, E.; Pavia-Ruz, N.; Vázquez-Prokopec, G.; Ayora-Talavera, G.; Manrique-Saide, P. Dengue Immunopathogenesis: A Crosstalk between Host and Viral Factors Leading to Disease: PART II—DENV Infection, Adaptive Immune Responses, and NS1 Pathogenesis. In Dengue Fever in a One Health Perspective; IntechOpen Book Series; IntechOpen, Ed.; Intechopen: London, UK, 2020; Volume 5. [Google Scholar]
- Chiu, C.-F.; Chu, L.-W.; Liao, I.-C.; Simanjuntak, Y.; Lin, Y.-L.; Juan, C.-C.; Ping, Y.-H. The Mechanism of the Zika Virus Crossing the Placental Barrier and the Blood-Brain Barrier. Front. Microbiol. 2020, 11, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastogi, M.; Singh, S.K. Zika virus NS1 affects the junctional integrity of human brain microvascular endothelial cells. Biochimie 2020, 176, 52–61. [Google Scholar] [CrossRef]
- Xu, Z.; Waeckerlin, R.; Urbanowski, M.D.; van Marle, G.; Hobman, T.C. West Nile virus infection causes endocytosis of a specific subset of tight junction membrane proteins. PLoS ONE 2012, 7, e37886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Lo, Y.; Chapagain, M.; Lum, S.; Kumar, M.; Gurjav, U.; Luo, H.; Nakatsuka, A.; Nerurkar, V.R. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier. Virology 2009, 385, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.T.; Rathore, A.P.S.; Soundarajan, G.; St. John, A.L. Japanese encephalitis virus neuropenetrance is driven by mast cell chymase. Nat. Commun. 2019, 10, 706. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.-S.; Zou, Q.-C.; Xiong, W.-J.; Cui, N.-Y.; Wang, K.; Liu, H.-X.; Lou, W.-J.; Higazy, D.; Zhang, Y.-G.; Cui, M. Brain Microvascular Endothelial Cell-Derived HMGB1 Facilitates Monocyte Adhesion and Transmigration to Promote JEV Neuroinvasion. Front. Cell Infect. Microbiol. 2021, 11, 1820. [Google Scholar] [CrossRef]
- Agrawal, T.; Sharvani, V.; Nair, D.; Medigeshi, G.R. Japanese encephalitis virus disrupts cell-cell junctions and affects the epithelial permeability barrier functions. PLoS ONE 2013, 8, e69465. [Google Scholar] [CrossRef] [Green Version]
- Velandia-Romero, M.L.; Calderón-Peláez, M.-A.; Castellanos, J.E. In Vitro Infection with Dengue Virus Induces Changes in the Structure and Function of the Mouse Brain Endothelium. PLoS ONE 2016, 11, e0157786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, L.; Betanzos, A.; Raya-Sandino, A.; González-Mariscal, L.; Del Angel, R.M. Dengue virus enters and exits epithelial cells through both apical and basolateral surfaces and perturbs the apical junctional complex. Virus Res. 2018, 258, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Talavera, D.; Castillo, A.M.; Dominguez, M.C.; Gutierrez, A.E.; Meza, I. IL8 release, tight junction and cytoskeleton dynamic reorganization conducive to permeability increase are induced by dengue virus infection of microvascular endothelial monolayers. J. Gen. Virol. 2004, 85, 1801–1813. [Google Scholar] [CrossRef] [PubMed]
- Kanlaya, R.; Pattanakitsakul, S.-n.; Sinchaikul, S.; Chen, S.-T.; Thongboonkerd, V. Alterations in Actin Cytoskeletal Assembly and Junctional Protein Complexes in Human Endothelial Cells Induced by Dengue Virus Infection and Mimicry of Leukocyte Transendothelial Migration. J. Proteome Res. 2009, 8, 2551–2562. [Google Scholar] [CrossRef]
- Dewi, B.E.; Takasaki, T.; Kurane, I. Peripheral blood mononuclear cells increase the permeability of dengue virus-infected endothelial cells in association with downregulation of vascular endothelial cadherin. J. Gen. Virol 2008, 89, 642–652. [Google Scholar] [CrossRef]
- Carr, J.M.; Ashander, L.M.; Calvert, J.K.; Ma, Y.; Aloia, A.; Bracho, G.G.; Chee, S.-P.; Appukuttan, B.; Smith, J.R. Molecular Responses of Human Retinal Cells to Infection with Dengue Virus. Mediat. Inflamm. 2017, 2017, 3164375. [Google Scholar] [CrossRef] [Green Version]
- Bakoa, F.; Préhaud, C.; Beauclair, G.; Chazal, M.; Mantel, N.; Lafon, M.; Jouvenet, N. Genomic diversity contributes to the neuroinvasiveness of the Yellow fever French neurotropic vaccine. NPJ Vaccines 2021, 6, 64. [Google Scholar] [CrossRef]
- Jeewandara, C.; Gomes, L.; Wickramasinghe, N.; Gutowska-Owsiak, D.; Waithe, D.; Paranavitane, S.A.; Shyamali, N.L.A.; Ogg, G.S.; Malavige, G.N. Platelet activating factor contributes to vascular leak in acute dengue infection. PLoS Negl. Trop. Dis. 2015, 9, e0003459. [Google Scholar] [CrossRef]
- Appanna, R.; Wang, S.M.; Ponnampalavanar, S.A.; Lum, L.C.S.; Sekaran, S.D. Cytokine Factors Present in Dengue Patient Sera Induces Alterations of Junctional Proteins in Human Endothelial Cells. Am. J. Trop. Med. Hyg. 2012, 87, 936–942. [Google Scholar] [CrossRef] [Green Version]
- Tramontini Gomes de Sousa Cardozo, F.; Baimukanova, G.; Lanteri, M.C.; Keating, S.M.; Moraes Ferreira, F.; Heitman, J.; Pannuti, C.S.; Pati, S.; Romano, C.M.; Cerdeira Sabino, E. Serum from dengue virus-infected patients with and without plasma leakage differentially affects endothelial cells barrier function in vitro. PLoS ONE 2017, 12, e0178820. [Google Scholar] [CrossRef]
- Rathore, A.P.; Mantri, C.K.; Aman, S.A.; Syenina, A.; Ooi, J.; Jagaraj, C.J.; Goh, C.C.; Tissera, H.; Wilder-Smith, A.; Ng, L.G.; et al. Dengue virus-elicited tryptase induces endothelial permeability and shock. J. Clin. Investig. 2019, 129, 4180–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puerta-Guardo, H.; Raya-Sandino, A.; González-Mariscal, L.; Rosales, V.H.; Ayala-Dávila, J.; Chávez-Mungía, B.; Martínez-Fong, D.; Medina, F.; Ludert, J.E.; del Angel, R.M. The cytokine response of U937-derived macrophages infected through antibody-dependent enhancement of dengue virus disrupts cell apical-junction complexes and increases vascular permeability. J. Virol. 2013, 87, 7486–7501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Anupriya, M.G.; Modak, A.; Sreekumar, E. Dengue virus or NS1 protein induces trans-endothelial cell permeability associated with VE-Cadherin and RhoA phosphorylation in HMEC-1 cells preventable by Angiopoietin-1. J. Gen. Virol. 2018, 99, 1658–1670. [Google Scholar] [CrossRef] [PubMed]
- Conroy, A.L.; Gélvez, M.; Hawkes, M.; Rajwans, N.; Tran, V.; Liles, W.C.; Villar-Centeno, L.A.; Kain, K.C. Host biomarkers are associated with progression to dengue haemorrhagic fever: A nested case-control study. Int. J. Infect. Dis. 2015, 40, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, B.; Tang, Y.; Hu, F.; Zhou, W.; Yao, X.; Hong, W.; Wang, J.; Zhang, X.; Tang, X.; Zhang, F. Serum levels of soluble vascular cell adhesion molecules may correlate with the severity of dengue virus-1 infection in adults. Emerg. Microbes Infect. 2015, 4, e4. [Google Scholar] [CrossRef]
- Castanha, P.M.S.; Braga, C.; Cordeiro, M.T.; Souza, A.I.; Silva, C.D., Jr.; Martelli, C.M.T.; van Panhuis, W.G.; Nascimento, E.J.M.; Marques, E.T.A. Placental Transfer of Dengue Virus (DENV)–Specific Antibodies and Kinetics of DENV Infection–Enhancing Activity in Brazilian Infants. J. Infect. Dis. 2016, 214, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, C.F.; Lopes, V.G.S.; Brasil, P.; Pires, A.R.C.; Rohloff, R.; Nogueira, R.M.R. Dengue infection in pregnancy and its impact on the placenta. Int. J. Infect. Dis. 2017, 55, 109–112. [Google Scholar] [CrossRef] [Green Version]
- Tabata, T.; Petitt, M.; Puerta-Guardo, H.; Michlmayr, D.; Wang, C.; Fang-Hoover, J.; Harris, E.; Pereira, L. Zika Virus Targets Different Primary Human Placental Cells, Suggesting Two Routes for Vertical Transmission. Cell Host Microbe 2016, 20, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Tabata, T.; Petitt, M.; Puerta-Guardo, H.; Michlmayr, D.; Harris, E.; Pereira, L. Zika Virus Replicates in Proliferating Cells in Explants From First-Trimester Human Placentas, Potential Sites for Dissemination of Infection. J. Infect. Dis. 2017, 217, 1202–1213. [Google Scholar] [CrossRef] [Green Version]
- Petitt, M.; Tabata, T.; Puerta-Guardo, H.; Harris, E.; Pereira, L. Zika virus infection of first-trimester human placentas: Utility of an explant model of replication to evaluate correlates of immune protection ex vivo. Curr. Opin. Virol. 2017, 27, 48–56. [Google Scholar] [CrossRef]
- Akey, D.L.; Brown, W.C.; Dutta, S.; Konwerski, J.; Jose, J.; Jurkiw, T.J.; DelProposto, J.; Ogata, C.M.; Skiniotis, G.; Kuhn, R.J.; et al. Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 2014, 343, 881–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biering, S.B.; Akey, D.L.; Wong, M.P.; Brown, W.C.; Lo, N.T.N.; Puerta-Guardo, H.; Tramontini Gomes de Sousa, F.; Wang, C.; Konwerski, J.R.; Espinosa, D.A.; et al. Structural basis for antibody inhibition of flavivirus NS1-triggered endothelial dysfunction. Science 2021, 371, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Lo, N.T.N.; Roodsari, S.; Tin, N.R.; Biering, S.B.; Harris, E. Molecular Determinants of Tissue Specificity of Flavivirus Nonstructural Protein 1 Interaction with Endothelial Cells. bioRxiv 2022. [Google Scholar] [CrossRef]
- Alcon-LePoder, S.; Drouet, M.T.; Roux, P.; Frenkiel, M.P.; Arborio, M.; Durand-Schneider, A.M.; Maurice, M.; Le Blanc, I.; Gruenberg, J.; Flamand, M. The secreted form of dengue virus nonstructural protein NS1 is endocytosed by hepatocytes and accumulates in late endosomes: Implications for viral infectivity. J. Virol. 2005, 79, 11403–11411. [Google Scholar] [CrossRef] [Green Version]
- Beurel, E.; Michalek, S.M.; Jope, R.S. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol. 2010, 31, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Rayasam, G.V.; Tulasi, V.K.; Sodhi, R.; Davis, J.A.; Ray, A. Glycogen synthase kinase 3: More than a namesake. Br. J. Pharm. 2009, 156, 885–898. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.H.; Yeh, S.H.; Tsay, Y.G.; Shieh, Y.H.; Kao, C.L.; Chen, Y.S.; Wang, S.H.; Kuo, T.J.; Chen, D.S.; Chen, P.J. Glycogen synthase kinase-3 regulates the phosphorylation of severe acute respiratory syndrome coronavirus nucleocapsid protein and viral replication. J. Biol. Chem. 2009, 284, 5229–5239. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Verma, A.; Garcia, G.; Ramage, H.; Myers, R.L.; Lucas, A.; Michaelson, J.J.; Coryell, W.; Kumar, A.; Charney, A.W.; et al. Targeting the Coronavirus Nucleocapsid Protein through GSK-3 Inhibition. medRxiv 2021. [Google Scholar] [CrossRef]
- König, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef]
- Saleh, M.; Rüschenbaum, S.; Welsch, C.; Zeuzem, S.; Moradpour, D.; Gouttenoire, J.; Lange, C.M. Glycogen Synthase Kinase 3β Enhances Hepatitis C Virus Replication by Supporting miR-122. Front. Microbiol. 2018, 9, 2949. [Google Scholar] [CrossRef] [Green Version]
- Cuartas-López, A.M.; Gallego-Gómez, J.C. Glycogen synthase kinase 3ß participates in late stages of Dengue virus-2 infection. Mem. Inst. Oswaldo Cruz. 2020, 115, e190357. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, J.; Maier, C.; Pagano, J.S. Epstein–Barr virus activates β-catenin in type III latently infected B lymphocyte lines: Association with deubiquitinating enzymes. Proc. Natl. Acad. Sci. USA 2003, 100, 15572–15576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Zloza, A.; Moon, R.T.; Watts, J.; Tenorio, A.R.; Al-Harthi, L. Active beta-catenin signaling is an inhibitory pathway for human immunodeficiency virus replication in peripheral blood mononuclear cells. J. Virol. 2008, 82, 2813–2820. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Montes-Grajales, D.; Puerta-Guardo, H.; Espinosa, D.A.; Harris, E.; Caicedo-Torres, W.; Olivero-Verbel, J.; Martínez-Romero, E. In silico drug repurposing for the identification of potential candidate molecules against arboviruses infection. Antivir. Res. 2020, 173, 104668. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puerta-Guardo, H.; Biering, S.B.; de Sousa, F.T.G.; Shu, J.; Glasner, D.R.; Li, J.; Blanc, S.F.; Beatty, P.R.; Harris, E. Flavivirus NS1 Triggers Tissue-Specific Disassembly of Intercellular Junctions Leading to Barrier Dysfunction and Vascular Leak in a GSK-3β-Dependent Manner. Pathogens 2022, 11, 615. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11060615
Puerta-Guardo H, Biering SB, de Sousa FTG, Shu J, Glasner DR, Li J, Blanc SF, Beatty PR, Harris E. Flavivirus NS1 Triggers Tissue-Specific Disassembly of Intercellular Junctions Leading to Barrier Dysfunction and Vascular Leak in a GSK-3β-Dependent Manner. Pathogens. 2022; 11(6):615. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11060615
Chicago/Turabian StylePuerta-Guardo, Henry, Scott B. Biering, Francielle Tramontini Gomes de Sousa, Jeffrey Shu, Dustin R. Glasner, Jeffrey Li, Sophie F. Blanc, P. Robert Beatty, and Eva Harris. 2022. "Flavivirus NS1 Triggers Tissue-Specific Disassembly of Intercellular Junctions Leading to Barrier Dysfunction and Vascular Leak in a GSK-3β-Dependent Manner" Pathogens 11, no. 6: 615. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11060615