
In Silico, In Vitro, and Pharmacokinetic Studies of UBMC-4, a Potential Novel Compound for Treating against Trypanosoma cruzi

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
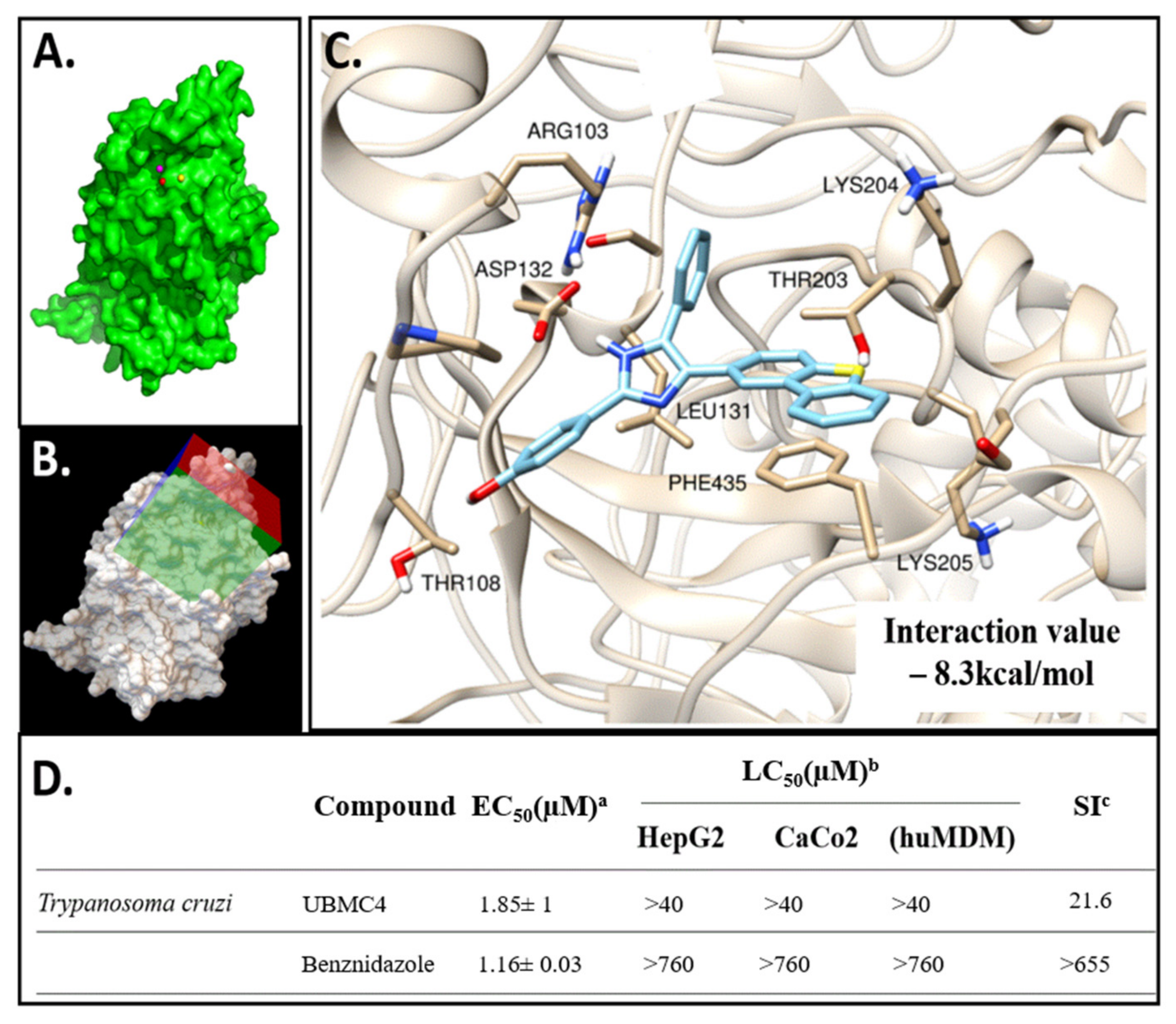
2.1. Molecular Docking of UBMC-4 and the T. cruzi AKT-like Pleckstrin Domain
2.2. UBMC-4 Cytotoxic and Trypanocidal Activity
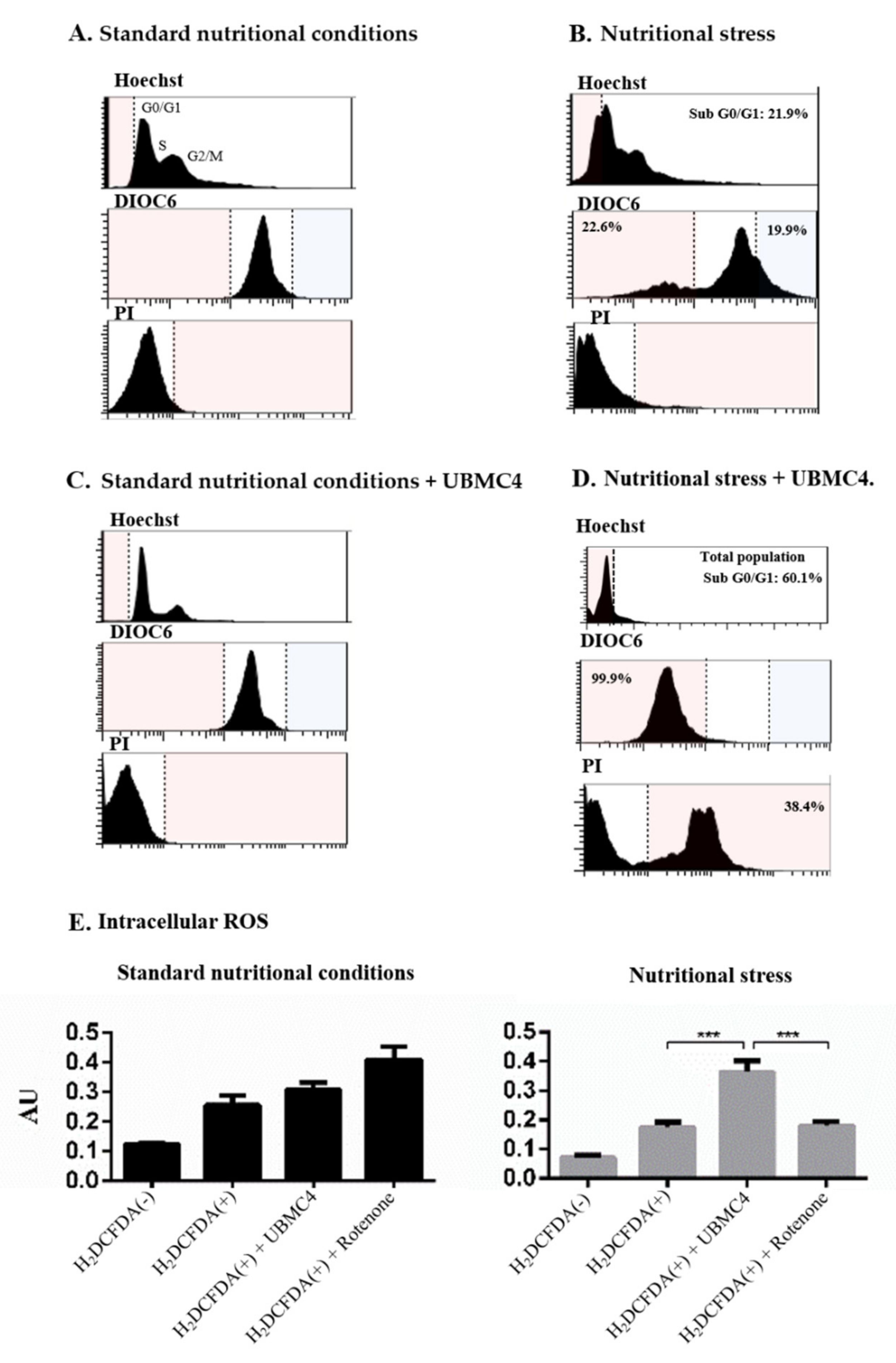
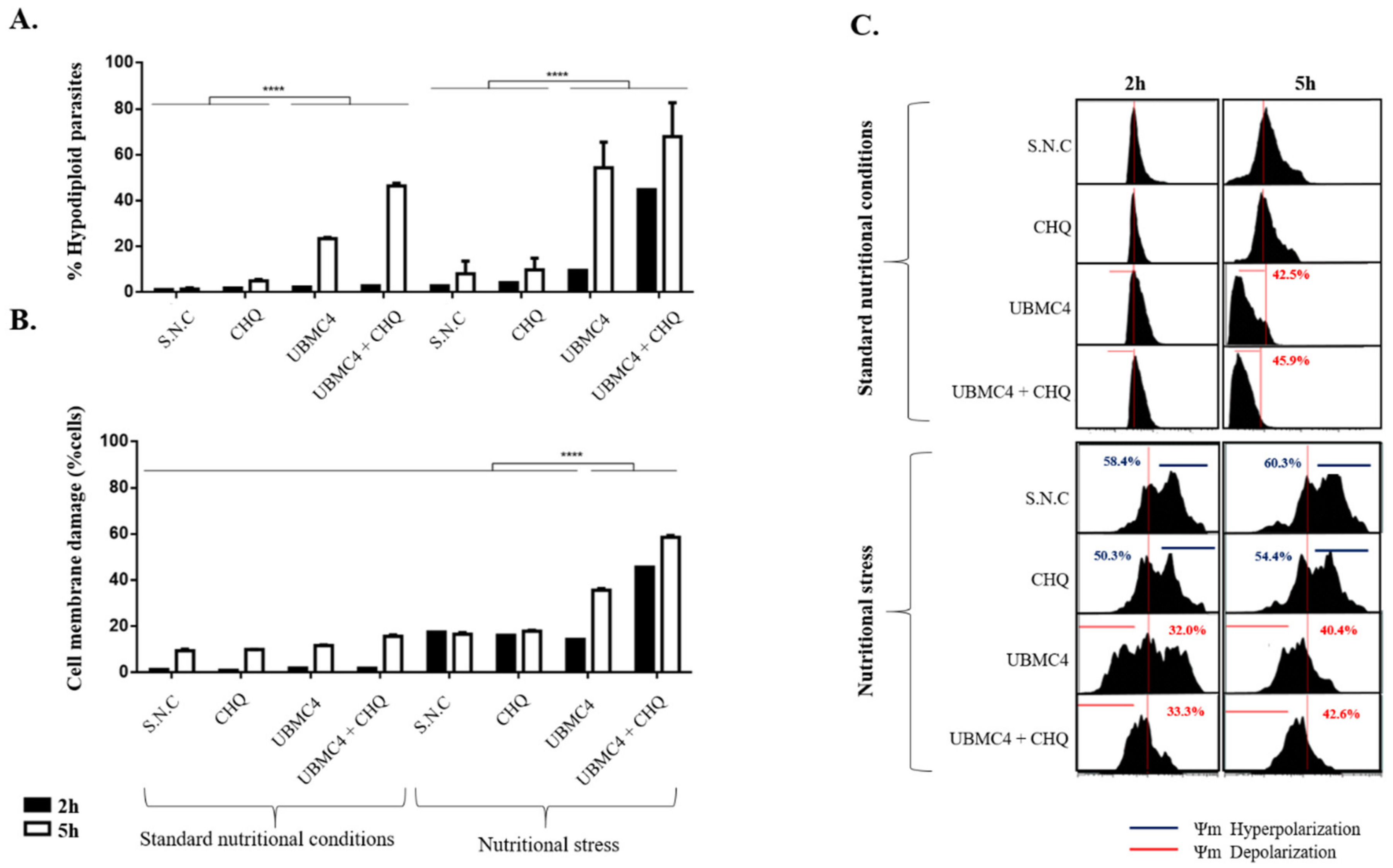
2.3. T. cruzi Is More Sensitive to Nutritional Stress in the Presence of AKT-like Inhibitor
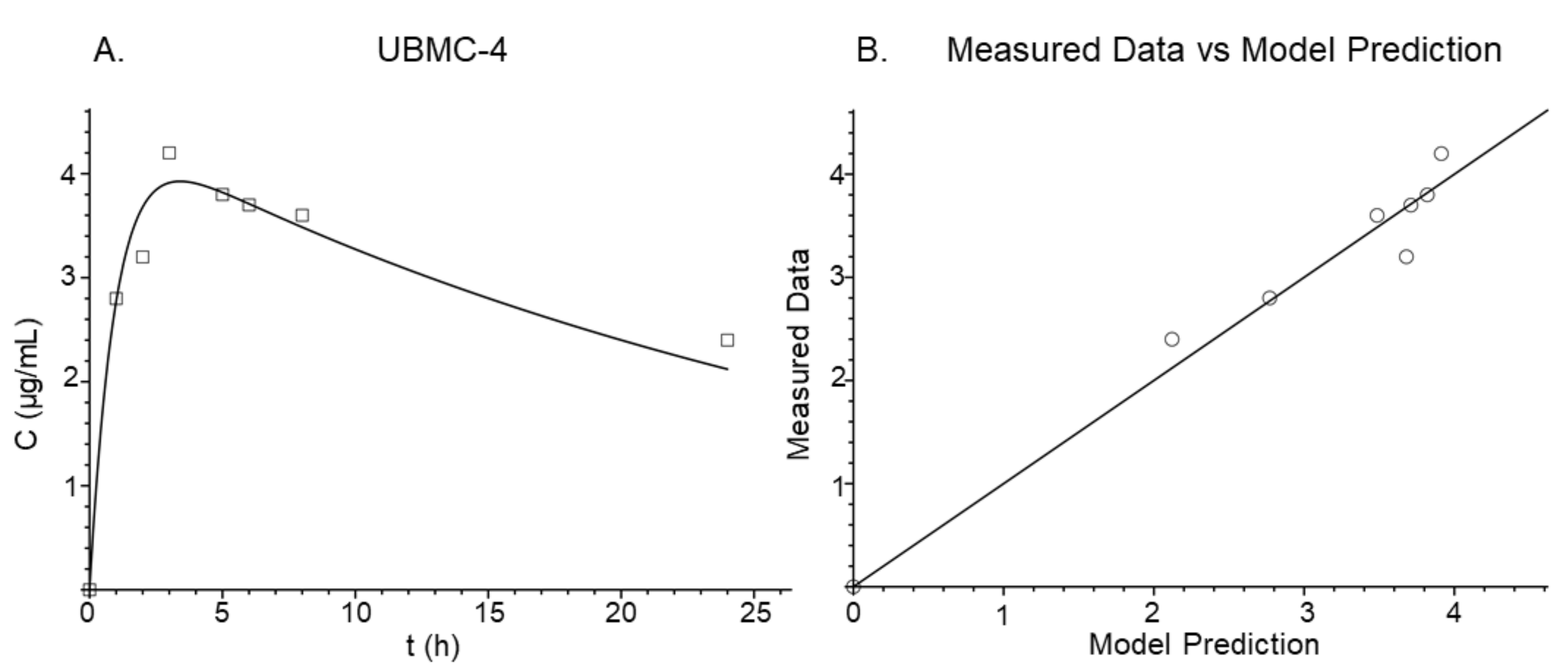
2.4. Plasma Concentration
2.5. Pharmacokinetic Modeling
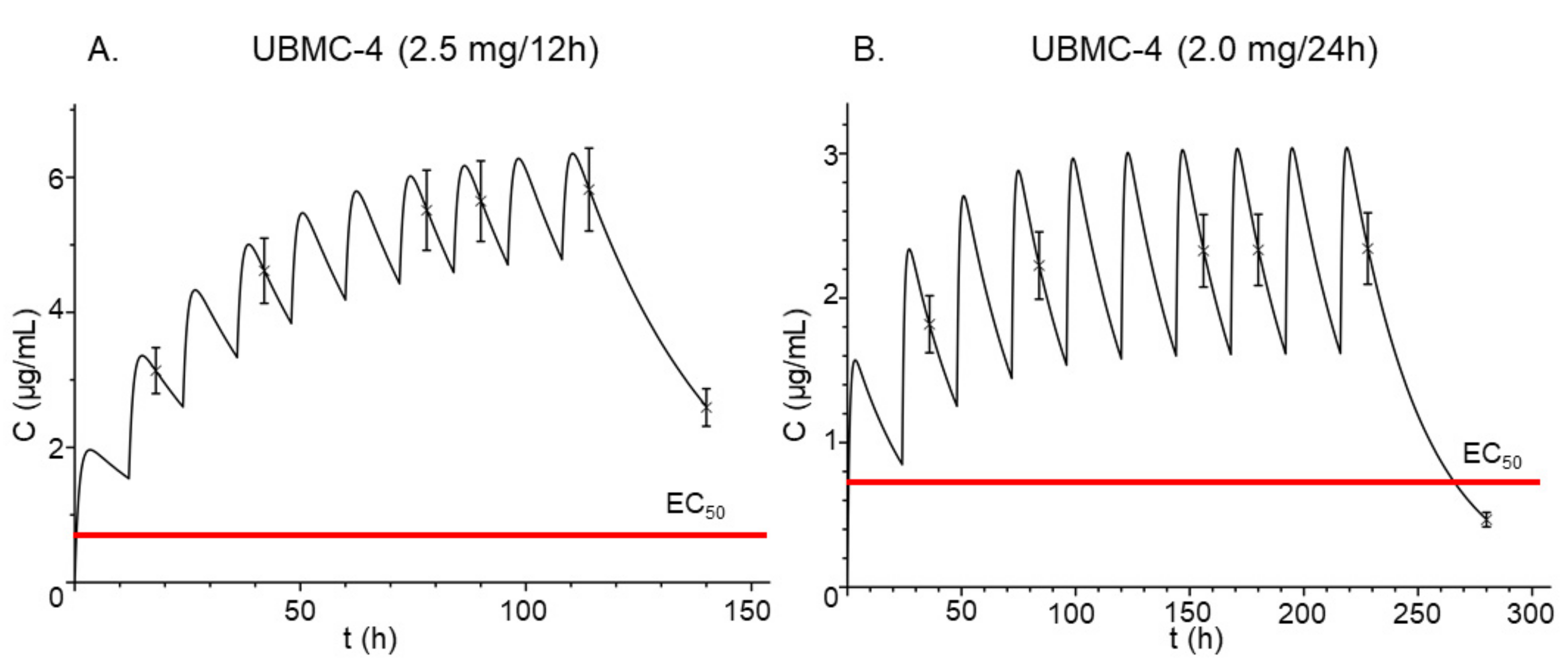
2.6. Pharmacokinetic Simulation
3. Discussion
4. Materials and Methods
4.1. Molecular Modeling of T. cruzi AKT-like Protein Structure and UBMC-4 Molecular Docking
4.2. Chemicals
4.3. Cell Lines and Parasite Strains
4.4. Cytotoxicity
4.5. Trypanocidal Activity of UBMC-4 in Intracellular Amastigotes
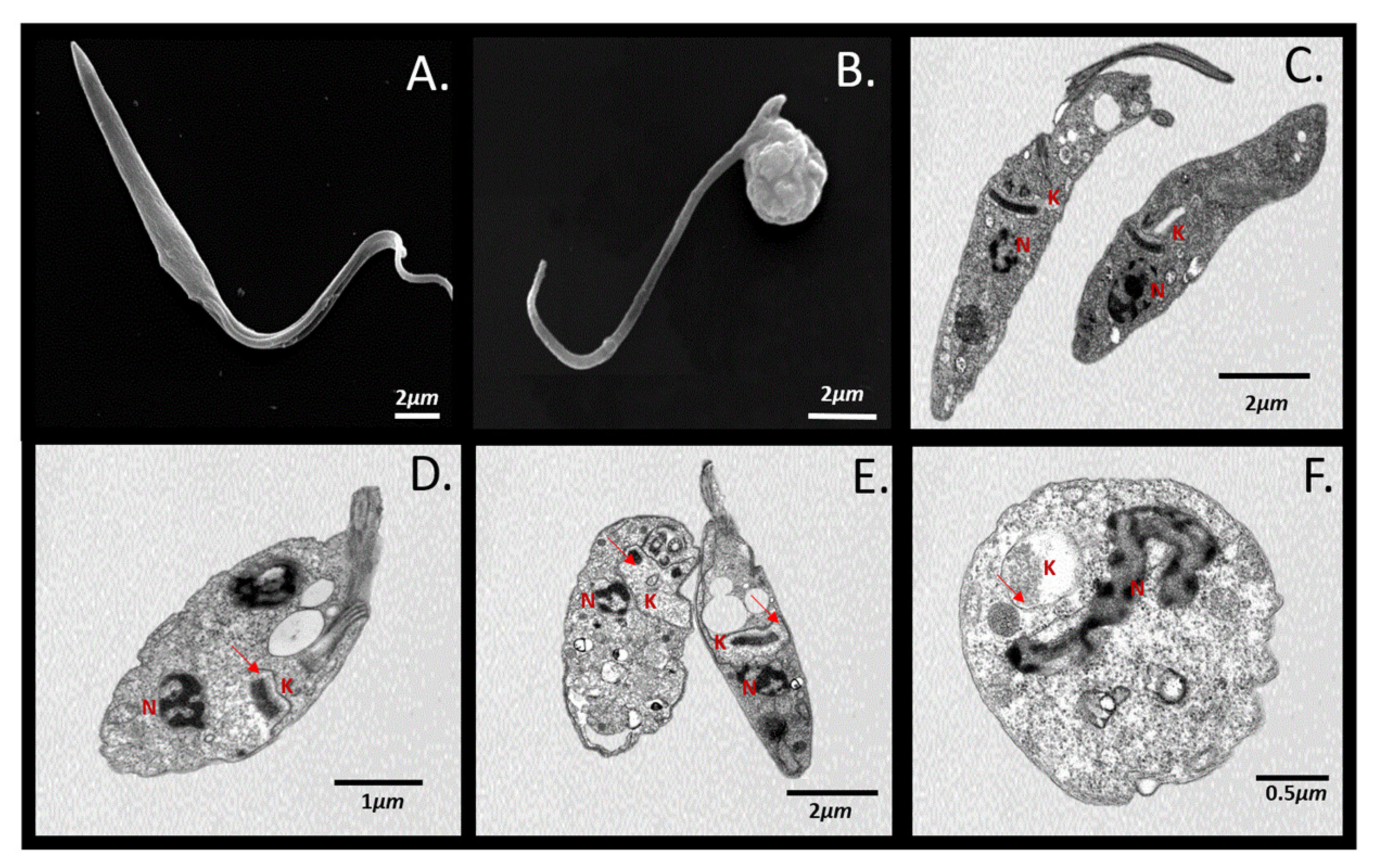
4.6. Scanning and Transmission Electron Microscopy
4.7. Flow Cytometry Analysis of Apoptosis-like Markers in T. cruzi
4.8. Analysis of Intracellular Production of Reactive Oxygen Species (ROS) in UBMC-4-Treated T. cruzi Epimastigotes
4.9. Formulation
4.10. Animals
4.11. In Vivo Pharmacokinetic Analysis of UBMC-4
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Q.; Chen, J.; Zhou, X.-N. Preparedness for Chagas disease spreading worldwide. Infect. Dis. Poverty 2020, 9, 4–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas Disease: From Discovery to a Worldwide Health Problem. Front. Public Health 2019, 7, 166. [Google Scholar] [CrossRef]
- Junior, P.A.S.; Molina, I.; Murta, S.M.F.; Sánchez-Montalvá, A.; Salvador, F.; Correa-Oliveira, R.; Carneiro, C.M. Experimental and Clinical Treatment of Chagas Disease: A Review. Am. J. Trop. Med. Hyg. 2017, 97, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Müller Kratz, J.; Garcia Bournissen, F.; Forsyth, C.J.; Sosa-Estani, S. Clinical and pharmacological profile of benznidazole for treatment of Chagas disease. Expert Rev. Clin. Pharmacol. 2018, 11, 943–957. [Google Scholar] [CrossRef] [Green Version]
- Altcheh, J.; Moscatelli, G.; Moroni, S.; Garcia-Bournissen, F.; Freilij, H. Adverse Events After the Use of Benznidazole in Infants and Children With Chagas Disease. Pediatrics 2011, 127, e212–e218. [Google Scholar] [CrossRef] [Green Version]
- Hasslocher-Moreno, A.M.; do Brasil, P.E.A.A.; de Sousa, A.S.; Xavier, S.S.; Chambela, M.C.; Sperandio da Silva, G.M. Safety of benznidazole use in the treatment of chronic Chagas’ disease. J. Antimicrob. Chemother. 2012, 67, 1261–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tornheim, J.A.; Beltran, D.F.L.; Gilman, R.H.; Castellon, M.; Mercado, M.A.S.; Sullca, W.; Torrico, F.; Bern, C. Improved Completion Rates and Characterization of Drug Reactions with an Intensive Chagas Disease Treatment Program in Rural Bolivia. PLoS Negl. Trop. Dis. 2013, 7, e2407. [Google Scholar] [CrossRef]
- Jackson, Y.; Wyssa, B.; Chappuis, F. Tolerance to nifurtimox and benznidazole in adult patients with chronic Chagas’ disease. J. Antimicrob. Chemother. 2020, 75, 690–696. [Google Scholar] [CrossRef] [Green Version]
- Maguire, J.H. Chagas’ disease—can we stop the deaths? N. Engl. J. Med. 2006, 355, 760–761. [Google Scholar] [CrossRef]
- Urbina, J.A. Recent clinical trials for the etiological treatment of chronic chagas disease: Advances, challenges and perspectives. J. Eukaryot. Microbiol. 2015, 62, 149–156. [Google Scholar] [CrossRef]
- Molina-Morant, D.; Fernández, M.L.; Bosch-Nicolau, P.; Sulleiro, E.; Bangher, M.; Salvador, F.; Sanchez-Montalva, A.; Ribeiro, A.L.P.; de Paula, A.M.B.; Eloi, S.; et al. Efficacy and safety assessment of different dosage of benznidazol for the treatment of Chagas disease in chronic phase in adults (MULTIBENZ study): Study protocol for a multicenter randomized Phase II non-inferiority clinical trial. Trials 2020, 21, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Peinado, N.; Cortes-Serra, N.; Losada-Galvan, I.; Alonso-Vega, C.; Urbina, J.A.; Rodríguez, A.; VandeBerg, J.L.; Pinazo, M.-J.; Gascon, J.; Alonso-Padilla, J. Emerging agents for the treatment of Chagas disease: What is in the preclinical and clinical development pipeline? Expert Opin. Investig. Drugs 2020, 29, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Padilla, J.; Cortés-Serra, N.; Pinazo, M.J.; Bottazzi, M.E.; Abril, M.; Barreira, F.; Sosa-Estani, S.; Hotez, P.J.; Gascón, J. Strategies to enhance access to diagnosis and treatment for Chagas disease patients in Latin America. Expert Rev. Anti. Infect. Ther. 2019, 17, 145–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, M.C.; Horn, D.; Fairlamb, A.H.; Ferguson, M.A.J.; Gray, D.W.; Read, K.D.; De Rycker, M.; Torrie, L.S.; Wyatt, P.G.; Wyllie, S.; et al. Anti-trypanosomatid drug discovery: An ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017, 15, 217–231. [Google Scholar] [CrossRef] [Green Version]
- De Koning, H.P. Drug resistance in protozoan parasites. Emerg. Top. life Sci. 2017, 1, 627–632. [Google Scholar] [CrossRef] [Green Version]
- Khare, S.; Nagle, A.S.; Biggart, A.; Lai, Y.H.; Liang, F.; Davis, L.C.; Barnes, S.W.; Mathison, C.J.N.; Myburgh, E.; Gao, M.-Y.; et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [Google Scholar] [CrossRef]
- Alvarez, V.E.; Iribarren, P.A.; Niemirowicz, G.T.; Cazzulo, J.J. Update on relevant trypanosome peptidases: Validated targets and future challenges. Biochim. Biophys. Acta—Proteins Proteom. 2021, 1869, 140577. [Google Scholar] [CrossRef]
- Beltrán-Hortelano, I.; Perez-Silanes, S.; Galiano, S. Trypanothione Reductase and Superoxide Dismutase as Current Drug Targets for Trypanosoma cruzi: An Overview of Compounds with Activity against Chagas Disease. Curr. Med. Chem. 2017, 24, 1066–1138. [Google Scholar] [CrossRef]
- Matoba, K.; Nara, T.; Aoki, T.; Honma, T.; Tanaka, A.; Inoue, M.; Matsuoka, S.; Inaoka, D.K.; Kita, K.; Harada, S. Crystallization and preliminary X-ray analysis of aspartate transcarbamoylase from the parasitic protist {\it Trypanosoma cruzi}. Acta Crystallogr. Sect. F 2009, 65, 933–936. [Google Scholar] [CrossRef] [Green Version]
- Trevisan, R.O.; Santos, M.M.; Desidério, C.S.; Alves, L.G.; Sousa, T.D.J.; Oliveira, L.D.C.; Jaiswal, A.K.; Tiwari, S.; Bovi, W.G.; De Oliveira-Silva, M.; et al. In Silico Identification of New Targets for Diagnosis, Vaccine, and Drug Candidates against Trypanosoma cruzi. Dis. Markers 2020, 2020, 9130719. [Google Scholar] [CrossRef]
- Lima, C.R.; Carels, N.; Guimaraes, A.C.R.; Tufféry, P.; Derreumaux, P. In silico structural characterization of protein targets for drug development against Trypanosoma cruzi. J. Mol. Model. 2016, 22, 244. [Google Scholar] [CrossRef] [PubMed]
- Capriles, P.V.S.Z.; Guimarães, A.C.R.; Otto, T.D.; Miranda, A.B.; Dardenne, L.E.; Degrave, W.M. Structural modelling and comparative analysis of homologous, analogous and specific proteins from Trypanosoma cruzi versus Homo sapiens: Putative drug targets for chagas’ disease treatment. BMC Genom. 2010, 11, 610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, E.; Ueda-Nakamura, T.; Dias Filho, B.P.; Veiga Júnior, V.F.; Nakamura, C.V. Natural products and Chagas{’} disease: A review of plant compounds studied for activity against Trypanosoma cruzi. Nat. Prod. Rep. 2011, 28, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.M.; Greco, G.M.Z.; Moreira, A.M.; Chagas, P.F.; Caldas, I.V.O.S.; Gonçalves, R.V.; Novaes, R.D. Applicability of plant-based products in the treatment of Trypanosoma cruzi and Trypanosoma brucei infections: A systematic review of preclinical in vivo evidence. Parasitology 2017, 144, 1275–1287. [Google Scholar] [CrossRef]
- Jones, A.J.; Grkovic, T.; Sykes, M.L.; Avery, V.M. Trypanocidal Activity of Marine Natural Products. Mar. Drugs 2013, 11, 4058–4082. [Google Scholar] [CrossRef] [Green Version]
- Torrico, F.; Gascón, J.; Barreira, F.; Blum, B.; Almeida, I.C.; Alonso-Vega, C.; Barboza, T.; Bilbe, G.; Correia, E.; Garcia, W.; et al. New regimens of benznidazole monotherapy and in combination with fosravuconazole for treatment of Chagas disease (BENDITA): A phase 2, double-blind, randomised trial. Lancet Infect. Dis. 2021, 21, 1129–1140. [Google Scholar] [CrossRef]
- Gashaw, I.; Ellinghaus, P.; Sommer, A.; Asadullah, K. What makes a good drug target? Drug Discov. Today 2012, 17, S24–S30. [Google Scholar] [CrossRef]
- Varela, R.E.M.; Ochoa, R.; Muskus, C.E.; Muro, A.; Mollinedo, F. Identification of a RAC/AKT-like gene in Leishmania parasites as a putative therapeutic target in leishmaniasis. Parasites Vectors 2017, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tirado-Duarte, D.; Marín-Villa, M.; Ochoa, R.; Blandón-Fuentes, G.; Soares, M.J.; Robledo, S.M.; Varela-Miranda, R.E. The Akt-like kinase of Leishmania panamensis: As a new molecular target for drug discovery. Acta Trop. 2018, 177, 171–178. [Google Scholar] [CrossRef]
- Ochoa, R.; Rocha-Roa, C.; Marín-Villa, M.; Robledo, S.M.; Varela-M, R.E. Search of allosteric inhibitors and associated proteins of an AKT-like kinase from trypanosoma cruzi. Int. J. Mol. Sci. 2018, 19, 3951. [Google Scholar] [CrossRef] [Green Version]
- Díez, A.F. Caracterización Funcional de la Quinasa AKT-like de Trypanosoma cruzi y Trypanosoma Brucei como Blanco para una Nueva Estrategia Terapéutica Contra la Tripanosomiasis. Master’s Thesis, University of Antioquia, Medellín, Colombia, 2019. [Google Scholar]
- Hanada, M.; Feng, J.; Hemmings, B.A. Structure, regulation and function of PKB/AKT—A major therapeutic target. Biochim. Biophys. Acta—Proteins Proteom. 2004, 1697, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Rahal, R.; Stransky, N.; Lengauer, C.; Hoeflich, K.P. Targeting cancer with kinase inhibitors. J. Clin. Investig. 2015, 125, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, C.W.; Barnett, S.F.; Yaroschak, M.; Bilodeau, M.T.; Layton, M.E. Recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Curr. Top. Med. Chem. 2007, 7, 1349–1363. [Google Scholar] [CrossRef] [PubMed]
- Digirolamo, F.; Miranda, M.; Bouvier, L.; Cámara, M.; Cánepa, G.; Claudio Pereira La Vía de Transducción de Señales tor de Mamíferos Está Presente en Trypanosoma Cruzi. Reconstrucción In Silico y Posibles Funciones. Medicina (Buenos Aires) 2012, 72, 221–226. [Google Scholar] [CrossRef]
- Bonate, P.L. Pharmacokinetic-Pharmacodynamic Modeling and Simulation, 2nd ed.; Springer: Deerfield, IL, USA, 2011; ISBN 978-1-4419-9485-1. [Google Scholar]
- Menna-Barreto, R.F.S. Between Armour and Weapons—Cell Death Mechanisms in Trypanosomatid Parasites. Available online: https://www.intechopen.com/chapters/48836 (accessed on 16 May 2022).
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Isaka, Y. Chloroquine in cancer therapy: A double-edged sword of autophagy. Cancer Res. 2013, 73, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Chuenkova, M.V.; PereiraPerrin, M. Trypanosoma cruzi Targets Akt in Host Cells as an Intracellular Antiapoptotic Strategy. Sci. Signal. 2009, 2, ra74. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Pascuccelli, V.; Labriola, C.; Téllez-Iñón, M.T.; Parodi, A.J. Molecular and biochemical characterization of a protein kinase B from Trypanosoma cruzi. Mol. Biochem. Parasitol. 1999, 102, 21–33. [Google Scholar] [CrossRef]
- Adejoro, I.A.; Waheed, S.O.; Adeboye, O.O. Molecular Docking Studies of Lonchocarpus cyanescens Triterpenoids as Inhibitors for Malaria. J. Phys. Chem. Biophys. 2016, 6, 2–5. [Google Scholar] [CrossRef] [Green Version]
- Dhorajiwala, T.M.; Halder, S.T.; Samant, L. Comparative in silico molecular docking analysis of l-threonine-3-dehydrogenase, a protein target against African trypanosomiasis using selected phytochemicals. J. Appl. Biotechnol. Rep. 2019, 6, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Araujo-Lima, C.F.; Peres, R.B.; Silva, P.B.; Batista, M.M.; Aiub, C.A.F.; Felzenszwalb, I.; Soeiro, M.N.C. Repurposing Strategy of Atorvastatin against Trypanosoma cruzi: In Vitro Monotherapy and Combined Therapy with Benznidazole Exhibit Synergistic Trypanocidal Activity. Antimicrob. Agents Chemother. 2018, 62, e00979-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menna-Barreto, R.F.S. Cell death pathways in pathogenic trypanosomatids: Lessons of (over)kill. Cell Death Dis. 2019, 10, 93. [Google Scholar] [CrossRef] [PubMed]
- Angus, B.J.; Thaiaporn, I.; Chanthapadith, K.; Suputtamongkol, Y.; White, N.J. Oral artesunate dose-response relationship in acute falciparum malaria. Antimicrob. Agents Chemother. 2002, 46, 778–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, M.; Bray, M.A.; Cal, M.; Trunz, B.B.; Torreele, E.; Brun, R. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 2011, 55, 5602–5608. [Google Scholar] [CrossRef] [Green Version]
- Levêque, D.; Becker, G.; Bilger, K.; Natarajan-Amé, S. Clinical Pharmacokinetics and Pharmacodynamics of Dasatinib. Clin. Pharmacokinet. 2020, 59, 849–856. [Google Scholar] [CrossRef] [PubMed]
- DNDi—Drugs for Neglected Diseases Initiative Target Product Profile for Chagas Disease. Available online: https://dndi.org/diseases/chagas/target-product-profile/ (accessed on 16 May 2022).
- Kratz, J.M.; Gonçalves, K.R.; Romera, L.M.D.; Moraes, C.B.; Bittencourt-Cunha, P.; Schenkman, S.; Chatelain, E.; Sosa-Estani, S. The translational challenge in chagas disease drug development. Mem. Inst. Oswaldo Cruz 2021, 116, 1–11. [Google Scholar] [CrossRef]
- Katsuno, K.; Burrows, J.N.; Duncan, K.; Hooft van Huijsduijnen, R.; Kaneko, T.; Kita, K.; Mowbray, C.E.; Schmatz, D.; Warner, P.; Slingsby, B.T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov. 2015, 14, 751–758. [Google Scholar] [CrossRef]
- El-sayed, N.M.; Myler, P.J.; Bartholomeu, D.C.; Nilsson, D.; Aggarwal, G.; Westenberger, S.J.; Tran, A.; Ghedin, E.; Worthey, E.A.; Delcher, A.L.; et al. The Genome Sequence of Trypanosoma cruzi, Etiologic Agent of Chagas Disease. Science 2005, 4975, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2011, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, 252–258. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa, R.; Watowich, S.J.; Flórez, A.; Mesa, C.V.; Robledo, S.M.; Muskus, C. Drug search for leishmaniasis: A virtual screening approach by grid computing. J. Comput. Aided. Mol. Des. 2016, 30, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Schroeder, M. LIGSITE csc: Predicting ligand binding sites using the Connolly surface and degree of conservation. BMC Struct. Biol. 2006, 11, 19. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. Wiley Intersci. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Daigneault, M.; Preston, J.A.; Marriott, H.M.; Whyte, M.K.B.; Dockrell, D.H. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS ONE 2010, 5, e8668. [Google Scholar] [CrossRef]
- Buckner, F.S.; Verlinde, C.L.M.J.; La Flamme, A.C.; Van Voorhis, W.C. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing β-galactosidase. Antimicrob. Agents Chemother. 1996, 40, 2592–2597. [Google Scholar] [CrossRef] [Green Version]
- Tolosa, L.; Donato, M.T.; Gómez-Lechón, M.J. General Cytotoxicity Assessment by Means of the MTT Assay. In Protocols in In Vitro Hepatocyte Research; Vinken, M., Rogiers, V., Eds.; Springer: New York, NY, USA, 2015; pp. 333–348. ISBN 978-1-4939-2074-7. [Google Scholar]
- Tavares, G.D.S.V.; Mendonça, D.V.C.; Lage, D.P.; Granato, J.D.T.; Ottoni, F.M.; Ludolf, F.; Chávez-Fumagalli, M.A.; Duarte, M.C.; Tavares, C.A.P.; Alves, R.J.; et al. Antileishmanial Activity, Cytotoxicity and Mechanism of Action of Clioquinol against Leishmania infantum and Leishmania amazonensis Species. Basic Clin. Pharmacol. Toxicol. 2018, 123, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Yotnda, P. Production and Detection of Reactive Oxygen Species (ROS) in Cancers. JoVE 2011, 57, e3357. [Google Scholar] [CrossRef]
- D’Argenio, D.Z.; Schumitzky, A.; Wang, X.A. ADAPT 5 User’s Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software. 2009. Available online: https://bmsr.usc.edu/software/adapt/users-guide/ (accessed on 10 August 2021).
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Automat. Contr. 1974, 19, 716–723. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (h) | Concentration (μg/mL) |
|---|---|
| 0 | 0 |
| 1 | 2.8 |
| 2 | 3.2 |
| 3 | 4.2 |
| 5 | 3.8 |
| 6 | 3.7 |
| 8 | 3.6 |
| 24 | 2.4 |
| Selection Parameters | One-Compartment | Two-Compartment | Three-Compartment |
|---|---|---|---|
| −Log-Likelihood (−LL) | 14.903 | −100.874 | 8.636 |
| Akaike Information Criterion (AIC) | 41.806 | −187.749 | 33.372 |
| r2 | 0.737 | 0.968 | 0.882 |
| Parameter | Estimated Value |
|---|---|
| CLt/F(L/h) | 0.0357 |
| Vc/F (L) | 0.6930 |
| Ka (h−1) | 0.6213 |
| CLd/F (L/h) | 0.2514 |
| Vp/F (L) | 0.4360 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustamante, C.; Díez-Mejía, A.F.; Arbeláez, N.; Soares, M.J.; Robledo, S.M.; Ochoa, R.; Varela-M., R.E.; Marín-Villa, M. In Silico, In Vitro, and Pharmacokinetic Studies of UBMC-4, a Potential Novel Compound for Treating against Trypanosoma cruzi. Pathogens 2022, 11, 616. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11060616
Bustamante C, Díez-Mejía AF, Arbeláez N, Soares MJ, Robledo SM, Ochoa R, Varela-M. RE, Marín-Villa M. In Silico, In Vitro, and Pharmacokinetic Studies of UBMC-4, a Potential Novel Compound for Treating against Trypanosoma cruzi. Pathogens. 2022; 11(6):616. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11060616
Chicago/Turabian StyleBustamante, Christian, Andrés Felipe Díez-Mejía, Natalia Arbeláez, Maurilio José Soares, Sara M. Robledo, Rodrigo Ochoa, Rubén E. Varela-M., and Marcel Marín-Villa. 2022. "In Silico, In Vitro, and Pharmacokinetic Studies of UBMC-4, a Potential Novel Compound for Treating against Trypanosoma cruzi" Pathogens 11, no. 6: 616. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11060616