Multiple Introductions and Predominance of Rotavirus Group A Genotype G3P[8] in Kilifi, Coastal Kenya, 4 Years after Nationwide Vaccine Introduction

 , and
, and
Abstract
:1. Introduction
2. Results
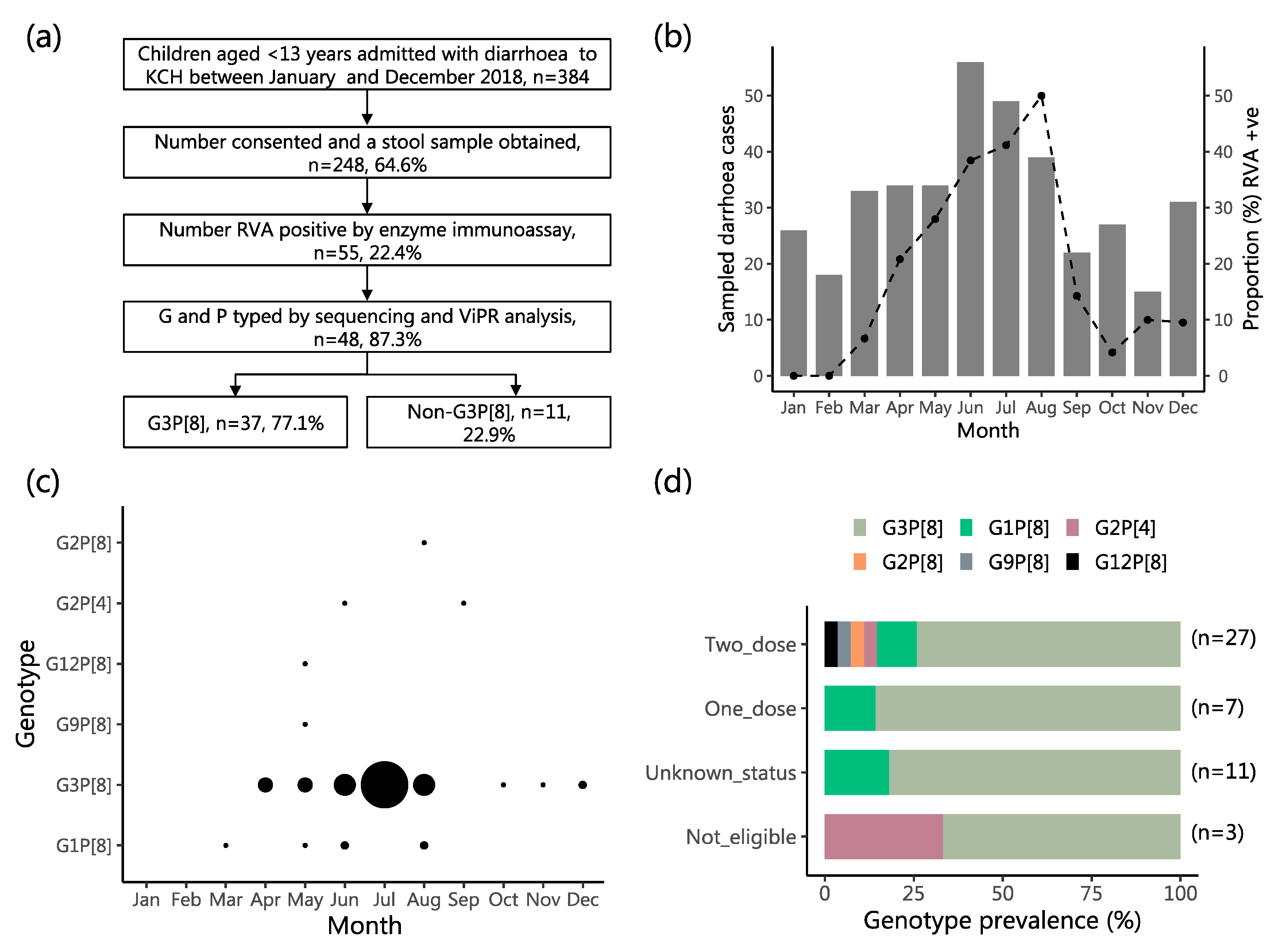
2.1. Study Population Characteristics
2.2. Characteristics of the RVA Infections and the Infected Children
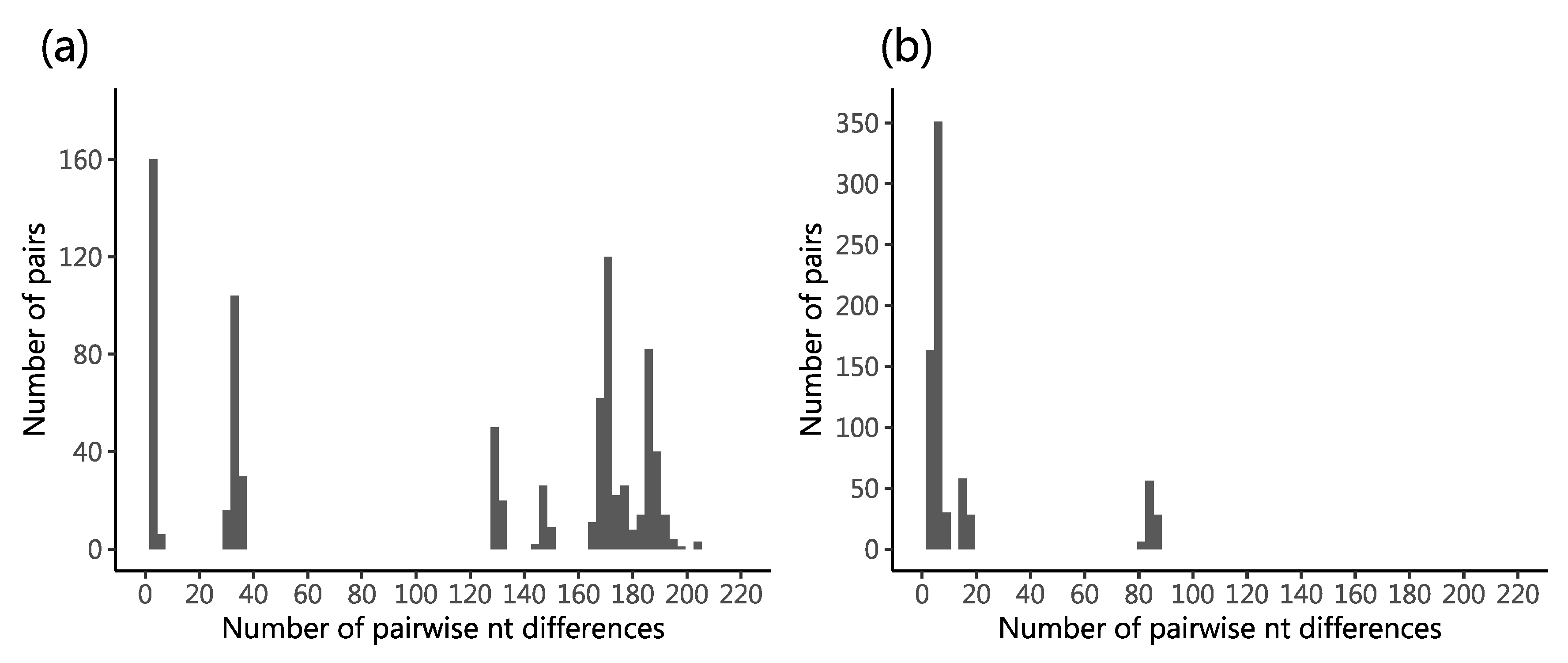
2.3. Genetic Diversity in the Sequenced Viruses
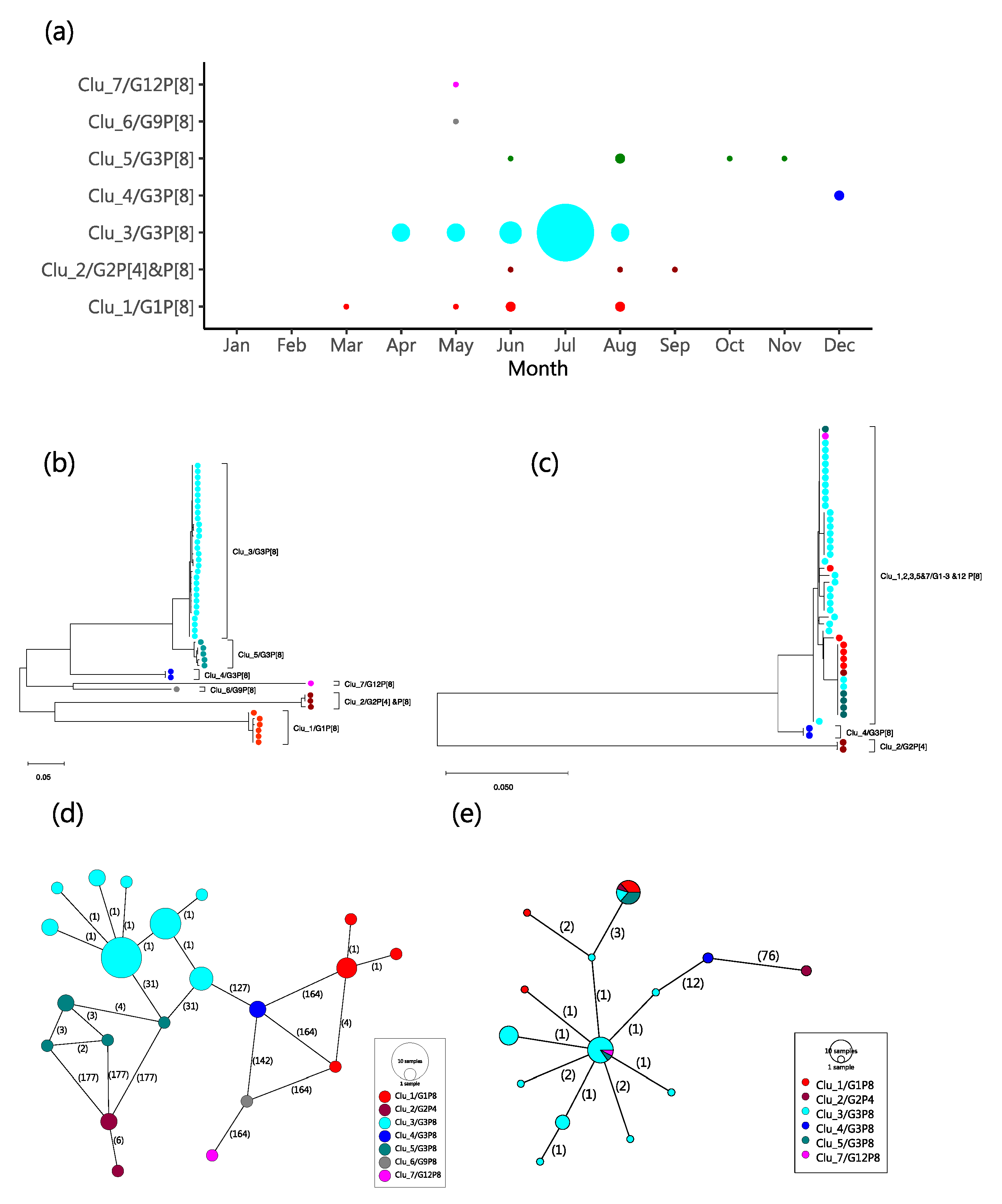
2.4. Molecular Genetic Clusters
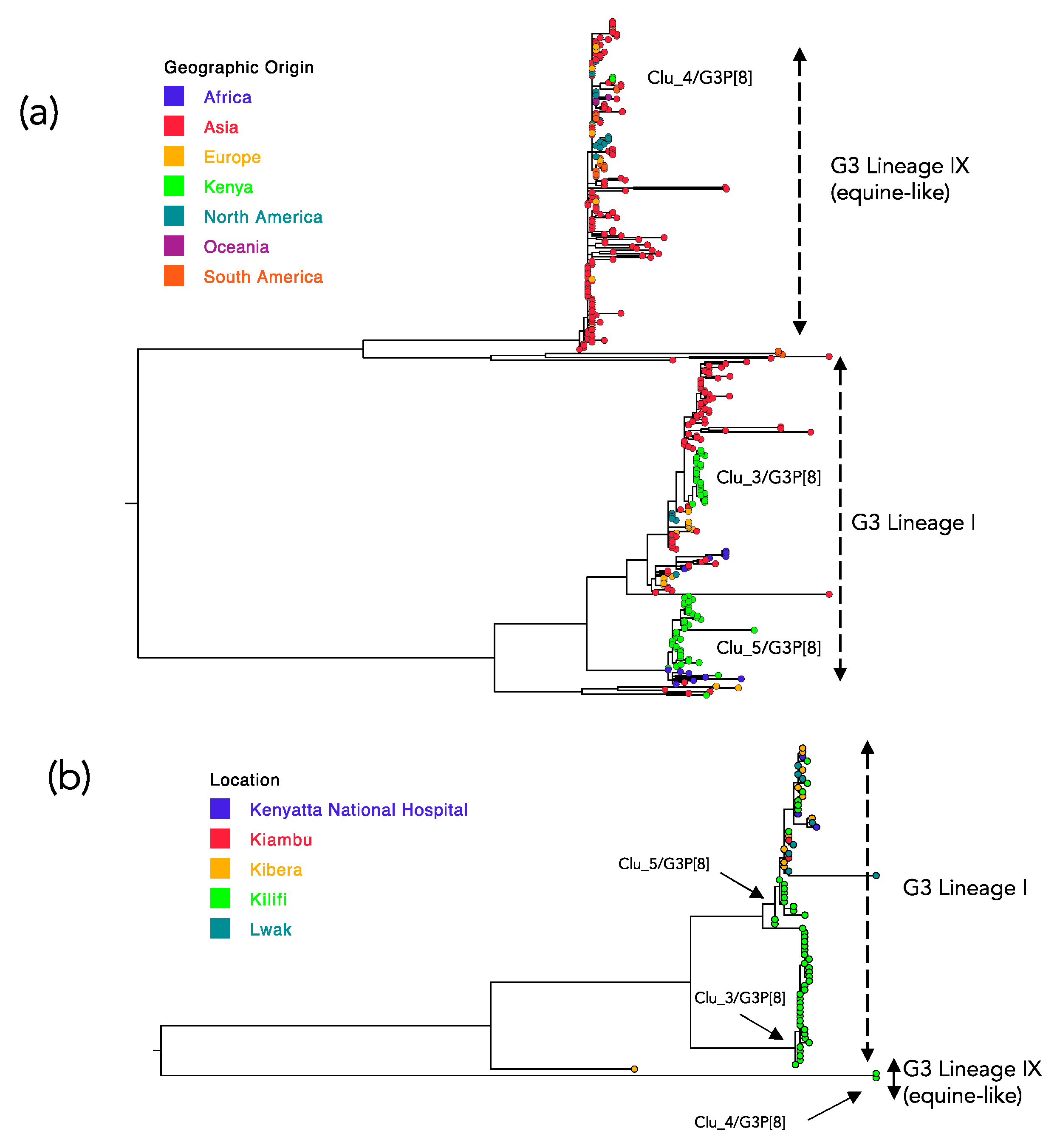
2.5. Spatial Distribution of the Kilifi G3 Genetic Clusters
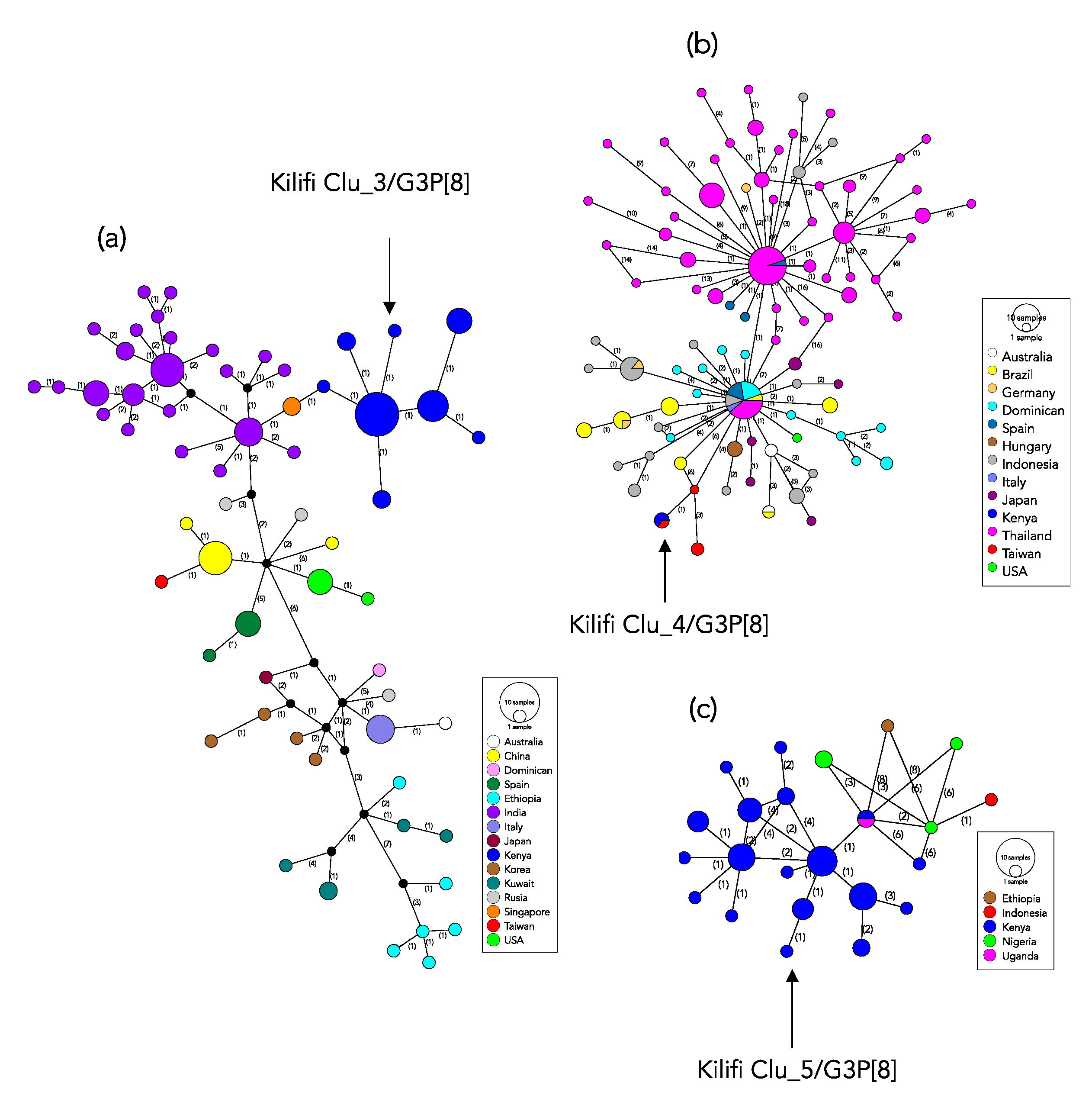
2.6. Global Genetic Context of the Kilifi 2018 G3 Strains
3. Discussion
4. Materials and Methods
4.1. Study Population and Location
4.2. Specimen Laboratory Processing
4.3. Genotyping and Phylogenetic Analysis
4.4. Genetic Clusters
4.5. Comparison Dataset
4.6. Statistical Analysis
4.7. Data Availability
4.8. Ethical Statement
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Burnett, E.; Jonesteller, C.L.; Tate, J.E.; Yen, C.; Parashar, U.D. Global Impact of Rotavirus Vaccination on Childhood Hospitalizations and Mortality From Diarrhea. J. Infect. Dis. 2017, 215, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.D.; Groome, M.J. Measuring Rotavirus Vaccine Impact in Sub-Saharan Africa. Clin. Infect. Dis. 2020, 70, 2314–2316. [Google Scholar] [CrossRef] [PubMed]
- Operario, D.J.; Platts-Mills, J.A.; Nadan, S.; Page, N.; Seheri, M.; Mphahlele, J.; Praharaj, I.; Kang, G.; Araujo, I.T.; Leite, J.P.G.; et al. Etiology of Severe Acute Watery Diarrhea in Children in the Global Rotavirus Surveillance Network Using Quantitative Polymerase Chain Reaction. J. Infect. Dis. 2017, 216, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Iturriza-Gómara, M.; Jere, K.C.; Hungerford, D.; Bar-Zeev, N.; Shioda, K.; Kanjerwa, O.; Houpt, E.R.; Operario, D.J.; Wachepa, R.; Pollock, L.; et al. Etiology of Diarrhea Among Hospitalized Children in Blantyre, Malawi, Following Rotavirus Vaccine Introduction: A Case-Control Study. J. Infect. Dis. 2019, 220, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus Vaccination and the Global Burden of Rotavirus Diarrhea Among Children Younger Than 5 Years. JAMA Pediatr. 2018, 172, 958–965. [Google Scholar] [CrossRef] [Green Version]
- Steele, D.; Victor, J.; Carey, M.; Tate, J.; Atherly, D.; Pecenka, C.; Diaz, Z.; Parashar, U.; Kirkwood, C. Experiences with rotavirus vaccines: Can we improve rotavirus vaccine impact in developing countries? Hum. Vaccines Immunother. 2019, 15, 1215–1227. [Google Scholar] [CrossRef] [Green Version]
- Willame, C.; Noordegraaf-Schouten, M.V.; Gvozdenović, E.; Kochems, K.; Oordt-Speets, A.; Praet, N.; Van Hoorn, R.; Rosillon, D. Effectiveness of the Oral Human Attenuated Rotavirus Vaccine: A Systematic Review and Meta-analysis—2006–2016. Open Forum Infect. Dis. 2018, 5, ofy292. [Google Scholar] [CrossRef] [Green Version]
- Khagayi, S.; Omore, R.; Otieno, G.P.; Ogwel, B.; Ochieng, J.B.; Juma, J.; Apondi, E.; Bigogo, G.; Onyango, C.; Ngama, M.; et al. Effectiveness of Monovalent Rotavirus Vaccine Against Hospitalization With Acute Rotavirus Gastroenteritis in Kenyan Children. Clin. Infect. Dis. 2019, 70, 2298–2305. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.L.; Andrews, N.J.; Atchison, C.J.; Collins, S.; Allen, D.J.; Ramsay, M.E.; Ladhani, S.N.; Thomas, S.L. Effectiveness of oral rotavirus vaccination in England against rotavirus-confirmed and all-cause acute gastroenteritis. Vaccine X 2019, 1, 100005. [Google Scholar] [CrossRef]
- Nair, N.; Feng, N.; Blum, L.K.; Sanyal, M.; Ding, S.; Jiang, B.; Sen, A.; Morton, J.M.; He, X.-S.; Robinson, W.H.; et al. VP4- and VP7-specific antibodies mediate heterotypic immunity to rotavirus in humans. Sci. Transl. Med. 2017, 9, eaam5434. [Google Scholar] [CrossRef] [Green Version]
- RCWG. Rotavirus Classification Working Group: Newly Assigned Genotypes. Available online: https://rega.kuleuven.be/cev/viralmetagenomics/virus-classification/rcwg (accessed on 7 January 2020).
- Sadiq, A.; Bostan, N.; Yinda, K.C.; Naseem, S.; Sattar, S. Rotavirus: Genetics, pathogenesis and vaccine advances. Rev. Med. Virol. 2018, 28, e2003. [Google Scholar] [CrossRef] [PubMed]
- Leshem, E.; Lopman, B.; Glass, R.; Gentsch, J.; Bányai, K.; Parashar, U.; Patel, M. Distribution of rotavirus strains and strain-specific effectiveness of the rotavirus vaccine after its introduction: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 847–856. [Google Scholar] [CrossRef]
- Roczo-Farkas, S.; Kirkwood, C.D.; Cowley, D.; Barnes, G.L.; Bishop, R.F.; Bogdanovic-Sakran, N.; Boniface, K.; Donato, C.M.; Bines, J.E. The Impact of Rotavirus Vaccines on Genotype Diversity: A Comprehensive Analysis of 2 Decades of Australian Surveillance Data. J. Infect. Dis. 2018, 218, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.M.; Tate, J.E.; Barin, N.; Bock, C.; Bowen, M.D.; Chang, D.; Gautam, R.; Han, G.; Holguin, J.; Huynh, T.; et al. Three Rotavirus Outbreaks in the Postvaccine Era—California, 2017. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 470–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitzer, V.E.; Bilcke, J.; Heylen, E.; Crawford, F.W.; Callens, M.; De Smet, F.; Van Ranst, M.; Zeller, M.; Matthijnssens, J. Did Large-Scale Vaccination Drive Changes in the Circulating Rotavirus Population in Belgium? Sci. Rep. 2015, 5, 18585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianiro, G.; Micolano, R.; Di Bartolo, I.; Scavia, G.; Monini, M.; RotaNet-Italy Study Group. Group A rotavirus surveillance before vaccine introduction in Italy, September 2014 to August 2017. Eurosurveillance 2019, 24, 1800418. [Google Scholar] [CrossRef]
- Ogden, K.M.; Tan, Y.; Akopov, A.; Stewart, L.S.; McHenry, R.; Fonnesbeck, C.J.; Piya, B.; Carter, M.H.; Fedorova, N.B.; Halpin, R.A.; et al. Multiple Introductions and Antigenic Mismatch with Vaccines May Contribute to Increased Predominance of G12P[8] Rotaviruses in the United States. J. Virol. 2018, 93, e01476-18. [Google Scholar] [CrossRef] [Green Version]
- Santos, N.; Hoshino, Y. Global distribution of rotavirus serotypes/genotypes and its implication for the development and implementation of an effective rotavirus vaccine. Rev. Med. Virol. 2005, 15, 29–56. [Google Scholar] [CrossRef]
- Arana, A.; Montes, M.; Jere, K.C.; Alkorta, M.; Iturriza-Gómara, M.; Cilla, G. Emergence and spread of G3P[8] rotaviruses possessing an equine-like VP7 and a DS-1-like genetic backbone in the Basque Country (North of Spain), 2015. Infect. Genet. Evol. 2016, 44, 137–144. [Google Scholar] [CrossRef]
- Cowley, D.; Donato, C.M.; Roczo-Farkas, S.; Kirkwood, C.D. Emergence of a novel equine-like G3P[8] inter-genogroup reassortant rotavirus strain associated with gastroenteritis in Australian children. J. Gen. Virol. 2016, 97, 403–410. [Google Scholar] [CrossRef]
- Dóró, R.; Marton, S.; Bartókné, A.H.; Lengyel, G.; Agócs, Z.; Jakab, F.; Bányai, K. Equine-like G3 rotavirus in Hungary, 2015—Is it a novel intergenogroup reassortant pandemic strain? Acta Microbiol. Immunol. Hung. 2016, 63, 243–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, S.; Camilloni, B.; Bianchini, S.; Ianiro, G.; Polinori, I.; Farinelli, E.; Monini, M.; Principi, N. First detection of a reassortant G3P[8] rotavirus A strain in Italy: A case report in an 8-year-old child. Virol. J. 2019, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- João, E.D.; Munlela, B.; Chissaque, A.; Chilaúle, J.; Langa, J.; Augusto, O.; Boene, S.S.; Anapakala, E.; Sambo, J.; Guimarães, E.; et al. Molecular Epidemiology of Rotavirus A Strains Pre- and Post-Vaccine (Rotarix®) Introduction in Mozambique, 2012–2019: Emergence of Genotypes G3P[4] and G3P[8]. Pathogens 2020, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.M.; Esona, M.D.; Betrapally, N.; Leon, L.A.D.L.C.D.; Neira, Y.R.; Rey, G.J.; Bowen, M.D. Whole-gene analysis of inter-genogroup reassortant rotaviruses from the Dominican Republic: Emergence of equine-like G3 strains and evidence of their reassortment with locally-circulating strains. Virology 2019, 534, 114–131. [Google Scholar] [CrossRef]
- Perkins, C.; Mijatovic-Rustempasic, S.; Ward, M.L.; Cortese, M.M.; Bowen, M.D. Genomic Characterization of the First Equine-Like G3P[8] Rotavirus Strain Detected in the United States. Genome Announc. 2017, 5, e01341-17. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, C.; Liebert, U. Molecular characterization of different equine-like G3 rotavirus strains from Germany. Infect. Genet. Evol. 2018, 57, 46–50. [Google Scholar] [CrossRef]
- Tacharoenmuang, R.; Komoto, S.; Guntapong, R.; Upachai, S.; Singchai, P.; Ide, T.; Fukuda, S.; Ruchusatsawast, K.; Sriwantana, B.; Tatsumi, M.; et al. High prevalence of equine-like G3P[8] rotavirus in children and adults with acute gastroenteritis in Thailand. J. Med. Virol. 2020, 92, 174–186. [Google Scholar] [CrossRef]
- Utsumi, T.; Wahyuni, R.M.; Doan, Y.H.; Dinana, Z.; Soegijanto, S.; Fujii, Y.; Juniastuti; Yamani, L.N.; Matsui, C.; Deng, L.; et al. Equine-like G3 rotavirus strains as predominant strains among children in Indonesia in 2015-2016. Infect. Genet. Evol. 2018, 61, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Mwanga, M.J.; Owor, B.E.; Ochieng, J.B.; Ngama, M.H.; Ogwel, B.; Onyango, C.; Juma, J.; Njeru, R.; Gicheru, E.; Otieno, G.P.; et al. Rotavirus group A genotype circulation patterns across Kenya before and after nationwide vaccine introduction, 2010–2018. BMC Infect. Dis. 2020, 20, 504. [Google Scholar] [CrossRef]
- Owor, B.E.; Mwanga, M.J.; Njeru, R.; Mugo, R.; Ngama, M.; Otieno, G.P.; Nokes, D.J.; Agoti, C.N. Molecular characterization of rotavirus group A strains circulating prior to vaccine introduction in rural coastal Kenya, 2002–2013. Wellcome Open Res. 2018, 3, 150. [Google Scholar] [CrossRef]
- Thongprachum, A.; Chan-It, W.; Khamrin, P.; Okitsu, S.; Nishimura, S.; Kikuta, H.; Yamamoto, A.; Sugita, K.; Baba, T.; Mizuguchi, M.; et al. Reemergence of new variant G3 rotavirus in Japanese pediatric patients, 2009–2011. Infect. Genet. Evol. 2013, 13, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Umair, M.; Abbasi, B.H.; Sharif, S.; Alam, M.M.; Rana, M.S.; Mujtaba, G.; Arshad, Y.; Fatmi, M.Q.; Zaidi, S.Z. High prevalence of G3 rotavirus in hospitalized children in Rawalpindi, Pakistan during 2014. PLoS ONE 2018, 13, e0195947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, S.F.S.; Soares, L.S.; Lobo, P.S.; Júnior, E.T.P.; Júnior, E.C.S.; Bezerra, D.A.M.; Vaz, L.R.; Linhares, A.C.; Mascarenhas, J.D.P. Detection of a novel equine-like G3 rotavirus associated with acute gastroenteritis in Brazil. J. Gen. Virol. 2016, 97, 3131–3138. [Google Scholar] [CrossRef] [PubMed]
- Mhango, C.; Mandolo, J.J.; Chinyama, E.; Wachepa, R.; Kanjerwa, O.; Malamba-Banda, C.; Matambo, P.B.; Barnes, K.G.; Chaguza, C.; Shawa, I.T.; et al. Rotavirus Genotypes in Hospitalized Children with Acute Gastroenteritis Before and After Rotavirus Vaccine Introduction in Blantyre, Malawi, 1997–2019. J. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Mokomane, M.; Esona, M.; Bowen, M.D.; Tate, J.; Steenhoff, A.; Lechiile, K.; Gaseitsiwe, S.; Seheri, L.; Magagula, N.; Weldegebriel, G.; et al. Diversity of Rotavirus Strains Circulating in Botswana before and after introduction of the Monovalent Rotavirus Vaccine. Vaccine 2019, 37, 6324–6328. [Google Scholar] [CrossRef]
- Abebe, A.; Getahun, M.; Mapaseka, S.L.; Beyene, B.; Assefa, E.; Teshome, B.; Tefera, M.; Kebede, F.; Habtamu, A.; Haile-Mariam, T.; et al. Impact of rotavirus vaccine introduction and genotypic characteristics of rotavirus strains in children less than 5 years of age with gastroenteritis in Ethiopia: 2011–2016. Vaccine 2018, 36, 7043–7047. [Google Scholar] [CrossRef]
- Wandera, E.A.; Komoto, S.; Mohammad, S.; Ide, T.; Bundi, M.; Nyangao, J.; Kathiiko, C.; Odoyo, E.; Galata, A.; Miring’U, G.; et al. Genomic characterization of uncommon human G3P[6] rotavirus strains that have emerged in Kenya after rotavirus vaccine introduction, and pre-vaccine human G8P[4] rotavirus strains. Infect. Genet. Evol. 2019, 68, 231–248. [Google Scholar] [CrossRef]
- Gelaw, A.; Pietsch, C.; Liebert, U.G. Molecular epidemiology of rotaviruses in Northwest Ethiopia after national vaccine introduction. Infect. Genet. Evol. 2018, 65, 300–307. [Google Scholar] [CrossRef]
- Bwogi, J.; Jere, K.C.; Karamagi, C.; Byarugaba, D.K.; Namuwulya, P.; Baliraine, F.N.; Desselberger, U.; Iturriza-Gomara, M. Whole genome analysis of selected human and animal rotaviruses identified in Uganda from 2012 to 2014 reveals complex genome reassortment events between human, bovine, caprine and porcine strains. PLoS ONE 2017, 12, e0178855. [Google Scholar] [CrossRef] [Green Version]
- Jere, K.C.; Chaguza, C.; Bar-Zeev, N.; Lowe, J.; Peno, C.; Kumwenda, B.; Nakagomi, O.; Tate, J.E.; Parashar, U.D.; Heyderman, R.S.; et al. Emergence of Double- and Triple-Gene Reassortant G1P[8] Rotaviruses Possessing a DS-1-Like Backbone after Rotavirus Vaccine Introduction in Malawi. J. Virol. 2017, 92, 92. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.A.G.; Bauni, E.; Moisi, J.C.; Ojal, J.; Gatakaa, H.; Nyundo, C.; Molyneux, C.S.; Kombe, F.; Tsofa, B.; Marsh, K.; et al. Profile: The Kilifi Health and Demographic Surveillance System (KHDSS). Int. J. Epidemiol. 2012, 41, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Otieno, G.P.; Bottomley, C.; Khagayi, S.; Adetifa, I.; Ngama, M.; Omore, R.; Ogwel, B.; Owor, B.E.; Bigogo, G.; Ochieng, J.B.; et al. Impact of the Introduction of Rotavirus Vaccine on Hospital Admissions for Diarrhea Among Children in Kenya: A Controlled Interrupted Time-Series Analysis. Clin. Infect. Dis. 2019, 70, 2306–2313. [Google Scholar] [CrossRef] [PubMed]
- Adetifa, I.M.; Bwanaali, T.; Wafula, J.; Mutuku, A.; Karia, B.; Makumi, A.; Mwatsuma, P.; Bauni, E.; Hammitt, L.L.; Nokes, D.J.; et al. Cohort Profile: The Kilifi Vaccine Monitoring Study. Int. J. Epidemiol. 2016, 46, 792–792h. [Google Scholar] [CrossRef]
- Gómara, M.I.; Cubitt, D.; Desselberger, U.; Gray, J. Amino Acid Substitution within the VP7 Protein of G2 Rotavirus Strains Associated with Failure To Serotype. J. Clin. Microbiol. 2001, 39, 3796–3798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmonds, M.K.; Armah, G.; Asmah, R.; Banerjee, I.; Damanka, S.; Esona, M.; Gentsch, J.R.; Gray, J.J.; Kirkwood, C.; Page, N.; et al. New oligonucleotide primers for P-typing of rotavirus strains: Strategies for typing previously untypeable strains. J. Clin. Virol. 2008, 42, 368–373. [Google Scholar] [CrossRef]
- Pickett, B.E.; Greer, D.; Zhang, Y.; Stewart, L.; Zhou, L.; Sun, G.; Gu, Z.; Kumar, S.; Zaremba, S.; Larsen, C.N.; et al. Virus Pathogen Database and Analysis Resource (ViPR): A Comprehensive Bioinformatics Database and Analysis Resource for the Coronavirus Research Community. Viruses 2012, 4, 3209–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Stamatakis, A. Using RAxML to Infer Phylogenies. Curr. Protoc. Bioinform. 2015, 51, 6–14. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Taylor, N.E.; Greene, E.A. PARSESNP: A tool for the analysis of nucleotide polymorphisms. Nucleic Acids Res. 2003, 31, 3808–3811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | All (%) | Sampled (%) | Unsampled (%) | p Value $ | RVA + ve (%) | RVA − ve (%) | p Value * |
|---|---|---|---|---|---|---|---|
| Number of patients | 384 | 248 (64.6) | 136 (35.4) | 55 (22.2) | 193 (77.8) | ||
| Sex | 0.728 | 0.008 | |||||
| Male | 210 (54.7) | 134 (54.0) | 76 (55.9) | 21 (38.2) | 113 (58.6) | ||
| Female | 174 (45.3) | 114 (46.0) | 60 (44.1) | 34 (61.8) | 80 (41.5) | ||
| Age | |||||||
| Mean (SD ¶) | 27.4 (29.9) | 26.4 (31.8) | 29.3 (26.1) | 0.352 | 19.6 (15.0) | 28.3 (35.0) | 0.073 |
| Median (IQR δ) | 16.8 (9.8–29.3) | 15.1(9.4–24.1) | 19.9 (12.0–39.0) | 0.025 | 15.4 (9.9–20.8) | 15.1(8.9–24.9) | 1.000 |
| Age group | 0.002 | 0.254 | |||||
| 0–11 months | 126 (32.8) | 92 (37.1) | 34 (25.0) | 19 (34.6) | 73 (37.8) | ||
| 12–23 months | 136 (35.4) | 92 (37.1) | 44 (32.4) | 25 (45.5) | 67 (34.7) | ||
| 24–59 months | 73 (19.0) | 34 (13.7) | 39 (28.7) | 8 (14.6) | 26 (14.0) | ||
| >60 months | 49 (12.8) | 30 (12.1) | 19 (14.0) | 3 (5.5) | 27 (14.0) | ||
| RVA vaccine eligibility | 0.327 | 0.063 | |||||
| Age eligible 2 dose | 317 (82.6) | 204 (82.3) | 113 (83.1) | 51 (92.7) | 153 (79.3) | ||
| Age eligible 1 dose | 4 (1.0) | 4 (1.6) | 0 (0.0) | 0 (0.0) | 4 (2.1) | ||
| Age ineligible | 63 (16.4) | 40 (16.1) | 23 (16.9) | 4 (7.3) | 36 (18.7) | ||
| Vaccination status (n = 321) | 0.273 | 0.209 | |||||
| Two dose eligible & received 2 doses | 165 (51.4) | 111 (53.4) | 54 (47.8) | 29 (56.9) | 82 (52.2) | ||
| Two dose eligible & received 1 dose | 24 (7.5) | 17 (8.2) | 7 (6.2) | 7 (13.7) | 10 (6.2) | ||
| One or 2 dose eligible but received none | 6 (1.8) | 2 (1.0) | 4 (3.5) | 0 (0.0) | 2 (1.3) | ||
| One or 2 dose eligible but status unknown | 126 (39.3) | 78 (37.5) | 48 (42.5) | 15 (29.4) | 63 (40.1) | ||
| Outcome (n = 379) | <0.001 | 0.194 | |||||
| Died | 38 (10.0) | 13 (5.3) | 25 (18.9) | 12 (6.3) | |||
| Alive | 341 (90.0) | 234 (94.7) | 133 (81.1) | 180 (93.8) |
| Characteristic | Genotyped RVA (%) | G3P[8] (%) | Non-G3P[8] (%) | p Value |
|---|---|---|---|---|
| Number of patients | 48 | 37 (77.1) | 11 (22.9) | |
| Sex | 0.248 | |||
| Male | 19 (39.6) | 13 (35.1) | 6 (55.6) | |
| Female | 29 (60.4) | 24 (64.9) | 5 (45.5) | |
| Age | ||||
| Mean (SD #) | 19.3 (14.8) | 19.2 (13.3) | 19.5 (18.6) | 0.946 |
| Median (IQR δ) | 15.7 (9.9–20.4) | 15.9 (9.8–20.4) | 15.4 (7.8–23.1) | 1.000 |
| Age group | 0.770 | |||
| 0–11 months | 17 (35.4) | 13 (35.1) | 4 (36.4) | |
| 12–23 months | 22 (45.8) | 17 (46.0) | 5 (45.6) | |
| 24–59 months | 7 (14.6) | 6 (16.2) | 1 (9.1) | |
| >60 months | 2 (4.2) | 1 (2.7) | 1 (9.1) | |
| RVA vaccine eligibility | 0.658 | |||
| Age eligible 2 dose | 45 (93.8) | 35 (94.6) | 10 (90.9) | |
| Age eligible 1 dose | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Age ineligible | 3 (6.3) | 2 (5.4) | 1 (9.1) | |
| RVA vaccination status among eligible (n = 45) | 0.751 | |||
| Two dose eligible & received two doses | 27 (60.0) | 20 (57.1) | 7 (70.0) | |
| Two dose eligible & received one dose | 7 (15.6) | 6 (17.1) | 1 (10.0) | |
| One or 2 dose eligible but received none | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| One or 2 dose eligible but status unknown | 11 (24.4) | 9 (25.7) | 2 (20.0) | |
| Outcome | 0.064 | |||
| Died | 1 (2.1) | 0 (0.0) | 1 (9.1) | |
| Alive | 47 (97.2) | 37 (100.0) | 10 (90.9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mwanga, M.J.; Verani, J.R.; Omore, R.; Tate, J.E.; Parashar, U.D.; Murunga, N.; Gicheru, E.; Breiman, R.F.; Nokes, D.J.; Agoti, C.N. Multiple Introductions and Predominance of Rotavirus Group A Genotype G3P[8] in Kilifi, Coastal Kenya, 4 Years after Nationwide Vaccine Introduction. Pathogens 2020, 9, 981. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9120981
Mwanga MJ, Verani JR, Omore R, Tate JE, Parashar UD, Murunga N, Gicheru E, Breiman RF, Nokes DJ, Agoti CN. Multiple Introductions and Predominance of Rotavirus Group A Genotype G3P[8] in Kilifi, Coastal Kenya, 4 Years after Nationwide Vaccine Introduction. Pathogens. 2020; 9(12):981. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9120981
Chicago/Turabian StyleMwanga, Mike J., Jennifer R. Verani, Richard Omore, Jacqueline E. Tate, Umesh D. Parashar, Nickson Murunga, Elijah Gicheru, Robert F. Breiman, D. James Nokes, and Charles N. Agoti. 2020. "Multiple Introductions and Predominance of Rotavirus Group A Genotype G3P[8] in Kilifi, Coastal Kenya, 4 Years after Nationwide Vaccine Introduction" Pathogens 9, no. 12: 981. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9120981