Spatial and Temporal Evolutionary Patterns in Puumala Orthohantavirus (PUUV) S Segment

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
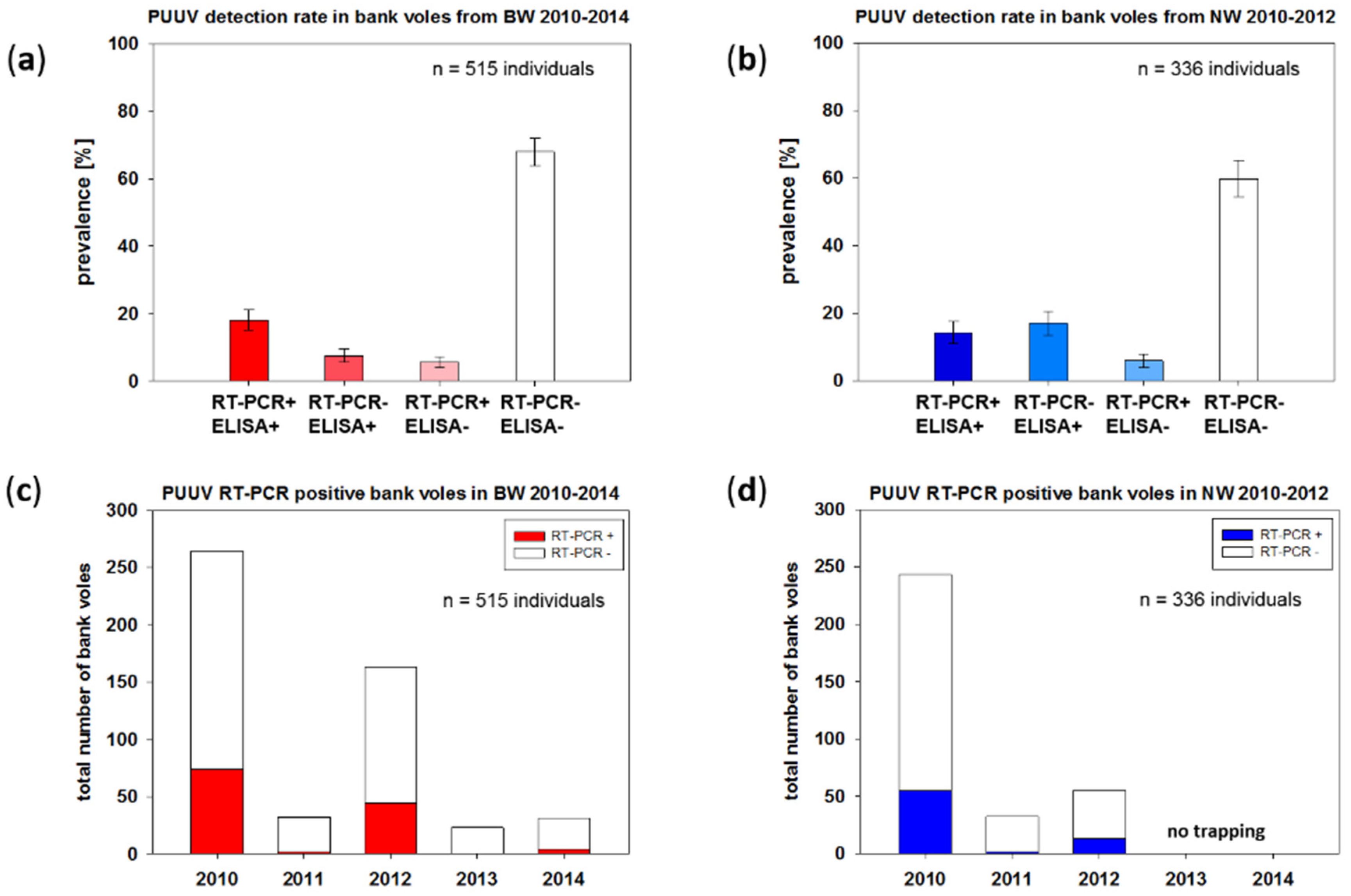
2.1. Temporal Fluctuation of PUUV Prevalence in Bank Voles from Baden-Wuerttemberg and North Rhine-Westphalia
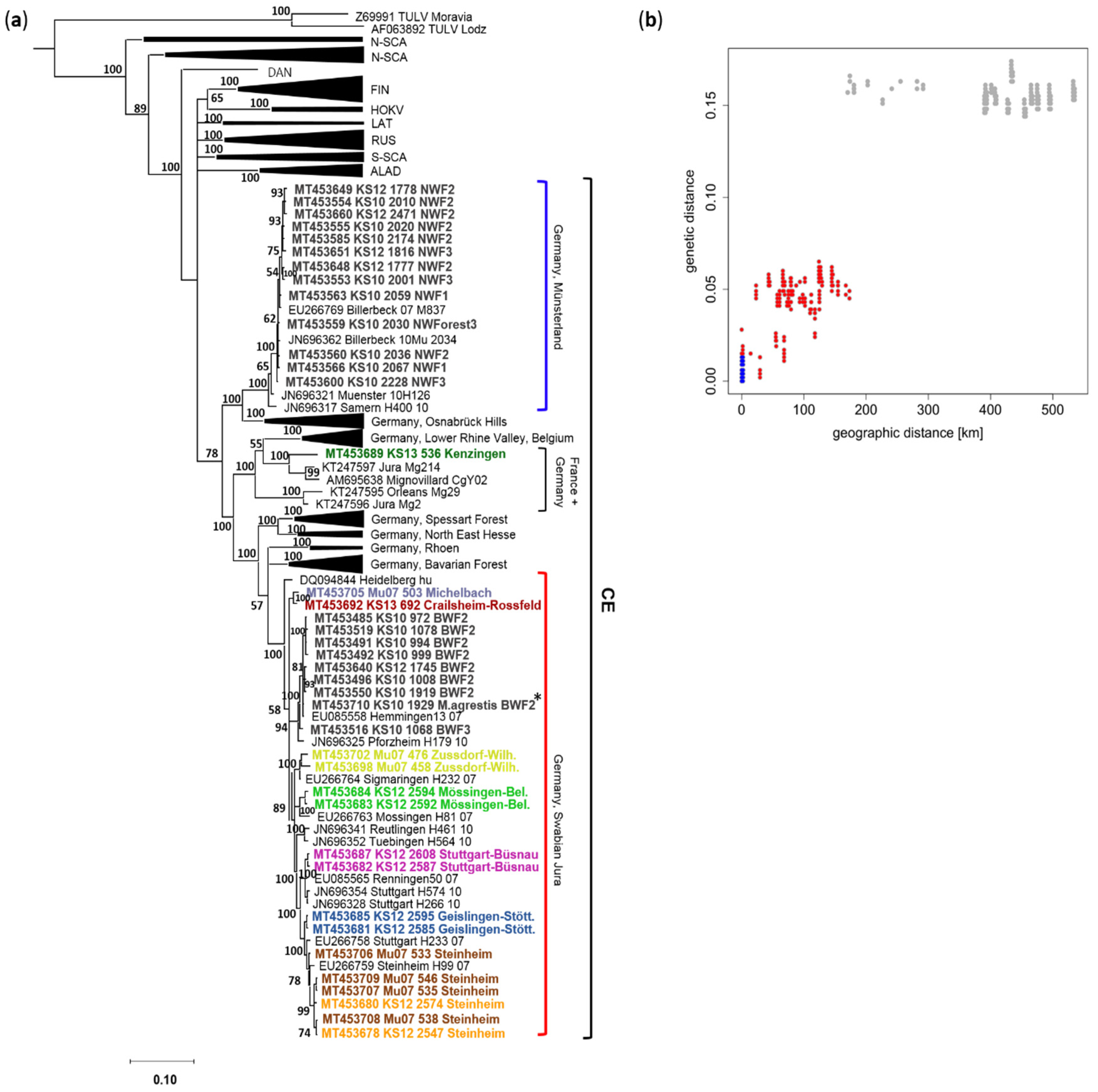
2.2. Phylogenetic and Isolation-by-Distance Analysis of Non-NSs-Overlapping S Segment Sequences
2.3. Frequency of Spillover Infections
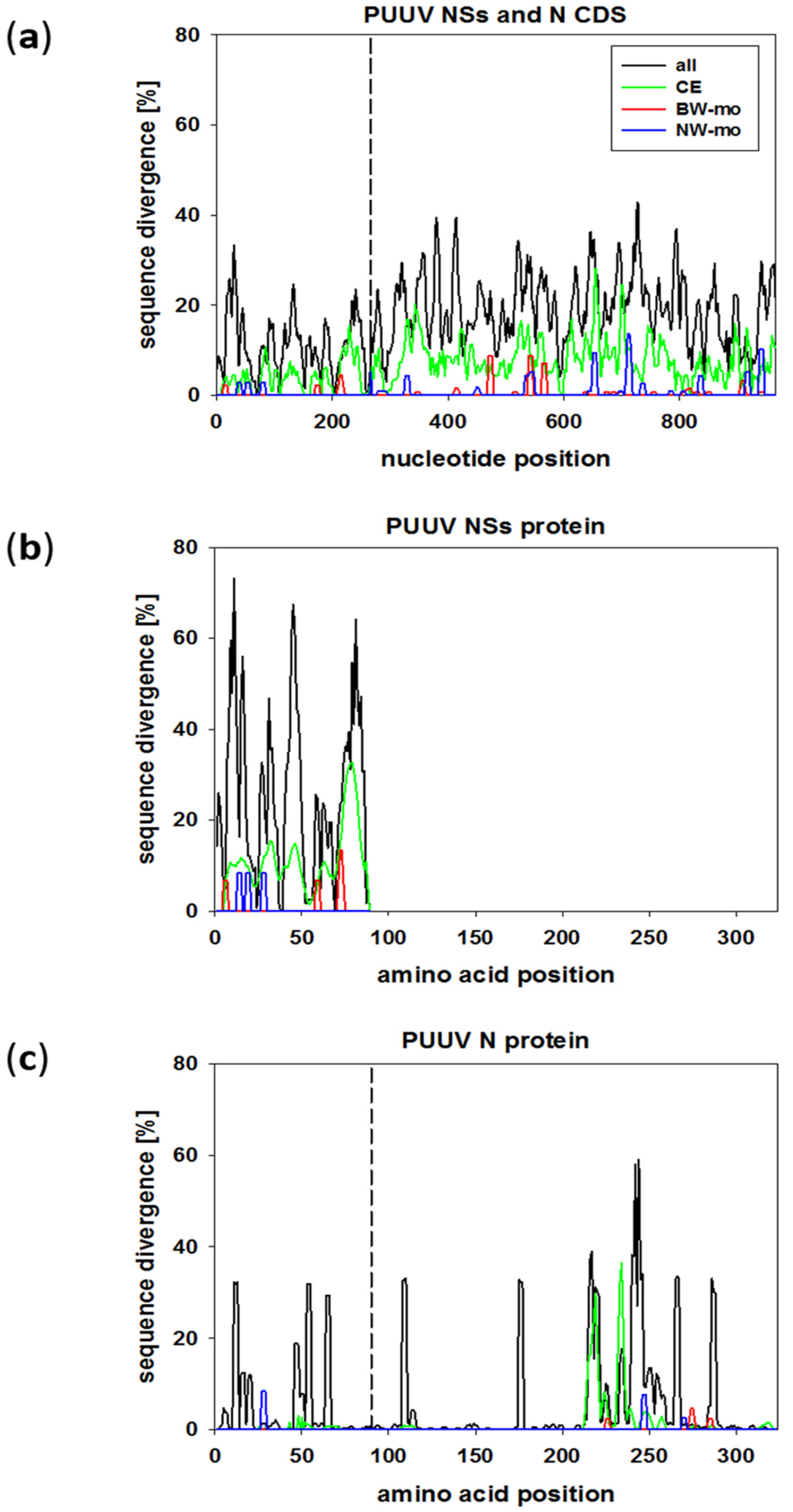
2.4. Sequence Variability in the N/NSs Overlapping and N Encoding Regions
2.5. Spatial and Temporal Evolution of Different Regions of the PUUV S Segment
3. Discussion
4. Materials and Methods
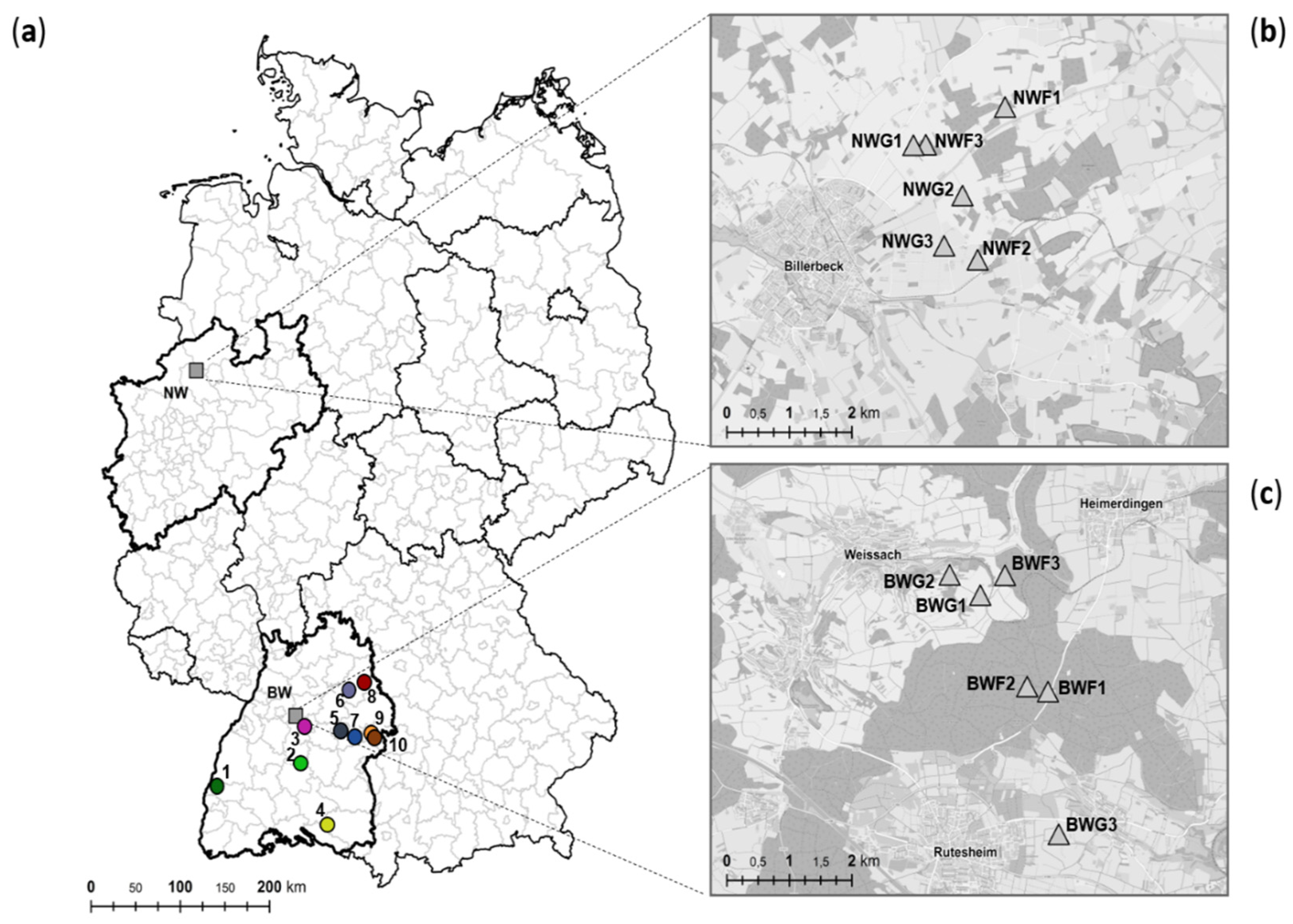
4.1. Study Animals
4.2. Serology
4.3. Detection of Hantavirus RNA
4.4. Sequence, Phylogenetic and Statistical Analysis
4.5. Ethical Statement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vaheri, A.; Strandin, T.; Hepojoki, J.; Sironen, T.; Henttonen, H.; Mäkelä, S.; Mustonen, J. Uncovering the mysteries of hantavirus infections. Nat. Rev. Microbiol. 2013, 11, 539–550. [Google Scholar] [CrossRef]
- Reuter, M.; Krüger, D.H. The nucleocapsid protein of hantaviruses: Much more than a genome-wrapping protein. Virus Genes 2017, 54, 5–16. [Google Scholar] [CrossRef]
- Jääskelainen, K.M.; Kaukinen, P.; Minskaya, E.S.; Plyusnina, A.; Vapalahti, O.; Elliott, R.M.; Weber, F.; Vaheri, A.; Plyusnin, A. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 2007, 79, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.S.; Drewes, S.; Weber de Melo, V.; Schlegel, M.; Freise, J.; Groschup, M.H.; Heckel, G.; Ulrich, R.G. Complete genome of a Puumala virus strain from Central Europe. Virus Genes 2015, 50, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Binder, F.; Reiche, S.; Roman-Sosa, G.; Saathoff, M.; Ryll, R.; Trimpert, J.; Kunec, D.; Hoper, D.; Ulrich, R.G. Isolation and characterization of new Puumala orthohantavirus strains from Germany. Virus Genes 2020. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.J.; Schmaljohn, C.S. Persistent hantavirus infections: Characteristics and mechanisms. Trends Microbiol. 2000, 8, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Kallio, E.R.; Voutilainen, L.; Vapalahti, O.; Vaheri, A.; Henttonen, H.; Koskela, E.; Mappes, T. Endemic hantavirus infection impairs the winter survival of its rodent host. Ecology 2007, 88, 1911–1916. [Google Scholar] [CrossRef] [Green Version]
- Vapalahti, O.; Mustonen, J.; Lundkvist, A.; Henttonen, H.; Plyusnin, A.; Vaheri, A. Hantavirus Infections in Europe. Lancet Infect. Dis. 2003, 3, 653–661. [Google Scholar] [CrossRef]
- Faber, M.; Krüger, D.H.; Auste, B.; Stark, K.; Hofmann, J.; Weiss, S. Molecular and epidemiological characteristics of human Puumala and Dobrava-Belgrade hantavirus infections, Germany, 2001 to 2017. Euro Surveill. 2019, 24, 32. [Google Scholar] [CrossRef]
- Schlegel, M.; Jacob, J.; Krüger, D.H.; Rang, A.; Ulrich, R.G. Hantavirus Emergence in Rodents, Insectivores and Bats: What Comes Next. In The Role of Animals in Emerging Viral Diseases; Johnson, N., Ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 235–292. [Google Scholar]
- Reil, D.; Imholt, C.; Eccard, J.A.; Jacob, J. Beech Fructification and Bank Vole Population Dynamics—Combined Analyses of Promoters of Human Puumala Virus Infections in Germany. PLoS ONE 2015, 10, e0134124. [Google Scholar] [CrossRef]
- Vanwambeke, S.O.; Zeimes, C.B.; Drewes, S.; Ulrich, R.G.; Reil, D.; Jacob, J. Spatial dynamics of a zoonotic orthohantavirus disease through heterogenous data on rodents, rodent infections, and human disease. Sci. Rep. 2019, 9, 2329. [Google Scholar] [CrossRef] [PubMed]
- Reil, D.; Rosenfeld, U.M.; Imholt, C.; Schmidt, S.; Ulrich, R.G.; Eccard, J.A.; Jacob, J. Puumala hantavirus infections in bank vole populations: Host and virus dynamics in Central Europe. BMC Ecol. 2017, 17, 9. [Google Scholar] [CrossRef] [Green Version]
- Kallio, E.R.; Begon, M.; Henttonen, H.; Koskela, E.; Mappes, T.; Vaheri, A.; Vapalahti, O. Hantavirus infections in fluctuating host populations: The role of maternal antibodies. Proc. R. Soc. B Biol. Sci. 2010, 277, 3783–3791. [Google Scholar] [CrossRef]
- Robert-Koch-Institut. Hantavirus Infektionen. Available online: https://www.rki.de/DE/Content/InfAZ/H/Hantavirus/Hantavirus.html (accessed on 15 April 2020).
- Drewes, S.; Turni, H.; Rosenfeld, U.M.; Obiegala, A.; Strakova, P.; Imholt, C.; Glatthaar, E.; Dressel, K.; Pfeffer, M.; Jacob, J.; et al. Reservoir-Driven Heterogeneous Distribution of Recorded Human Puumala virus Cases in South-West Germany. Zoonoses Public Health 2017, 64, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Drewes, S.; Ali, H.S.; Saxenhofer, M.; Rosenfeld, U.M.; Binder, F.; Cuypers, F.; Schlegel, M.; Rohrs, S.; Heckel, G.; Ulrich, R.G. Host-Associated Absence of Human Puumala Virus Infections in Northern and Eastern Germany. Emerg. Infect. Dis. 2017, 23, 83–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettinger, J.; Hofmann, J.; Enders, M.; Tewald, F.; Oehme, R.M.; Rosenfeld, U.M.; Ali, H.S.; Schlegel, M.; Essbauer, S.; Osterberg, A.; et al. Multiple synchronous outbreaks of Puumala virus, Germany, 2010. Emerg. Infect. Dis. 2012, 18, 1461–1464. [Google Scholar] [CrossRef]
- Saxenhofer, M.; Weber de Melo, V.; Ulrich, R.G.; Heckel, G. Revised time scales of RNA virus evolution based on spatial information. Proc. R. Soc. B Biol. Sci. 2017, 284. [Google Scholar] [CrossRef] [Green Version]
- Weber de Melo, V.; Sheikh Ali, H.; Freise, J.; Kuhnert, D.; Essbauer, S.; Mertens, M.; Wanka, K.M.; Drewes, S.; Ulrich, R.G.; Heckel, G. Spatiotemporal dynamics of Puumala hantavirus associated with its rodent host. Myodes Glareolus. Evol. Appl. 2015, 8, 545–559. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Schwartz, T.; Pickett, B.E.; He, S.; Klem, E.B.; Scheuermann, R.H.; Passarotti, M.; Kaufman, S.; O’Leary, M.A. A RESTful API for Access to Phylogenetic Tools via the CIPRES Science Gateway. Evol. Bioinform. Online 2015, 11, 43–48. [Google Scholar] [CrossRef]
- Essbauer, S.; Schmidt, J.; Conraths, F.J.; Friedrich, R.; Koch, J.; Hautmann, W.; Pfeffer, M.; Wolfel, R.; Finke, J.; Dobler, G.; et al. A new Puumala hantavirus subtype in rodents associated with an outbreak of Nephropathia epidemica in South-East Germany in 2004. Epidemiol. Infect. 2006, 134, 1333–1344. [Google Scholar] [CrossRef]
- Faber, M.; Wollny, T.; Schlegel, M.; Wanka, K.M.; Thiel, J.; Frank, C.; Rimek, D.; Ulrich, R.G.; Stark, K. Puumala virus outbreak in Western Thuringia, Germany, 2010: Epidemiology and strain identification. Zoonoses Public Health 2013, 60, 549–554. [Google Scholar] [CrossRef]
- Voutilainen, L.; Sironen, T.; Tonteri, E.; Back, A.T.; Razzauti, M.; Karlsson, M.; Wahlstrom, M.; Niemimaa, J.; Henttonen, H.; Lundkvist, A. Life-long shedding of Puumala hantavirus in wild bank voles (Myodes glareolus). J. Gen. Virol. 2015, 96, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Klempa, B.; Tenner, B.; Kruger, D.H.; Hofmann, J. Prediction of the Spatial Origin of Puumala Virus Infections Using L Segment Sequences Derived from a Generic Screening PCR. Viruses 2019, 11, 694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyman, P.; Ceianu, S.; Christova, I.; Tordo, N.; Vaheri, A. A five-year perspective on the situation of haemorrhagic fever with renal syndrome and status of the hantavirus reservoirs in Europe, 2005–2010. Euro Surveill. 2011, 16, 19961. [Google Scholar] [CrossRef] [Green Version]
- Krakauer, D.C. Stability and evolution of overlapping genes. Evol. Int. J. Org. Evol. 2000, 54, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Laenen, L.; Vergote, V.; Vanmechelen, B.; Tersago, K.; Baele, G.; Lemey, P.; Leirs, H.; Dellicour, S.; Vrancken, B.; Maes, P. Identifying the patterns and drivers of Puumala hantavirus enzootic dynamics using reservoir sampling. Virus Evol. 2019, 5, vez009. [Google Scholar] [CrossRef] [Green Version]
- Plyusnin, A.; Vapalahti, O.; Lankinen, H.; Lehväslaiho, H.; Apekina, N.; Myasnikov, Y.; Kallio-Kokko, H.; Henttonen, H.; Lundkvist, A.; Brummer-Korvenkontio, M.; et al. Tula Virus: A Newly Detected Hantavirus Carried by European Common Voles. J. Virol. 1994, 68, 7833–7839. [Google Scholar] [CrossRef] [Green Version]
- Sibold, C.; Sparr, S.; Schulz, A.; Labuda, M.; Kozuch, O.; Lysy, J.; Kruger, D.H.; Meisel, H. Genetic characterization of a new hantavirus detected in Microtus arvalis from Slovakia. Virus Genes 1995, 10, 277–281. [Google Scholar] [CrossRef]
- Schmidt-Chanasit, J.; Essbauer, S.; Petraityte, R.; Yoshimatsu, K.; Tackmann, K.; Conraths, F.J.; Sasnauskas, K.; Arikawa, J.; Thomas, A.; Pfeffer, M.; et al. Extensive host sharing of central European Tula virus. J. Virol. 2010, 84, 459–474. [Google Scholar] [CrossRef] [Green Version]
- Scharninghausen, J.J.; Pfeffer, M.; Meyer, H.; Davis, D.S.; Honeycutt, R.L.; Faulde, M. Genetic evidence for Tula virus in Microtus arvalis and Microtus agrestis populations in Croatia. Vector Borne Zoonotic Dis. (Larchmt. N. Y.) 2002, 2, 19–27. [Google Scholar] [CrossRef]
- Schlegel, M.; Kindler, E.; Essbauer, S.S.; Wolf, R.; Thiel, J.; Groschup, M.H.; Heckel, G.; Oehme, R.M.; Ulrich, R.G. Tula virus infections in the Eurasian water vole in Central Europe. Vector Borne Zoonotic Dis. (Larchmt. N. Y.) 2012, 12, 503–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, F.; Lenk, M.; Weber, S.; Stoek, F.; Dill, V.; Reiche, S.; Riebe, R.; Wernike, K.; Hoffmann, D.; Ziegler, U.; et al. Common vole (Microtus arvalis) and bank vole (Myodes glareolus) derived permanent cell lines differ in their susceptibility and replication kinetics of animal and zoonotic viruses. J. Virol. Methods 2019, 113729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingström, J.; Heyman, P.; Escutenaire, S.; Sjolander, K.B.; De Jaegere, F.; Henttonen, H.; Lundkvist, A. Rodent host specificity of European hantaviruses: Evidence of Puumala virus interspecific spillover. J. Med. Virol. 2002, 68, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Essbauer, S.S.; Schmidt-Chanasit, J.; Madeja, E.L.; Wegener, W.; Friedrich, R.; Petraityte, R.; Sasnauskas, K.; Jacob, J.; Koch, J.; Dobler, G.; et al. Nephropathia epidemica in metropolitan area, Germany. Emerg. Infect. Dis. 2007, 13, 1271–1273. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C.; Zhang, Y.Z. The evolution and emergence of hantaviruses. Curr. Opin. Virol. 2015, 10, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Vera-Otarola, J.; Solis, L.; Soto-Rifo, R.; Ricci, E.P.; Pino, K.; Tischler, N.D.; Ohlmann, T.; Darlix, J.L.; Lopez-Lastra, M. The Andes hantavirus NSs protein is expressed from the viral small mRNA by a leaky scanning mechanism. J. Virol. 2012, 86, 2176–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severson, W.; Xu, X.; Kuhn, M.; Senutovitch, N.; Thokala, M.; Ferron, F.; Longhi, S.; Canard, B.; Jonsson, C.B. Essential amino acids of the Hantaan virus N protein in its interaction with RNA. J. Virol. 2005, 79, 10032–10039. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Severson, W.; Villegas, N.; Schmaljohn, C.S.; Jonsson, C.B. The RNA binding domain of the Hantaan virus N protein maps to a central, conserved region. J. Virol. 2002, 76, 3301–3308. [Google Scholar] [CrossRef] [Green Version]
- Cheng, E.; Wang, Z.; Mir, M.A. Interaction between hantavirus nucleocapsid protein (N) and RNA-dependent RNA polymerase (RdRp) mutants reveals the requirement of an N-RdRp interaction for viral RNA synthesis. J. Virol. 2014, 88, 8706–8712. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Yoshimatsu, K.; Koma, T.; Yasuda, S.P.; Arikawa, J. Role of nucleocapsid protein of hantaviruses in intracellular traffic of viral glycoproteins. Virus Res. 2013, 178, 349–356. [Google Scholar] [CrossRef]
- Plyusnin, A. Genetics of hantaviruses: Implications to taxonomy. Arch. Virol. 2002, 147, 665–682. [Google Scholar] [CrossRef]
- Plyusnin, A.; Cheng, Y.; Lehvaslaiho, H.; Vaheri, A. Quasispecies in wild-type Tula hantavirus populations. J. Virol. 1996, 70, 9060–9063. [Google Scholar] [CrossRef] [Green Version]
- Mertens, M.; Kindler, E.; Emmerich, P.; Esser, J.; Wagner-Wiening, C.; Wolfel, R.; Petraityte-Burneikiene, R.; Schmidt-Chanasit, J.; Zvirbliene, A.; Groschup, M.H.; et al. Phylogenetic analysis of Puumala virus subtype Bavaria, characterization and diagnostic use of its recombinant nucleocapsid protein. Virus Genes 2011, 43, 177–191. [Google Scholar] [CrossRef]
- Kucinskaite-Kodze, I.; Petraityte-Burneikiene, R.; Zvirbliene, A.; Hjelle, B.; Medina, R.A.; Gedvilaite, A.; Razanskiene, A.; Schmidt-Chanasit, J.; Mertens, M.; Padula, P.; et al. Characterization of monoclonal antibodies against hantavirus nucleocapsid protein and their use for immunohistochemistry on rodent and human samples. Arch. Virol. 2011, 156, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Ersts, P.J. Geographic Distance Matrix Generator (Version 1.2.3). Available online: http://biodiversityinformatics.amnh.org/open_source/gdmg (accessed on 26 March 2020).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Binder, F.; Ryll, R.; Drewes, S.; Jagdmann, S.; Reil, D.; Hiltbrunner, M.; Rosenfeld, U.M.; Imholt, C.; Jacob, J.; Heckel, G.; et al. Spatial and Temporal Evolutionary Patterns in Puumala Orthohantavirus (PUUV) S Segment. Pathogens 2020, 9, 548. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9070548
Binder F, Ryll R, Drewes S, Jagdmann S, Reil D, Hiltbrunner M, Rosenfeld UM, Imholt C, Jacob J, Heckel G, et al. Spatial and Temporal Evolutionary Patterns in Puumala Orthohantavirus (PUUV) S Segment. Pathogens. 2020; 9(7):548. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9070548
Chicago/Turabian StyleBinder, Florian, René Ryll, Stephan Drewes, Sandra Jagdmann, Daniela Reil, Melanie Hiltbrunner, Ulrike M. Rosenfeld, Christian Imholt, Jens Jacob, Gerald Heckel, and et al. 2020. "Spatial and Temporal Evolutionary Patterns in Puumala Orthohantavirus (PUUV) S Segment" Pathogens 9, no. 7: 548. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9070548