Genetic Diversity Among SARS-CoV2 Strains in South America may Impact Performance of Molecular Detection

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
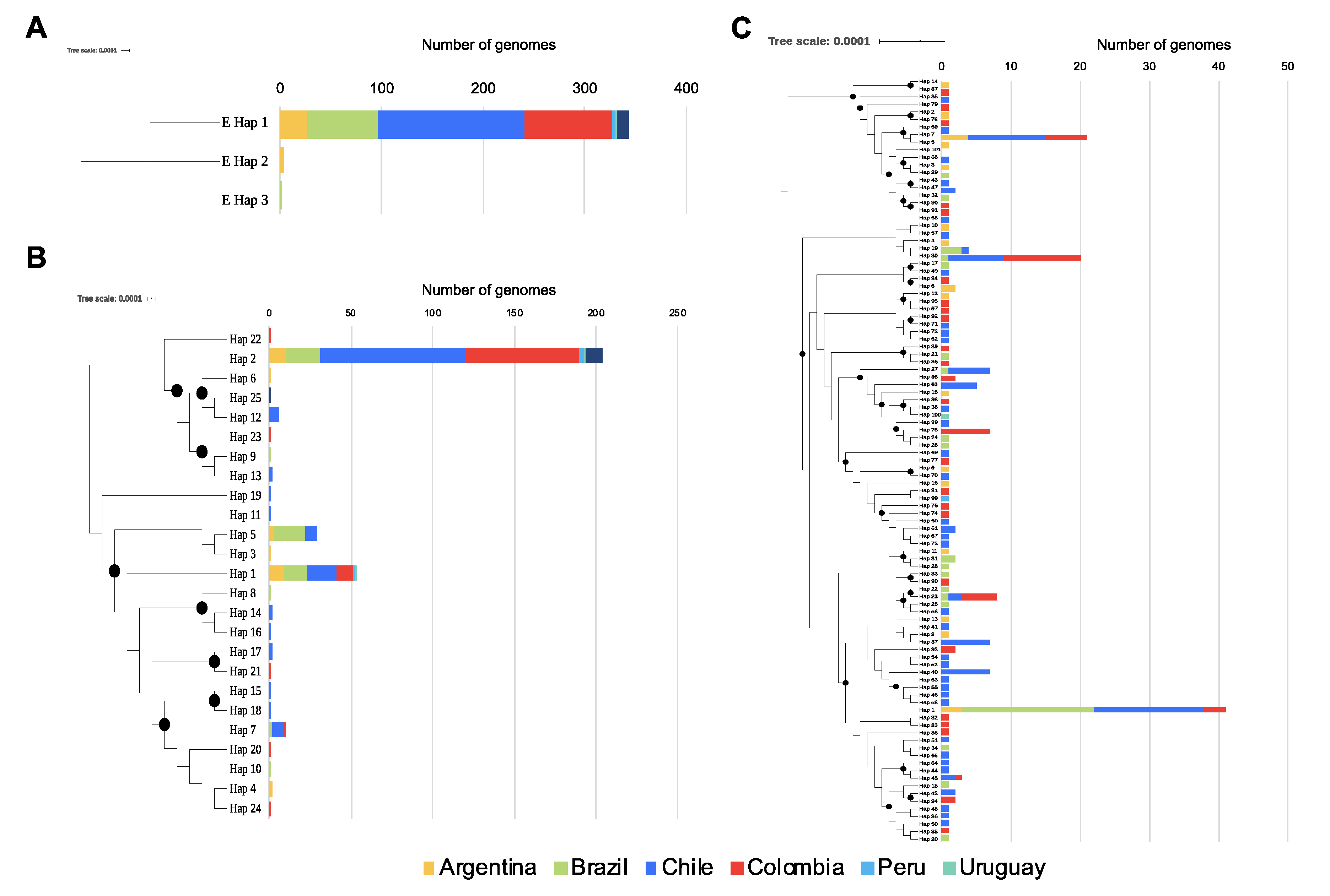
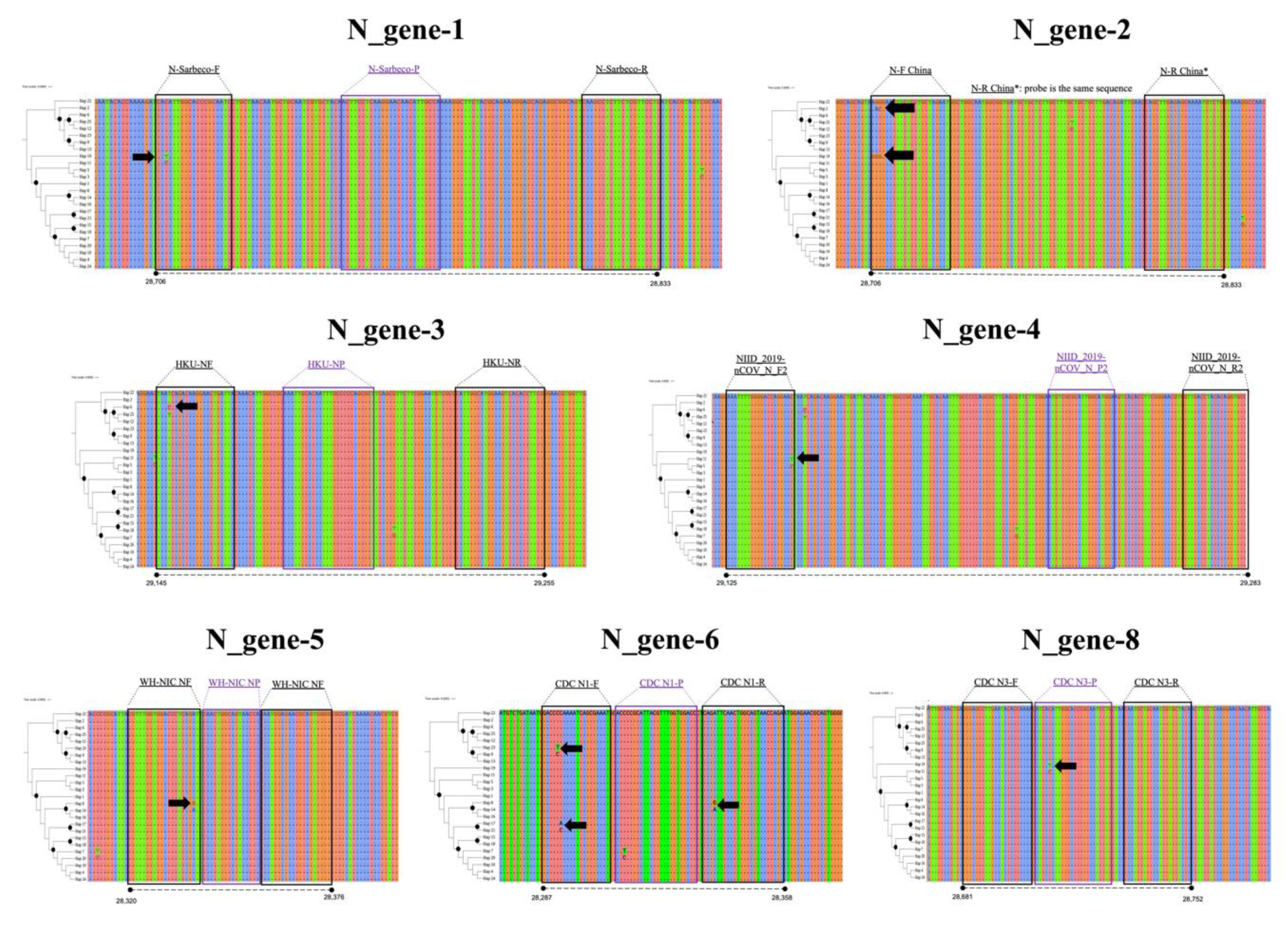
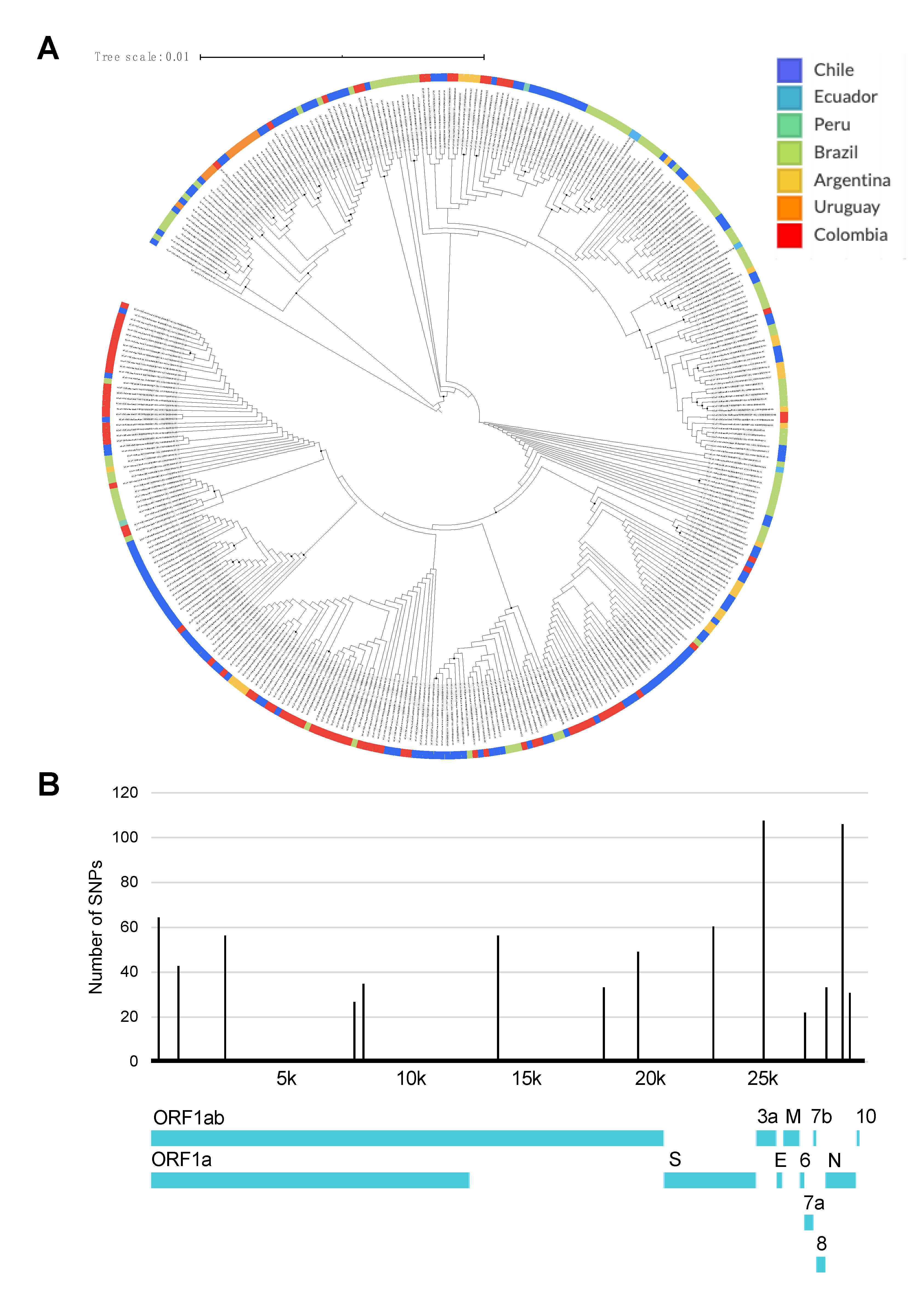
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.P.; Yuen, K.-Y. Coronavirus Genomics and Bioinformatics Analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luk, H.K.H.; Li, X.; Fung, J.; Lau, S.K.P.; Woo, P.C.Y. Molecular epidemiology, evolution and phylogeny of SARS coronavirus. Infect. Genet. Evol. 2019, 71, 21–30. [Google Scholar] [CrossRef] [PubMed]
- De Groot, R.J. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Ed.; Academic Press: London, UK; Waltham, MA, USA, 2012; ISBN 978-0-12-384684-6. [Google Scholar]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Novel Coronavirus (2019-nCoV); Situation Report-1; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Miller, M.J.; Loaiza, J.R.; Takyar, A.; Gilman, R.H. COVID-19 in Latin America: Novel transmission dynamics for a global pandemic? PLoS Negl. Trop. Dis. 2020, 14, e0008265. [Google Scholar] [CrossRef]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene Reports 2020, 19, 100682. [Google Scholar] [CrossRef]
- Sawicki, S.G. Coronavirus Genome Replication. In Viral Genome Replication; Raney, K.D., Gotte, M., Cameron, C.E., Eds.; Springer: Boston, MA, USA, 2009; pp. 25–39. ISBN 978-0-387-89425-6. [Google Scholar]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25. [Google Scholar] [CrossRef] [Green Version]
- Udugama, B.; Kadhiresan, P.; Kozlowski, H.N.; Malekjahani, A.; Osborne, M.; Li, V.Y.C.; Chen, H.; Mubareka, S.; Gubbay, J.B.; Chan, W.C.W. Diagnosing COVID-19: The Disease and Tools for Detection. ACS Nano 2020, 14, 3822–3835. [Google Scholar] [CrossRef] [Green Version]
- Poterico, J.A.; Mestanza, O. Genetic variants and source of introduction of SARS-CoV-2 in South America. J. Med. Virol. 2020, jmv.26001. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eur. Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.J.; Park, G.-S.; Moon, J.H.; Ku, K.; Beak, S.-H.; Kim, S.; Park, E.C.; Park, D.; Lee, J.-H.; Byeon, C.W.; et al. Comparative Analysis of Primer-Probe Sets for the Laboratory Confirmation of SARS-CoV-2. Microbiology: 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.02.25.964775v1 (accessed on 9 July 2020).
- Institut Pasteur Protocol. Real-time RT-PCR assays for the detection of SARS-CoV-2. Available online: https://www.who.int/docs/default-source/coronaviruse/real-time-rt-pcr-assays-for-the-detection-of-sars-cov-2-institut-pasteur-paris.pdf?sfvrsn=3662fcb6_2 (accessed on 9 July 2020).
- CDC Research Use Only 2019-Novel Coronavirus (2019-nCoV) Real-time RT-PCR Primers and Probes. Available online: https://www.cdc.gov/coronavirus/2019-ncov/lab/rt-pcr-panel-primer-probes.html (accessed on 9 July 2020).
- Taubenberger, J.K.; Morens, D.M. 1918 Influenza: The Mother of All Pandemics. Emerg. Infect. Dis. 2006, 12, 15–22. [Google Scholar] [CrossRef]
- Worldometer COVID-19 CORONAVIRUS PANDEMIC. Available online: https://www.worldometers.info/coronavirus/? (accessed on 9 June 2020).
- Burki, T. COVID-19 in Latin America. Lancet Infect. Dis. 2020, 20, 547–548. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Z.; Chen, Z.; Huang, X.; Xu, M.; He, T.; Zhang, Z. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J. Med. Virol. 2020, 92, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011, 8, 270–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagliani, R.; Forni, D.; Clerici, M.; Sironi, M. Computational Inference of Selection Underlying the Evolution of the Novel Coronavirus, Severe Acute Respiratory Syndrome Coronavirus 2. J. Virol. 2020, 94, e00411-20. [Google Scholar] [CrossRef] [Green Version]
- Nalla, A.K.; Casto, A.M.; Huang, M.-L.W.; Perchetti, G.A.; Sampoleo, R.; Shrestha, L.; Wei, Y.; Zhu, H.; Jerome, K.R.; Greninger, A.L. Comparative Performance of SARS-CoV-2 Detection Assays Using Seven Different Primer-Probe Sets and One Assay Kit. J. Clin. Microbiol. 2020, 58, e00557-20. [Google Scholar] [CrossRef] [Green Version]
- Artesi, M.; Bontems, S.; Gobbels, P.; Franckh, M.; Boreux, R.; Meex, C.; Melin, P.; Hayette, M.-P.; Bours, V.; Durkin, K. Failure of the cobas® SARS-CoV-2 (Roche) E-gene assay is associated with a C-to-T transition at position 26340 of the SARS-CoV-2 genome. MedRxiv 2020. [Google Scholar] [CrossRef]
- Pan American Health Organization. P. Laboratory Guidelines for the Detection and Diagnosis of COVID-19 Virus Infection, 30 March 2020; Pan American Health Organization: Washington, DC, USA, 2020. [Google Scholar]
- Astuti, I. Ysrafil Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020. [Google Scholar] [CrossRef]
- Drosten, C.; Chiu, L.-L.; Panning, M.; Leong, H.N.; Preiser, W.; Tam, J.S.; Gunther, S.; Kramme, S.; Emmerich, P.; Ng, W.L.; et al. Evaluation of Advanced Reverse Transcription-PCR Assays and an Alternative PCR Target Region for Detection of Severe Acute Respiratory Syndrome-Associated Coronavirus. J. Clin. Microbiol. 2004, 42, 2043–2047. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Pei, F.; Wang, L.; Zhao, H.; Li, H.; Ji, M.; Yang, W.; Wang, Q.; Zhao, Q.; Wang, Y. Sensitivity Evaluation of 2019 Novel Coronavirus (SARS-CoV-2) RT-PCR Detection Kits and Strategy to Reduce False Negative; Infectious Diseases (except HIV/AIDS), 2020. Available online: https://www.medrxiv.org/content/10.1101/2020.04.28.20083956v1 (accessed on 9 July 2020).
- Su, Y.C.; Anderson, D.E.; Young, B.E.; Zhu, F.; Linster, M.; Kalimuddin, S.; Low, J.G.; Yan, Z.; Jayakumar, J.; Sun, L.; et al. Discovery of a 382-nt Deletion During the Early Evolution of SARS-CoV-2. Microbiology. 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.03.11.987222v1 (accessed on 9 July 2020).
- Vogels, C.B.F.; Brito, A.F.; Wyllie, A.L.; Fauver, J.R.; Ott, I.M.; Kalinich, C.C.; Petrone, M.E.; Casanovas-Massana, A.; Muenker, M.C.; Moore, A.J.; et al. Analytical Sensitivity and Efficiency Comparisons of SARS-COV-2 qRT-PCR Primer-Probe Sets. Infectious Diseases (except HIV/AIDS). 2020. Available online: https://www.medrxiv.org/content/10.1101/2020.03.30.20048108v3 (accessed on 9 July 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Name | Sequences (5’-3’) | Reference | |
|---|---|---|---|---|
| 1 | RdRP_SARSr-F | GTGARATGGTCATGTGTGGCGG | [13] | |
| RdRp | RdRP_SARSr-R | CARATGTTAAASACACTATTAGCATA | ||
| RdRP_SARSr-P | FAM- CAGGTGGAACCTCATCAGGAGATGC-BHQ1 | |||
| 2 | ORF1ab-F (China) | CCCTGTGGGTTTTACACTTAA | [23] | |
| ORF1ab-R (China) | ACGATTGTGCATCAGCTGA | |||
| ORF1ab-Probe (China) | CCGTCTGCGGTATGTGGAAAGGTTATGG | |||
| 3 | HKU-ORF1b-nsp14F (Hong Kong) | TGGGGYTTTACRGGTAACCT | ||
| HKU-ORF1b-nsp14R Hong Kong | AACRCGCTTAACAAAGCACTC | |||
| HKU-ORF1b-nsp14P Hong Kong | TAGTTGTGATGCWATCATGACTAG | |||
| 4 | nCoV_IP2-12669Fw | ATGAGCTTAGTCCTGTTG | [24] | |
| nCoV_IP2-12759Rv | CTCCCTTTGTTGTGTTGT | |||
| nCoV_IP2-12696 Probe (+) | AGATGTCTTGTGCTGCCGGTA [5’]Hex [3’]BHQ-1 | |||
| 5 | nCoV_IP4-14059Fw | GGTAACTGGTATGATTTCG | ||
| nCoV_IP4-14146Rv | CTGGTCAAGGTTAATATAGG | |||
| nCoV_IP4-14084 Probe (+) | TCATACAAACCACGCCAGG [5’]Fam [3’]BHQ-1 | |||
| nCoV_2019 Forward | CAAATTCTATGGTGGTTGGCACA | [25] | ||
| 6 | nCoV_2019 Reverse | GGCATGGCTCTATCACATTTAGG | ||
| nCoV_2019 Probe | FAM- ATAATCCCAACCCATRAG-MGB | |||
| N-gene | 1 | N_Sarbeco_F | CACATTGGCACCCGCAATC | [13] |
| N_Sarbeco_R | GAGGAACGAGAAGAGGCTTG | |||
| N_Sarbeco_P | FAM- ACTTCCTCAAGGAACAACATTGCCA-BHQ1 | |||
| 2 | N-F (China) | GGGGAACTTCTCCTGCTAGAAT | [23] | |
| N-R (China) | CAGACATTTTGCTCTCAAGCTG | |||
| N-probe (China) | CAGACATTTTGCTCTCAAGCTG | |||
| 3 | HKU-NF (Hong Kong) | TAATCAGACAAGGAACTGATTA | ||
| HKU-NR (Hong Kong) | CGAAGGTGTGACTTCCATG | |||
| HKU-NP (Hong Kong) | GCAAATTGTGCAATTTGCGG | |||
| 4 | NIID_2019-nCOV_N_F2 (Japan) | AAATTTTGGGGACCAGGAAC | ||
| NIID_2019-nCOV_N_R2 (Japan) | TGGCAGCTGTGTAGGTCAAC | |||
| NIID_2019-nCOV_N_P2 (Japan) | ATGTCGCGCATTGGCATGGA | |||
| 5 | WH-NIC N-F Thailand | CGTTTGGTGGACCCTCAGAT | ||
| WH-NIC N-R Thailand | CCCCACTGCGTTCTCCATT | |||
| WH-NIC N-P Thailand | CAACTGGCAGTAACCA | |||
| 6 | CDC N1 Forward | GACCCCAAAATCAGCGAAAT | [26] | |
| CDC N1 Reverse | TCTGGTTACTGCCAGTTGAATCTG | |||
| CDC N1 Probe | FAM- ACCCCGCATTACGTTTGGTGGACC-BHQ1 | |||
| 7 | CDC N2 Forward | TTACAAACATTGGCCGCAAA | ||
| CDC N2 Reverse | GCGCGACATTCCGAAGAA | |||
| CDC N2 Probe | FAM- ACAATTTGCCCCCAGCGCTTCAG-BHQ1 | |||
| 8 | CDC N3 Forward | GGGAGCCTTGAATACACCAAAA | ||
| CDC N3 Reverse | TGTAGCACGATTGCAGCATTG | |||
| CDC N3 Probe | FAM- AYCACATTGGCACCCGCAATCCTG-BHQ1 | |||
| E-gene | E_Sarbeco_Forward | ACAGGTACGTTAATAGTTAATAGCGT | [13] | |
| E_Sarbeco_Reverse | ATATTGCAGCAGTACGCACACA | |||
| E_Sarbeco_Probe | FAM- ACACTAGCCATCCTTACTGCGCTTCG-BHQ1 | |||
| Parameter | E-Gene | N-Gene | RdRp (ORF1ab Polyprotein Region) |
|---|---|---|---|
| Number of sequences | 348 | 327 | 228 |
| Number of sites | 228 | 1260 | 21332 |
| Number of variable sites | 2 | 25 | 131 |
| Number of haplotypes (h) | 3 | 25 | 101 |
| Haplotype diversity (Hd) | 0.023 | 0.573 | 0.9475 |
| Nucleotide diversity (Pi) | 0.00010 | 0.00135 | 0.00015 |
| Theta (per site) from Eta | 0.00136 | 0.00312 | 0.00102 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez, J.D.; Muñoz, M.; Hernández, C.; Flórez, C.; Gomez, S.; Rico, A.; Pardo, L.; Barros, E.C.; Paniz-Mondolfi, A.E. Genetic Diversity Among SARS-CoV2 Strains in South America may Impact Performance of Molecular Detection. Pathogens 2020, 9, 580. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9070580
Ramírez JD, Muñoz M, Hernández C, Flórez C, Gomez S, Rico A, Pardo L, Barros EC, Paniz-Mondolfi AE. Genetic Diversity Among SARS-CoV2 Strains in South America may Impact Performance of Molecular Detection. Pathogens. 2020; 9(7):580. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9070580
Chicago/Turabian StyleRamírez, Juan David, Marina Muñoz, Carolina Hernández, Carolina Flórez, Sergio Gomez, Angelica Rico, Lisseth Pardo, Esther C. Barros, and Alberto E. Paniz-Mondolfi. 2020. "Genetic Diversity Among SARS-CoV2 Strains in South America may Impact Performance of Molecular Detection" Pathogens 9, no. 7: 580. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9070580