Cystatin C and α-1-Microglobulin Predict Severe Acute Kidney Injury in Patients with Hemorrhagic Fever with Renal Syndrome

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Characteristics of HFRS Patients Included in the Study
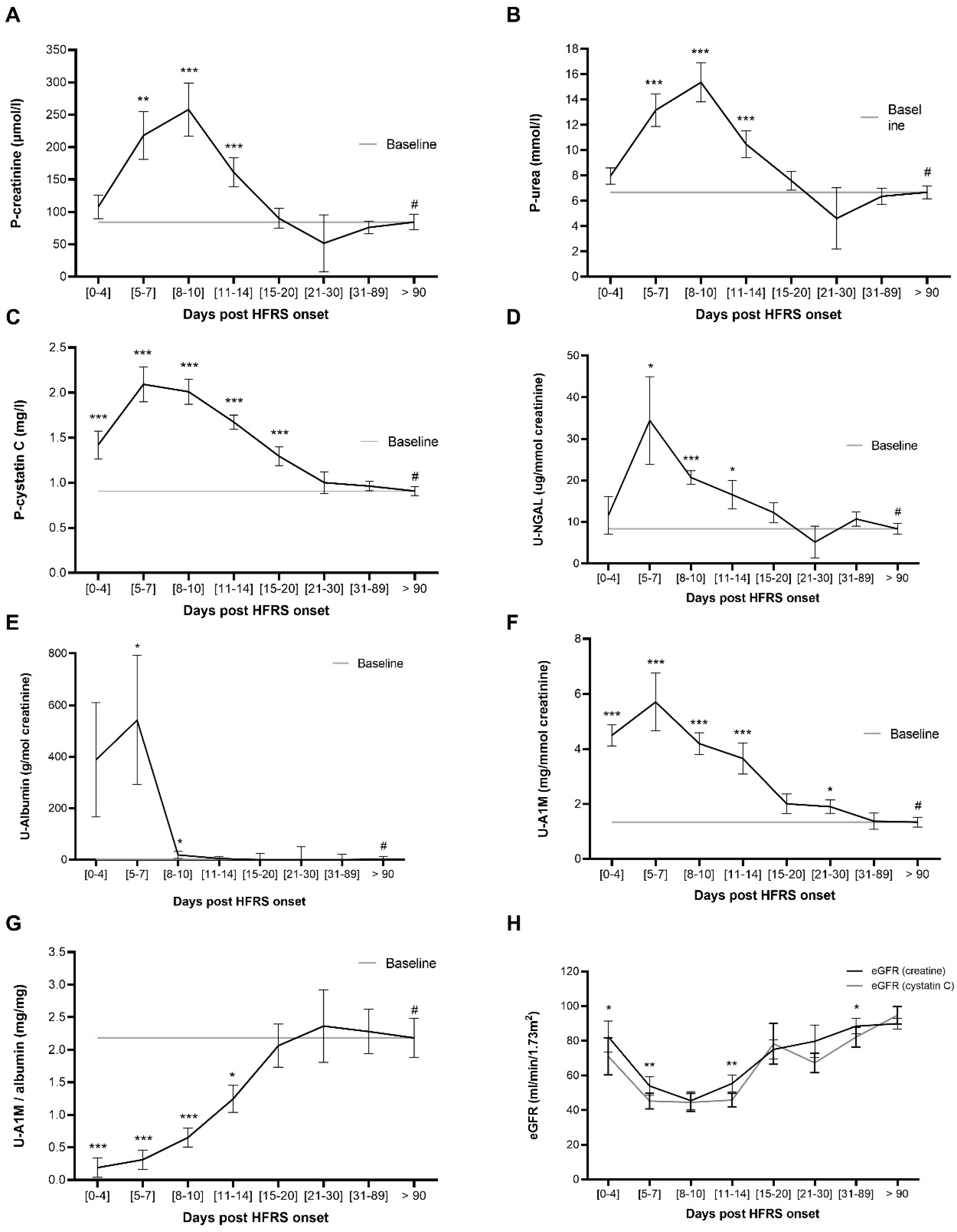
2.2. P-Cystatin C and U-A1M Levels Increase Earlier than P-Urea and P-Creatine during HFRS
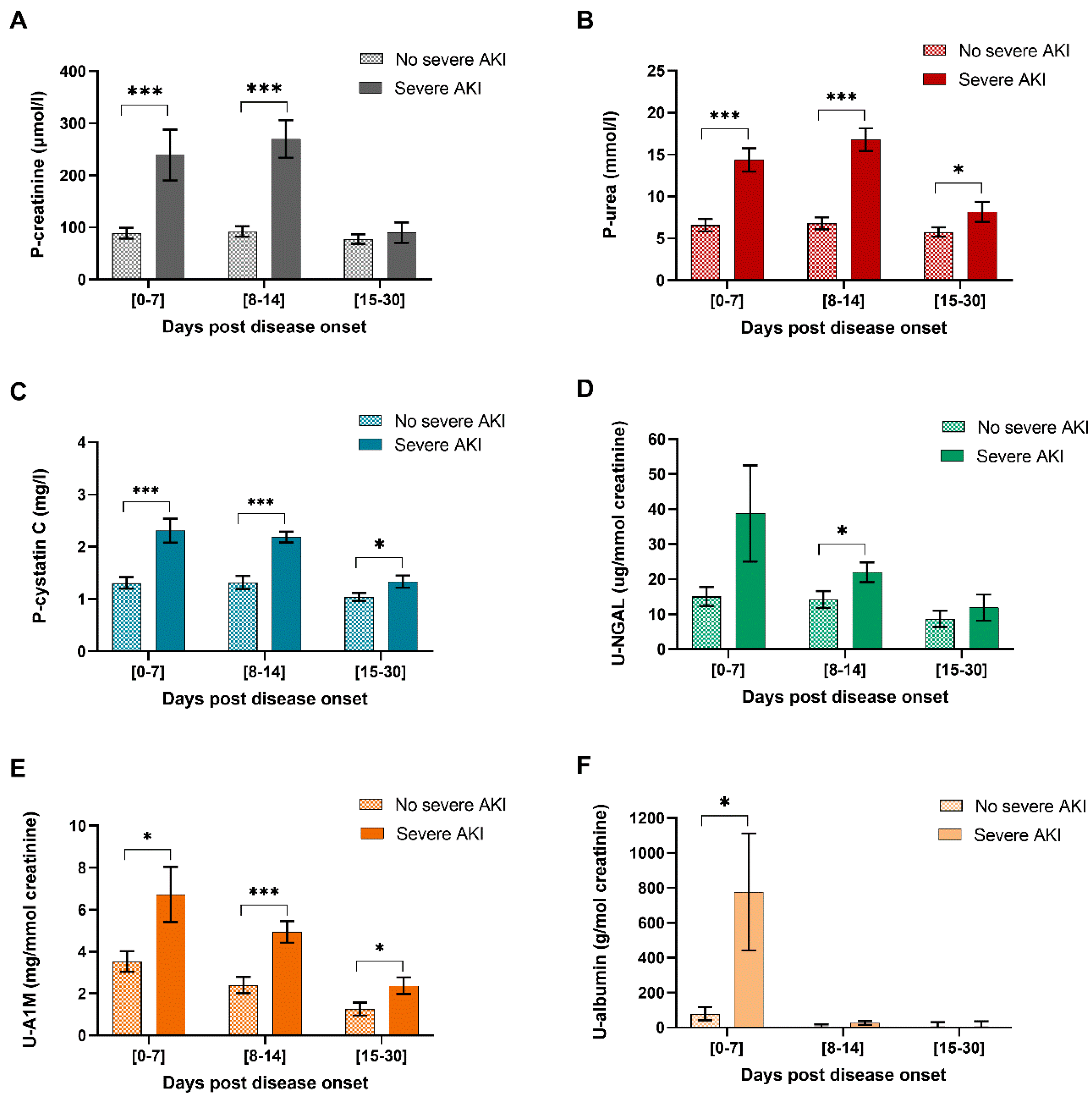
2.3. P-Cystatin C, U-A1M and U-NGAL Levels are Significantly Increased in HFRS Patients with Severe AKI
2.4. P-Cystatin C Levels Associate Significantly with the Levels of the Traditional Kidney Injury Markers P-Creatinine and P-Urea during HFRS
2.5. P-Cystatin C, U-A1M and P-Urea Predict Severe AKI in a ROC Curve Analysis
2.6. Shrunken Pore Syndrome in HFRS Patients
3. Discussion
4. Materials and Methods
4.1. Study Cohort
4.2. Routine Clinically Laboratory Analyses
4.3. Kidney Injury Markers
4.4. Severity of Acute Kidney Injury
4.5. Glomerular Filtration Rate and Shrunken Pore Syndrome
4.6. Statistics
4.7. Study Approval
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Peters, C.J.; Khan, A.S. Hantavirus pulmonary syndrome: The new American hemorrhagic fever. Clin. Infect. Dis. 2002, 34, 1224–1231. [Google Scholar] [CrossRef]
- Vaheri, A.; Strandin, T.; Hepojoki, J.; Sironen, T.; Henttonen, H.; Makela, S.; Mustonen, J. Uncovering the mysteries of hantavirus infections. Nat. Rev. Microbiol. 2013, 11, 539–550. [Google Scholar] [CrossRef]
- Rasmuson, J.; Andersson, C.; Norrman, E.; Haney, M.; Evander, M.; Ahlm, C. Time to revise the paradigm of hantavirus syndromes? Hantavirus pulmonary syndrome caused by European hantavirus. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 685–690. [Google Scholar] [CrossRef] [Green Version]
- Rasmuson, J.; Lindqvist, P.; Sorensen, K.; Hedstrom, M.; Blomberg, A.; Ahlm, C. Cardiopulmonary involvement in Puumala hantavirus infection. BMC Infect. Dis. 2013, 13, 501. [Google Scholar] [CrossRef]
- Hjertqvist, M.; Klein, S.L.; Ahlm, C.; Klingstrom, J. Mortality rate patterns for hemorrhagic fever with renal syndrome caused by Puumala virus. Emerg. Infect. Dis. 2010, 16, 1584–1586. [Google Scholar] [CrossRef]
- Bergstedt Oscarsson, K.; Brorstad, A.; Baudin, M.; Lindberg, A.; Forssén, A.; Evander, M.; Eriksson, M.; Ahlm, C. Human Puumala hantavirus infection in northern Sweden; increased seroprevalence and association to risk and health factors. BMC Infect. Dis. 2016, 16, 566. [Google Scholar] [CrossRef] [Green Version]
- Settergren, B.; Trollfors, B.; Fasth, A.; Hultberg, B.; Norrby, S.R. Glomerular filtration rate and tubular involvement during acute disease and convalescence in patients with nephropathia epidemica. J. Infect. Dis. 1990, 161, 716–720. [Google Scholar] [CrossRef]
- Kellum, J.A.; Prowle, J.R. Paradigms of acute kidney injury in the intensive care setting. Nat. Rev. Nephrol. 2018, 14, 217–230. [Google Scholar] [CrossRef]
- Khwaja, A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin. Pract. 2012, 120, c179–c184. [Google Scholar] [CrossRef]
- Mustonen, J.; Helin, H.; Pietila, K.; Brummer-Korvenkontio, M.; Hedman, K.; Vaheri, A.; Pasternack, A. Renal biopsy findings and clinicopathologic correlations in nephropathia epidemica. Clin. Nephrol. 1994, 41, 121–126. [Google Scholar]
- Delanaye, P.; Cavalier, E.; Pottel, H. Serum Creatinine: Not So Simple! Nephron 2017, 136, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Zbinden, S.; Bourquin, V. Understanding renal functional reserve. Rev. Med. Suisse 2018, 14, 276–278. [Google Scholar] [PubMed]
- Stevens, L.A.; Coresh, J.; Greene, T.; Levey, A.S. Assessing kidney function-measured and estimated glomerular filtration rate. N. Engl. J. Med. 2006, 354, 2473–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahamson, M.; Barrett, A.J.; Salvesen, G.; Grubb, A. Isolation of six cysteine proteinase inhibitors from human urine. Their physicochemical and enzyme kinetic properties and concentrations in biological fluids. J. Biol. Chem. 1986, 261, 11282–11289. [Google Scholar]
- Hoek, F.J.; Kemperman, F.A.; Krediet, R.T. A comparison between cystatin C, plasma creatinine and the Cockcroft and Gault formula for the estimation of glomerular filtration rate. Nephrol. Dial. Transpl. 2003, 18, 2024–2031. [Google Scholar] [CrossRef]
- Vinge, E.; Lindergard, B.; Nilsson-Ehle, P.; Grubb, A. Relationships among serum cystatin C, serum creatinine, lean tissue mass and glomerular filtration rate in healthy adults. Scand. J. Clin. Lab. Investig. 1999, 59, 587–592. [Google Scholar] [CrossRef]
- Finney, H.; Newman, D.J.; Price, C.P. Adult reference ranges for serum cystatin C, creatinine and predicted creatinine clearance. Ann. Clin. Biochem. 2000, 37, 9–59. [Google Scholar] [CrossRef] [Green Version]
- Norlund, L.; Fex, G.; Lanke, J.; Von Schenck, H.; Nilsson, J.E.; Leksell, H.; Grubb, A. Reference intervals for the glomerular filtration rate and cell-proliferation markers: Serum cystatin C and serum beta 2-microglobulin/cystatin C-ratio. Scand. J. Clin. Lab. Investig. 1997, 57, 463–470. [Google Scholar] [CrossRef]
- Dharnidharka, V.R.; Kwon, C.; Stevens, G. Serum cystatin C is superior to serum creatinine as a marker of kidney function: A meta-analysis. Am. J. Kidney Dis. 2002, 40, 221–226. [Google Scholar] [CrossRef]
- Penders, J.; Delanghe, J.R. Alpha 1-microglobulin: Clinical laboratory aspects and applications. Clin. Chim. Acta 2004, 346, 107–118. [Google Scholar] [CrossRef]
- Martensson, J.; Bellomo, R. The rise and fall of NGAL in acute kidney injury. Blood Purif. 2014, 37, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.H.; Verwiebe, R. Alpha 1-microglobulin (protein HC): Features of a promising indicator of proximal tubular dysfunction. Eur. J. Clin. Chem. Clin. Biochem. 1992, 30, 683–691. [Google Scholar]
- Makela, S.; Mustonen, J.; Ala-Houhala, I.; Hurme, M.; Koivisto, A.M.; Vaheri, A.; Pasternack, A. Urinary excretion of interleukin-6 correlates with proteinuria in acute Puumala hantavirus-induced nephritis. Am. J. Kidney Dis. 2004, 43, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, Q.; Wang, J.; Xu, Z.; Song, C.; Zhuang, R.; Yang, K.; Yang, A.; Jin, B. Cystatin C, a novel urinary biomarker for sensitive detection of acute kidney injury during haemorrhagic fever with renal syndrome. Biomarkers 2010, 15, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Bunz, H.; Weyrich, P.; Peter, A.; Baumann, D.; Tschritter, O.; Guthoff, M.; Beck, R.; Jahn, G.; Artunc, F.; Haring, H.U.; et al. Urinary Neutrophil Gelatinase-Associated Lipocalin (NGAL) and proteinuria predict severity of acute kidney injury in Puumala virus infection. BMC Infect. Dis. 2015, 15, 464. [Google Scholar] [CrossRef] [Green Version]
- Mantula, P.S.; Outinen, T.K.; Clement, J.P.G.; Huhtala, H.S.A.; Porsti, I.H.; Vaheri, A.; Mustonen, J.T.; Makela, S.M. Glomerular Proteinuria predicts the severity of acute kidney injury in puumala hantavirus-induced tubulointerstitial nephritis. Nephron 2017, 136, 193–201. [Google Scholar] [CrossRef]
- Bjork, J.; Grubb, A.; Sterner, G.; Nyman, U. Revised equations for estimating glomerular filtration rate based on the Lund-Malmo Study cohort. Scand. J. Clin. Lab. Investig. 2011, 71, 232–239. [Google Scholar] [CrossRef]
- Nyman, U.; Grubb, A.; Larsson, A.; Hansson, L.O.; Flodin, M.; Nordin, G.; Lindstrom, V.; Bjork, J. The revised Lund-Malmo GFR estimating equation outperforms MDRD and CKD-EPI across GFR, age and BMI intervals in a large Swedish population. Clin. Chem. Lab. Med. 2014, 52, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Grubb, A. Shrunken pore syndrome—A common kidney disorder with high mortality. Diagnosis, prevalence, pathophysiology and treatment options. Clin. Biochem. 2020. [Google Scholar] [CrossRef]
- Zhai, J.L.; Ge, N.; Zhen, Y.; Zhao, Q.; Liu, C. Corticosteroids significantly increase serum Cystatin C concentration without affecting renal function in symptomatic heart failure. Clin. Lab. 2016, 62, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Ala-Houhala, I.; Koskinen, M.; Ahola, T.; Harmoinen, A.; Kouri, T.; Laurila, K.; Mustonen, J.; Pasternack, A. Increased glomerular permeability in patients with nephropathia epidemica caused by Puumala hantavirus. Nephrol. Dial. Transpl. 2002, 17, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Meier, M.; Kramer, J.; Jabs, W.J.; Nolte, C.; Hofmann, J.; Kruger, D.H.; Lehnert, H.; Nitschke, M. Proteinuria and the clinical course of Dobrava-Belgrade hantavirus infection. Nephron Extra 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, E.; Grouls, S.; Stein, N.; Reiser, J.; Zeier, M. Pathogenic old world hantaviruses infect renal glomerular and tubular cells and induce disassembling of cell-to-cell contacts. J. Virol. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groen, J.; Bruijn, J.A.; Gerding, M.N.; Jordans, J.G.; Moll van Charante, A.W.; Osterhaus, A.D. Hantavirus antigen detection in kidney biopsies from patients with nephropathia epidemica. Clin. Nephrol. 1996, 46, 379–383. [Google Scholar] [PubMed]
- Nejat, M.; Pickering, J.W.; Walker, R.J.; Endre, Z.H. Rapid detection of acute kidney injury by plasma cystatin C in the intensive care unit. Nephrol. Dial. Transpl. 2010, 25, 3283–3289. [Google Scholar] [CrossRef]
- Coll, E.; Botey, A.; Alvarez, L.; Poch, E.; Quinto, L.; Saurina, A.; Vera, M.; Piera, C.; Darnell, A. Serum cystatin C as a new marker for noninvasive estimation of glomerular filtration rate and as a marker for early renal impairment. Am. J. Kidney Dis. 2000, 36, 29–34. [Google Scholar] [CrossRef]
- Zhu, J.; Yin, R.; Wu, H.; Yi, J.; Luo, L.; Dong, G.; Jing, H. Cystatin C as a reliable marker of renal function following heart valve replacement surgery with cardiopulmonary bypass. Clin. Chim. Acta 2006, 374, 116–121. [Google Scholar] [CrossRef]
- Lagos-Arevalo, P.; Palijan, A.; Vertullo, L.; Devarajan, P.; Bennett, M.R.; Sabbisetti, V.; Bonventre, J.V.; Ma, Q.; Gottesman, R.D.; Zappitelli, M. Cystatin C in acute kidney injury diagnosis: Early biomarker or alternative to serum creatinine? Pediatr. Nephrol. 2015, 30, 665–676. [Google Scholar] [CrossRef]
- Leem, A.Y.; Park, M.S.; Park, B.H.; Jung, W.J.; Chung, K.S.; Kim, S.Y.; Kim, E.Y.; Jung, J.Y.; Kang, Y.A.; Kim, Y.S.; et al. Value of serum Cystatin C measurement in the diagnosis of sepsis-induced kidney injury and prediction of renal function recovery. Yonsei Med. J. 2017, 58, 604–612. [Google Scholar] [CrossRef]
- Akesson, A.; Lindstrom, V.; Nyman, U.; Jonsson, M.; Abrahamson, M.; Christensson, A.; Bjork, J.; Grubb, A. Shrunken pore syndrome and mortality: A cohort study of patients with measured GFR and known comorbidities. Scand. J. Clin. Lab. Investig. 2020, 1–12. [Google Scholar] [CrossRef]
- Herou, E.; Dardashti, A.; Nozohoor, S.; Zindovic, I.; Ederoth, P.; Grubb, A.; Bjursten, H. The mortality increase in cardiac surgery patients associated with shrunken pore syndrome correlates with the eGFRcystatin C/eGFRcreatinine-ratio. Scand. J. Clin. Lab. Investig. 2019, 79, 167–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almen, M.S.; Bjork, J.; Nyman, U.; Lindstrom, V.; Jonsson, M.; Abrahamson, M.; Vestergren, A.S.; Lindhe, O.; Franklin, G.; Christensson, A.; et al. Shrunken pore syndrome is associated with increased levels of atherosclerosis-promoting proteins. Kidney Int. Rep. 2019, 4, 67–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.L.; Castro, A.F., 3rd; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Grubb, A.; Horio, M.; Hansson, L.O.; Bjork, J.; Nyman, U.; Flodin, M.; Larsson, A.; Bokenkamp, A.; Yasuda, Y.; Blufpand, H.; et al. Generation of a new cystatin C-based estimating equation for glomerular filtration rate by use of 7 assays standardized to the international calibrator. Clin. Chem. 2014, 60, 974–986. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| HFRS Patientsn = 44 * | Reference Values | |
|---|---|---|
| Demographic data | ||
| Age, years | 53 (41–64) | NA |
| Sex, n female/male (%) | 29 (66%)/15 (34%) | NA |
| Hospital care, n (%) | 36 (82%) | NA |
| Days of hospital care | 6 (4–7.75) | NA |
| Day of first sample | 6 (5–7) | NA |
| Estimated glomerular filtration rate (eGFR) a | ||
| eGFR—Creatinine (CKD-EPI) (mL/min/1.73 m2) | 32 (15–67) | >60 |
| eGFR—Creatinine (LM-Rev) (mL/min/1.73 m2) | 28 (15–63) | >60 |
| eGFR—Cystatin C (CAPA) (mL/min/1.73 m2) | 27 (21–58) | >60 |
| Acute kidney injury (AKI) b | ||
| No AKI | 10 (22.7%) | NA |
| Stage 1 | 7 (15.9%) | NA |
| Stage 2 | 10 (22.7%) | NA |
| Stage 3 | 17 (38:6%) | NA |
| Severe AKI c | 27 (61.4%) | NA |
| Shrunken Pore Syndrome d | 18 (41%) | NA |
| Kidney injury markers e | ||
| Creatinine (μmol/L) | 103 (65.75–163.5) | 45–105 |
| Urea (mmol/L) | 8 (5–14.9) | 2.6–8.1 |
| Cystatin C (mg/L) | 1.3 (0.98–2.28) | NA |
| U-A1M (mg/mmol creatinine) | 4.4 (2.6–5.4) | NA |
| U-NGAL (µg/mmol creatinine) | 16.4 (7.3–31) | NA |
| U-Albumin (g/mol creatinine) | 47.4 (17.8–244.1) | <3 |
| Kidney Injury Markers | U-A1M (μg/mmol Creatinine) | U-NGAL (ng/mmol Creatinine) | P-cystatin C (mg/mL) | |||
|---|---|---|---|---|---|---|
| β | p-Value | β | p-Value | β | p-Value | |
| P-Creatinine (μmol/L) | 0.01 | 0.036 | 0.78 | 0.058 | 150.4 | <0.001 |
| P-Urea (nmol/L) | 0.2 | 0.083 | 0.016 | 0.174 | 6789 | <0.001 |
| Kidney Injury Markers | AUC | 95% CI | p-Value | Cut-off Value | Sensitivity | Specificity |
|---|---|---|---|---|---|---|
| P-Urea (mmol/L) | 0.71 | 0.5–0.92 | 0.049 | 6.3 | 0.77 | 0.77 |
| P-Creatinine | 0.65 | 0.45–0.85 | 0.17 | - | - | - |
| U-Albumin (g/mol creatinine) | 0.53 | 0.3–0.77 | 0.77 | - | - | - |
| P-Cystatin C (mg/L) | 0.72 | 0.51–0.92 | 0.047 | 1.1 | 0.77 | 0.71 |
| U-A1M (mg/mmol creatinine) | 0.73 | 0.51–0.94 | 0.048 | 3.5 | 0.82 | 0.69 |
| U-NGAL (µg/mmol creatinine) | 0.27 | 0.05–0.46 | 0.034 | - | - | - |
| Kidney Injury Markers | No SPS (n = 26) | SPS (n = 18) | p-Value |
|---|---|---|---|
| P-Creatinine (μmol/L) | 273 (45) | 230 (56) | 0.561 |
| P-Urea (nmol/L) | 14 (2) | 16 (2) | 0.499 |
| P-Cystatin C (mg/L) | 2 (0.2) | 2.6 (0.3) | 0.087 |
| U-Albumin (g/mol crea) | 859 (332) | 110 (377) | 0.136 |
| U-A1M (mg/mmol crea) | 7.2 (1.6) | 5.8 (1.8) | 0.572 |
| U-NGAL (ng/mmol crea) | 56.5 (14.8) | 23 (17) | 0.133 |
| CRP (mg/L) | 79 (10) | 125 (13) | 0.005 |
| Hospitalization time (days) | 5.6 (1.3) | 8 (1.3) | 0.209 |
| Age (years) | 48 (3) | 56 (3) | 0.065 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansson, M.; Gustafsson, R.; Jacquet, C.; Chebaane, N.; Satchell, S.; Thunberg, T.; Ahlm, C.; Fors Connolly, A.-M. Cystatin C and α-1-Microglobulin Predict Severe Acute Kidney Injury in Patients with Hemorrhagic Fever with Renal Syndrome. Pathogens 2020, 9, 666. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9080666
Hansson M, Gustafsson R, Jacquet C, Chebaane N, Satchell S, Thunberg T, Ahlm C, Fors Connolly A-M. Cystatin C and α-1-Microglobulin Predict Severe Acute Kidney Injury in Patients with Hemorrhagic Fever with Renal Syndrome. Pathogens. 2020; 9(8):666. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9080666
Chicago/Turabian StyleHansson, Magnus, Rasmus Gustafsson, Chloé Jacquet, Nedia Chebaane, Simon Satchell, Therese Thunberg, Clas Ahlm, and Anne-Marie Fors Connolly. 2020. "Cystatin C and α-1-Microglobulin Predict Severe Acute Kidney Injury in Patients with Hemorrhagic Fever with Renal Syndrome" Pathogens 9, no. 8: 666. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9080666