Differences in the Endophytic Microbiome of Olive Cultivars Infected by Xylella fastidiosa across Seasons


 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
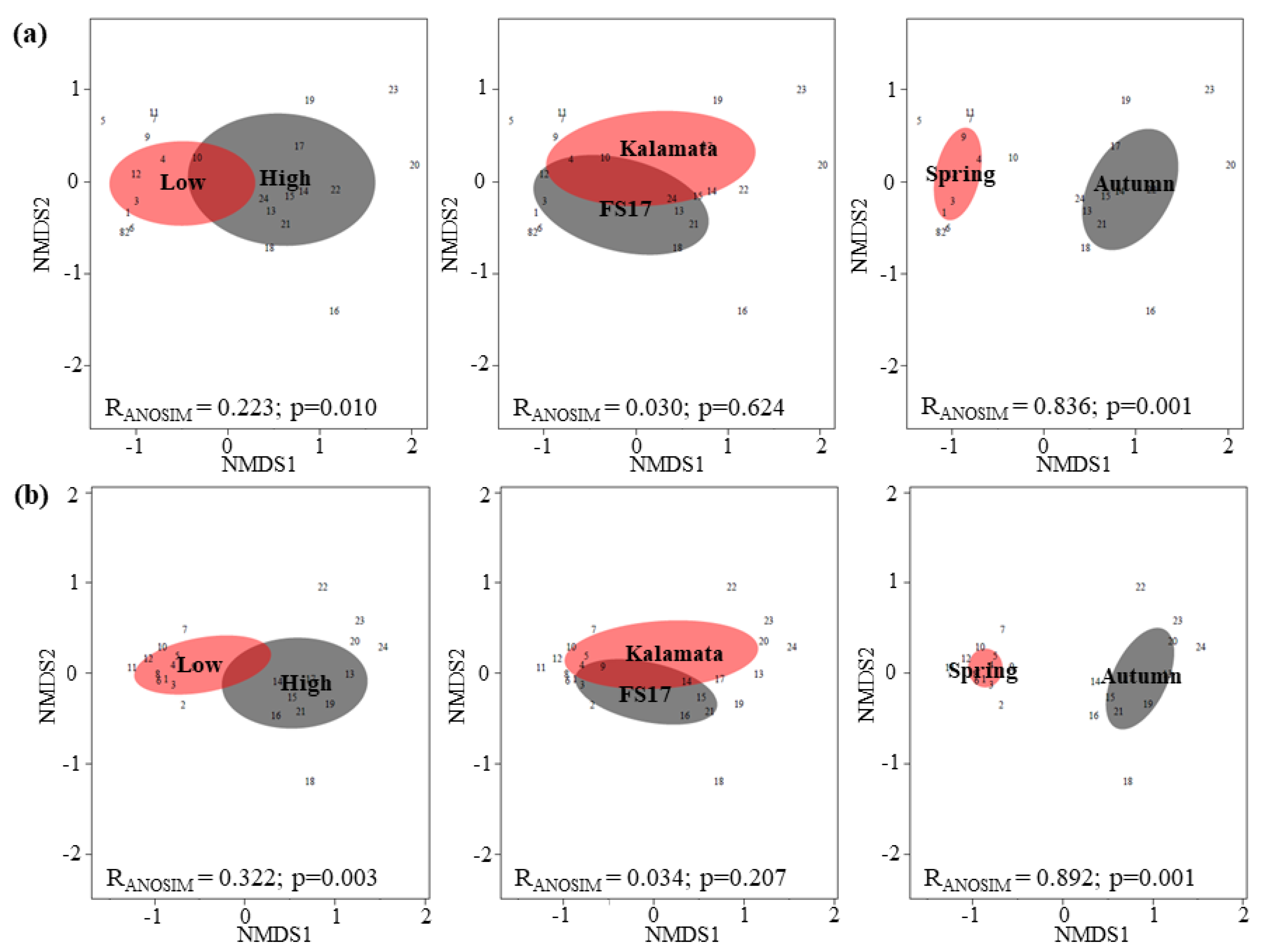
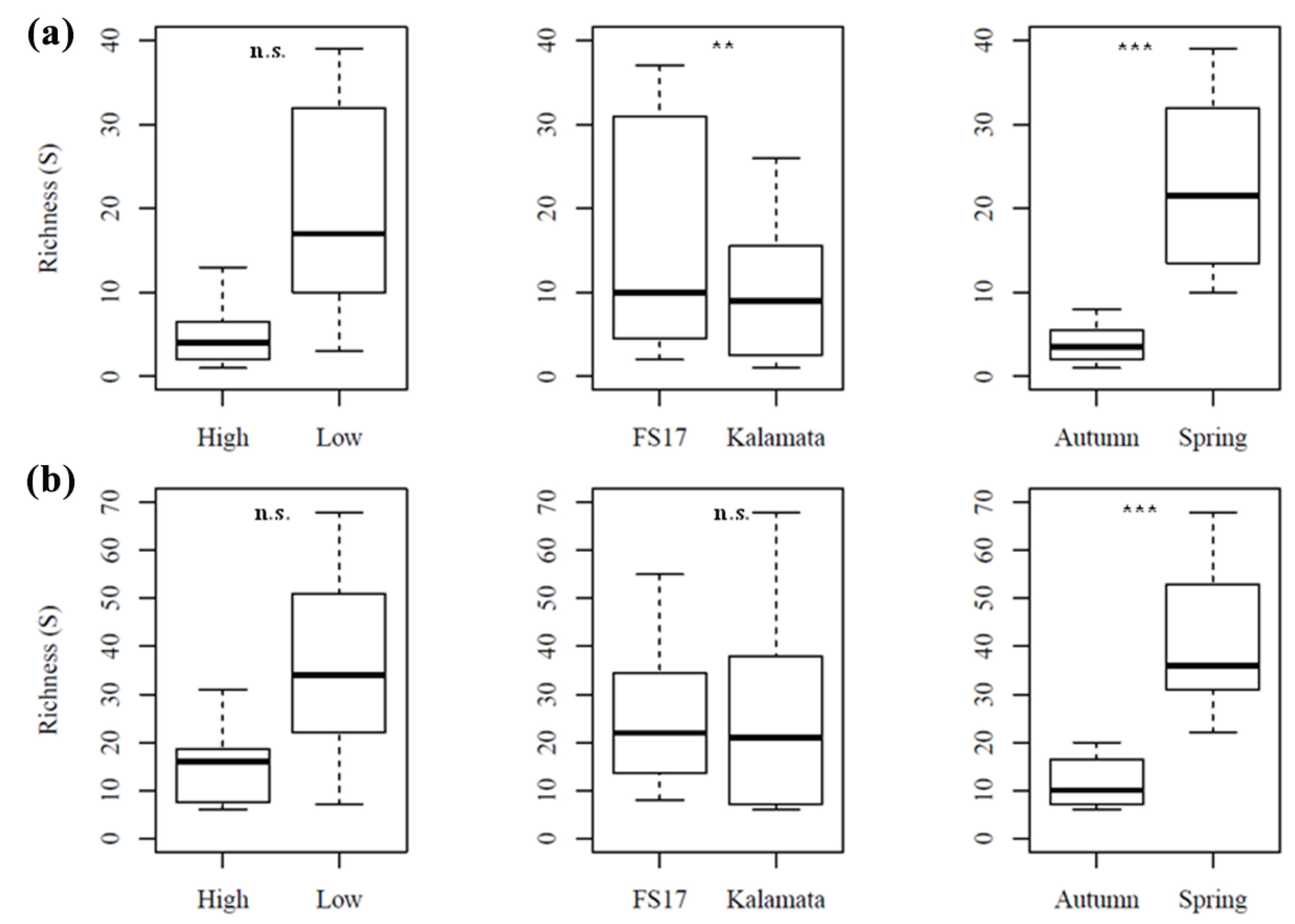
2. Results
2.1. Description of the Microbiome by Whole Metagenome Shotgun Sequencing (WMSS)
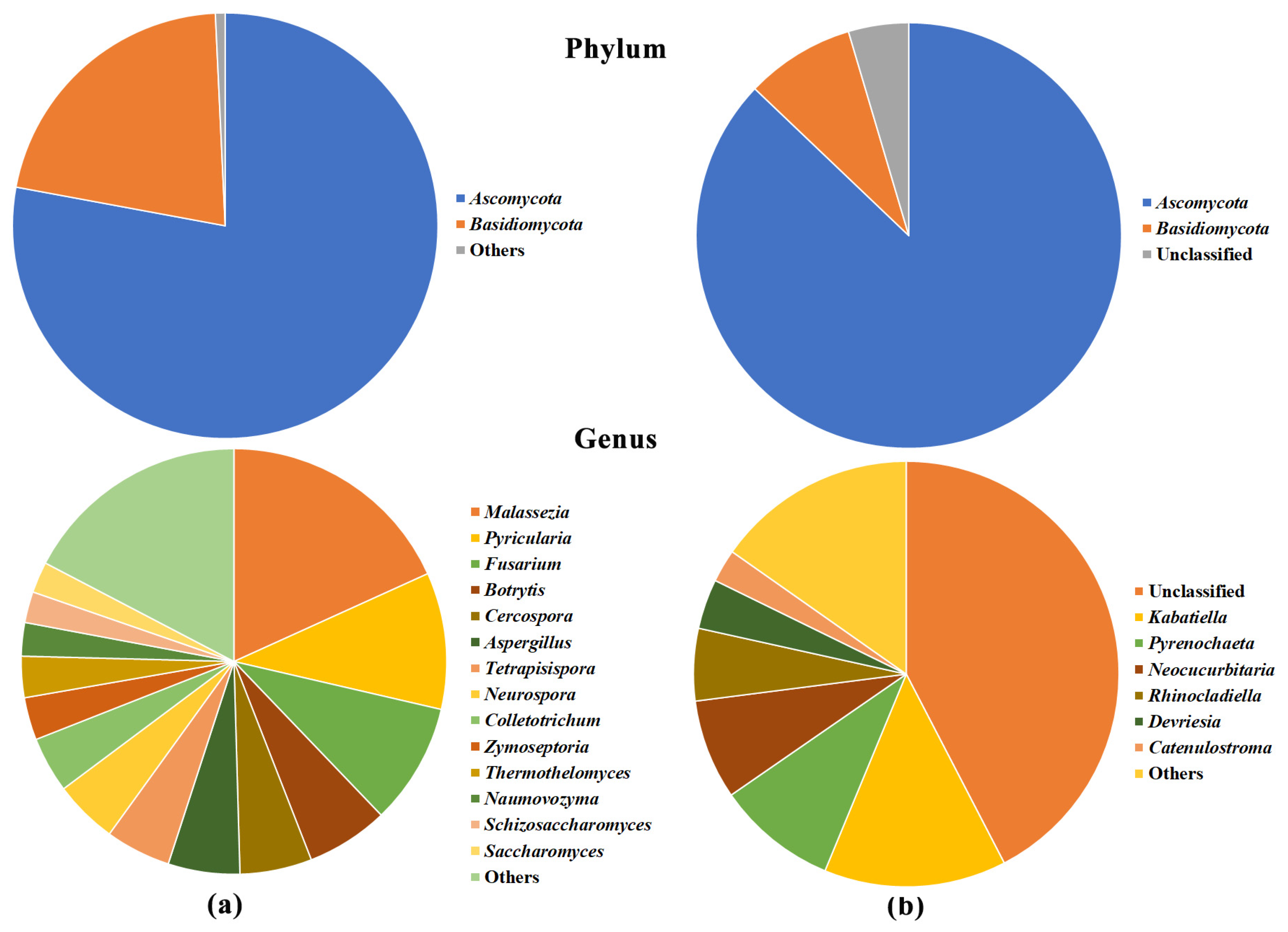
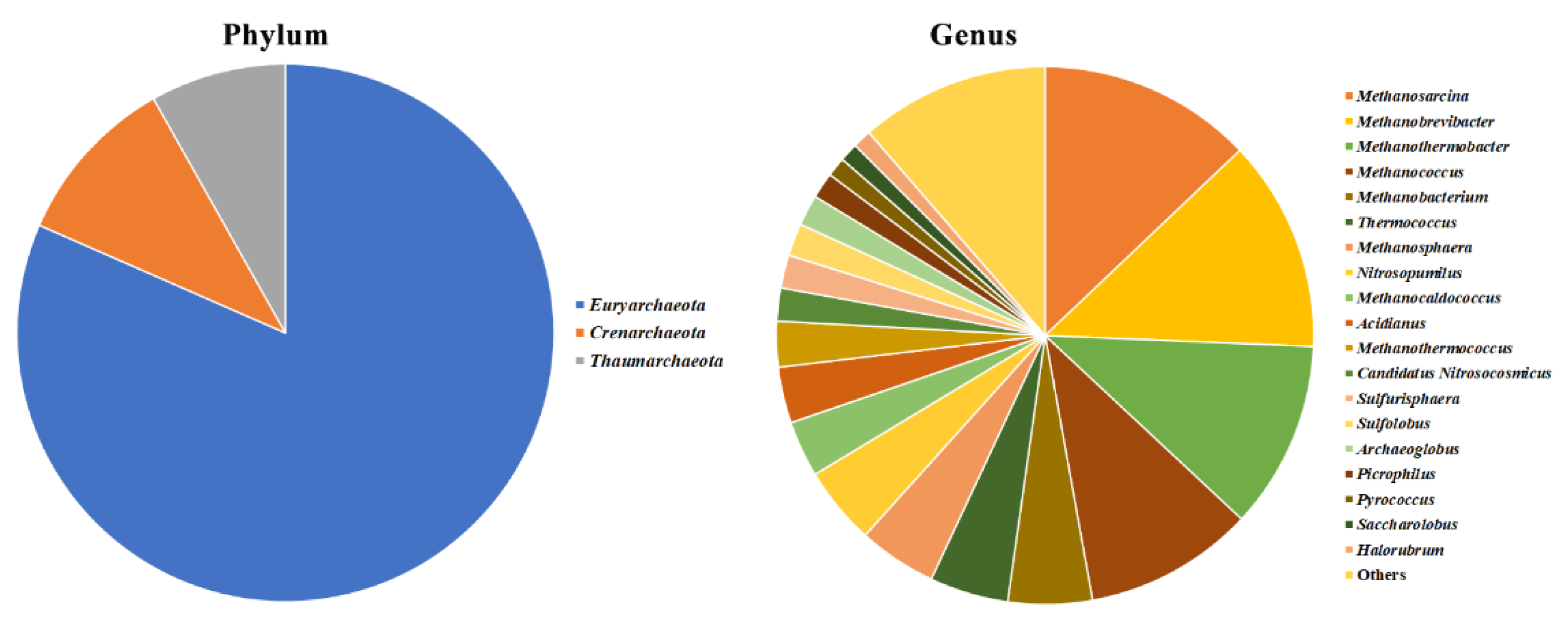
2.2. Description of the Microbiome by 16S and ITS1 rRNA Gene Sequencing
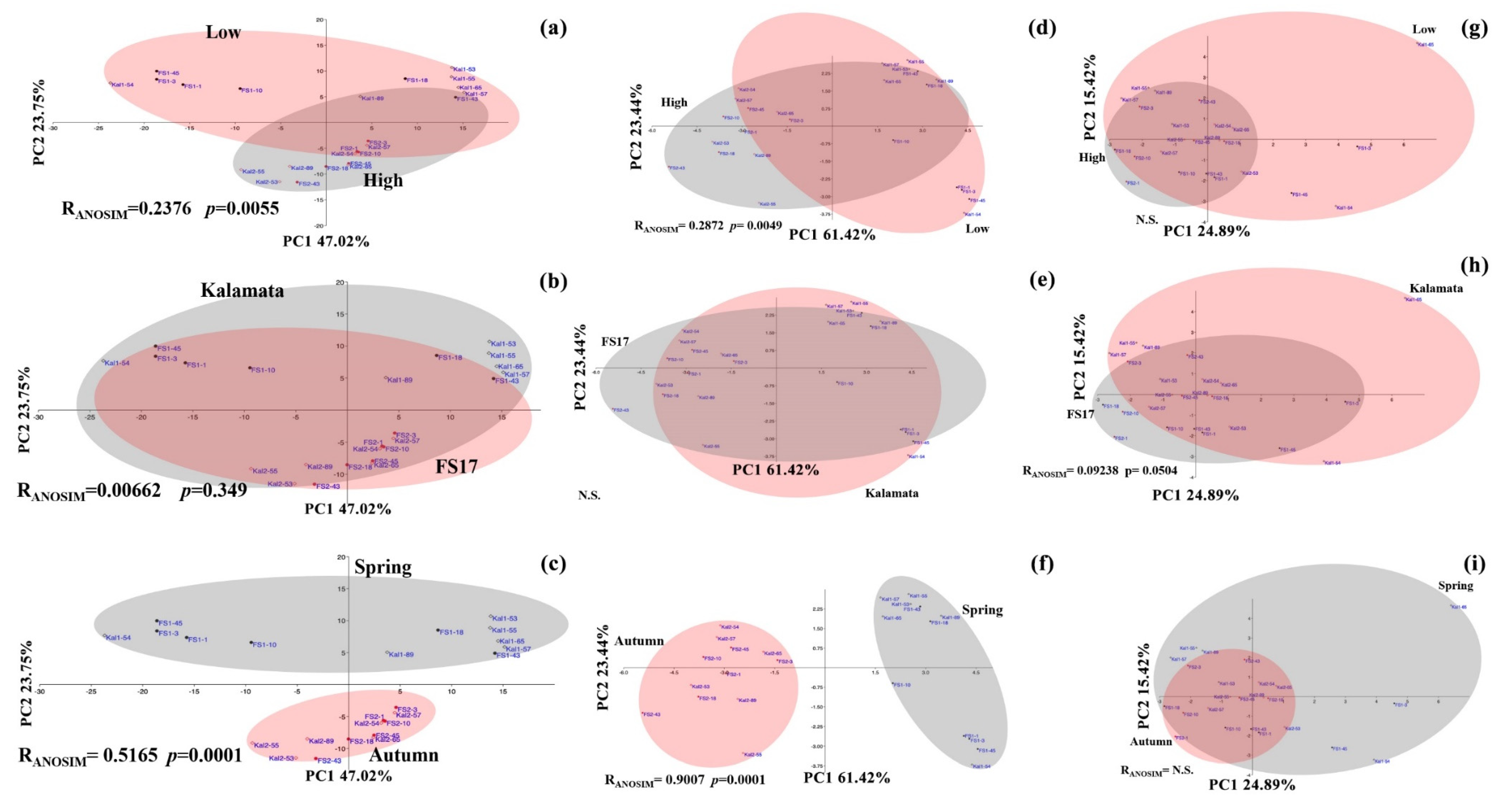
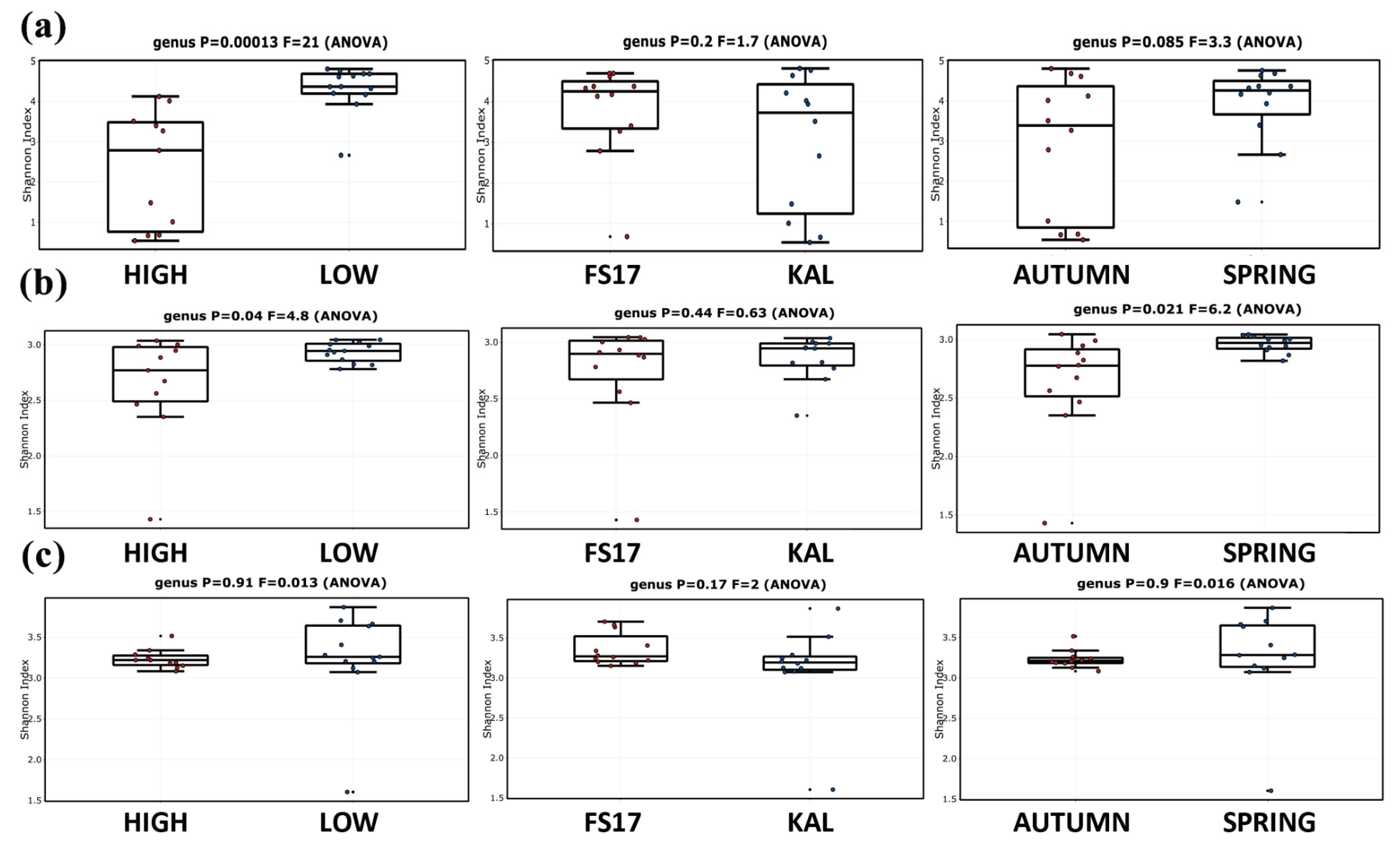
2.3. Olive Xylem Microbiome Composition by WMSS Analysis
2.4. Olive Xylem Microbiome Composition by 16S and ITS1 rRNA Gene Analysis
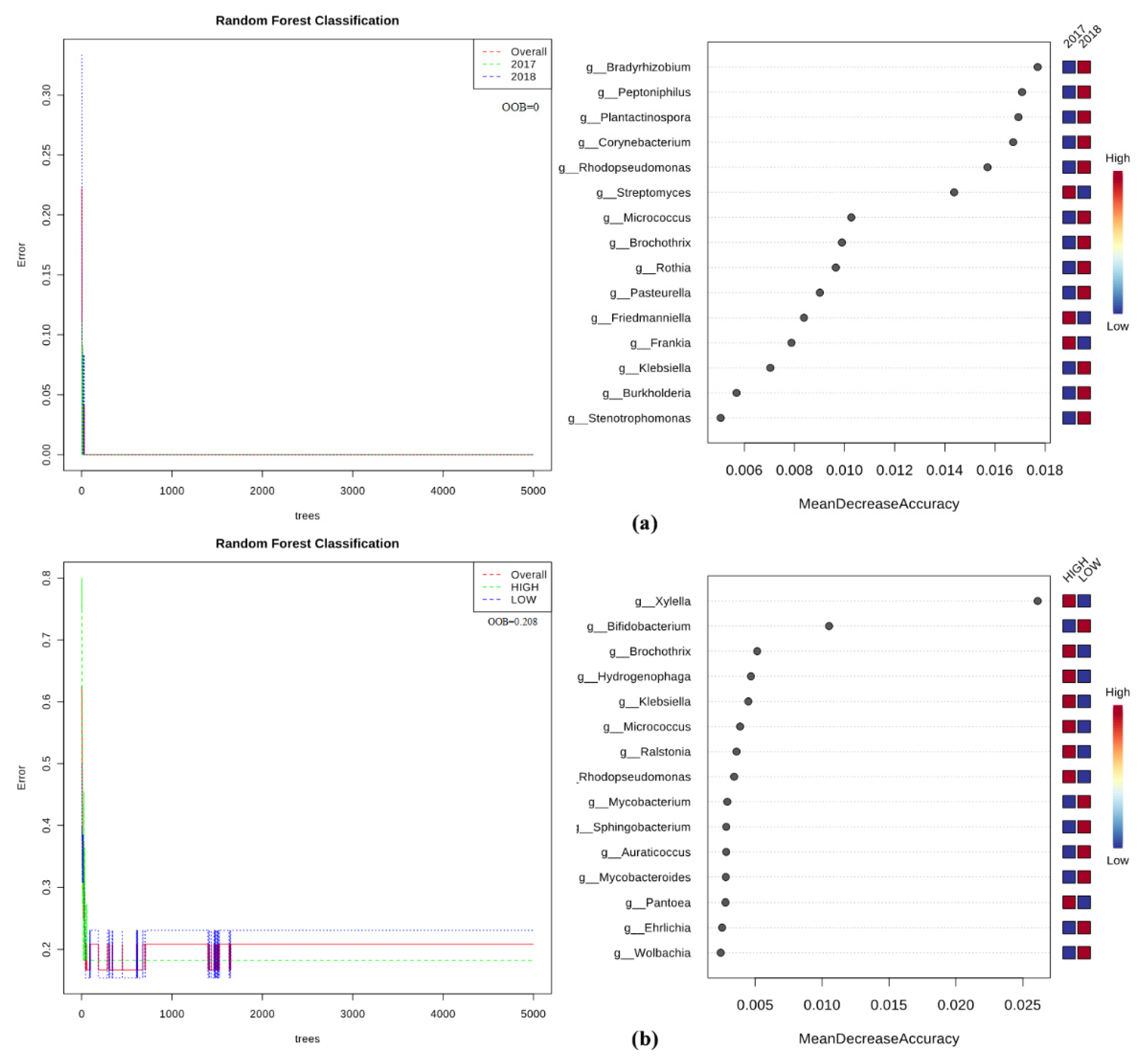
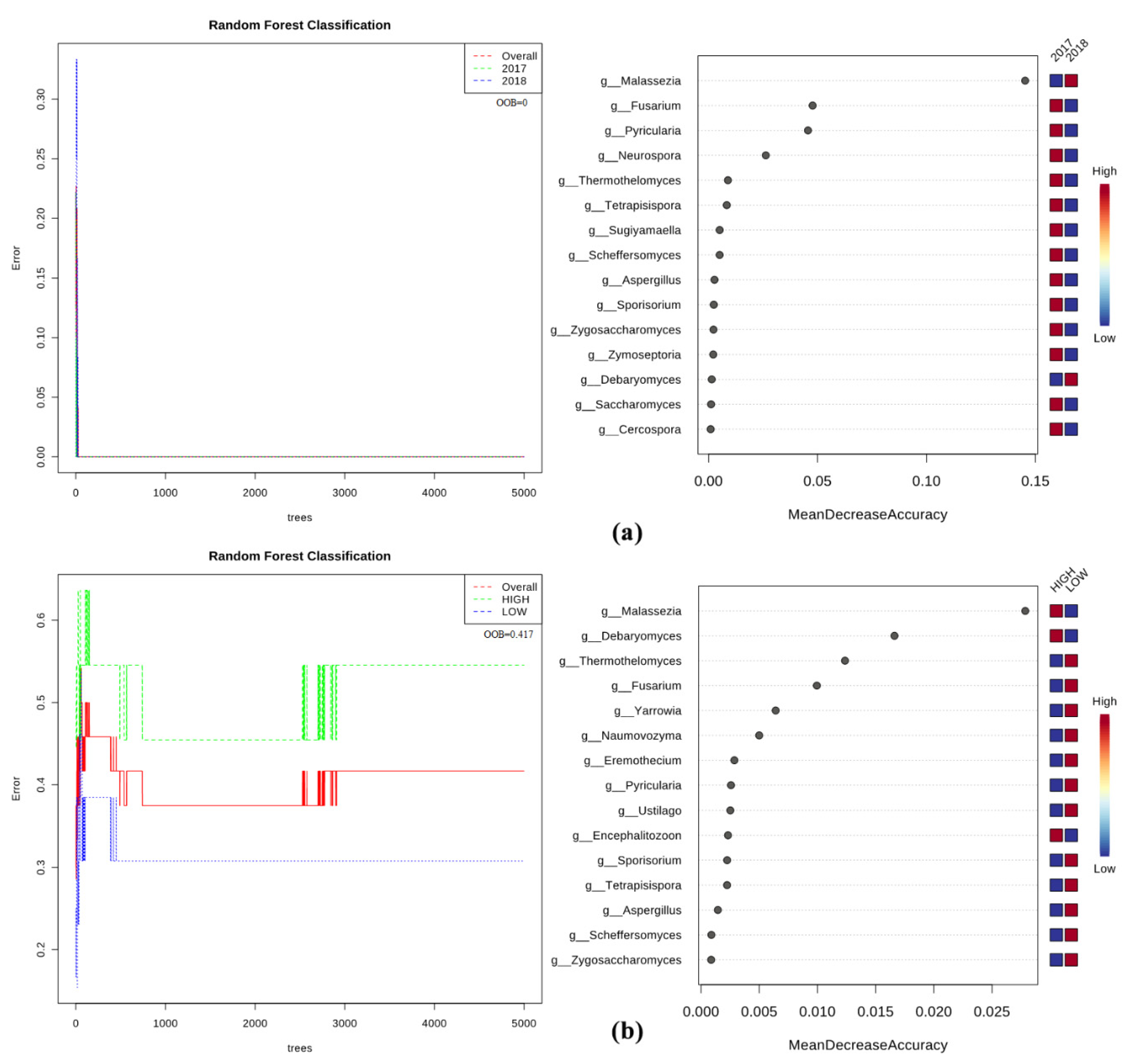
2.5. Bacteria/Fungi Genera Shaping the Olive Xylem Microbiome
3. Discussion
4. Materials and Methods
4.1. Collection of Plant Samples
4.2. Extraction of Total DNA and Detection of Xylella Fastidiosa
4.3. Whole Metagenome Shotgun Sequencing and Bioinformatic Analysis
4.4. 16S and ITS1 rRNA Gene Library Sequencing and Bioinformatic Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schaad, N.W.; Postnikova, E.; Lacy, G.; Chang, C.-J. Xylella fastidiosa subspecies: X. fastidiosa subsp piercei, subsp. nov., X. fastidiosa subsp. multiplex subsp. nov., and X. fastidiosa subsp. pauca subsp. nov. Syst. Appl. Microbiol. 2004, 27, 290–300. [Google Scholar] [CrossRef]
- Saponari, M.; Giampetruzzi, A.; Loconsole, G.; Boscia, D.; Saldarelli, P. Xylella fastidiosa in olive in Apulia: Where we stand. Phytopathology 2019, 109, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Rapicavoli, J.; Ingel, B.; Blanco-Ulate, B.; Cantu, D.; Roper, C. Xylella fastidiosa: An examination of a re-emerging plant pathogen. Mol. Plant Pathol. 2018, 19, 786–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saponari, M.; Boscia, D.; Nigro, F.; Martelli, G.P. Identification of DNA sequences related to Xylella fastidiosa in oleander, almond and olive trees exhibiting leaf schorch symptoms in Apulia (Southern Italy). J. Plant Pathol. 2013, 95. [Google Scholar] [CrossRef]
- Authority, E.F.S. Update of the Xylella spp. host plant database—Systematic literature search up to 30 June 2019. EFSA J. 2020, 18, e06114. [Google Scholar]
- Cavalieri, V.; Altamura, G.; Fumarola, G.; di Carolo, M.; Saponari, M.; Cornara, D.; Bosco, D.; Dongiovanni, C. Transmission of Xylella fastidiosa subspecies pauca sequence type 53 by different insect species. Insects 2019, 10, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deyett, E.; Pouzoulet, J.; Yang, J.I.; Ashworth, V.; Castro, C.; Roper, M.; Rolshausen, P. Assessment of Pierce’s disease susceptibility in Vitis vinifera cultivars with different pedigrees. Plant Pathol. 2019, 68, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Riaz, S.; Huerta-Acosta, K.; Tenscher, A.; Walker, M. Genetic characterization of Vitis germplasm collected from the southwestern US and Mexico to expedite Pierce’s disease-resistance breeding. Theor. Appl. Genet. 2018, 131, 1589–1602. [Google Scholar] [CrossRef] [PubMed]
- Coletta-Filho, H.; Pereira, E.; Souza, A.; Takita, M.; Cristofani-Yale, M.; Machado, M. Analysis of resistance to Xylella fastidiosa within a hybrid population of Pera sweet orangex Murcott tangor. Plant Pathol. 2007, 56, 661–668. [Google Scholar] [CrossRef]
- Rodrigues, C.M.; de Souza, A.A.; Takita, M.A.; Kishi, L.T.; Machado, M.A. RNA-Seq analysis of Citrus reticulata in the early stages of Xylella fastidiosa infection reveals auxin-related genes as a defense response. BMC Genom. 2013, 14, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niza, B.; Coletta-Filho, H.; Merfa, M.; Takita, M.; de Souza, A. Differential colonization patterns of Xylella fastidiosa infecting citrus genotypes. Plant Pathol. 2015, 64, 1259–1269. [Google Scholar] [CrossRef]
- Mauricio, F.; Soratto, T.; Diogo, J.; Boscariol-Camargo, R.; De Souza, A.; Coletta-Filho, H.; Silva, J.; Medeiros, A.; Machado, M.; Cristofani-Yaly, M. Analysis of defense-related gene expression in citrus hybrids infected by Xylella fastidiosa. Phytopathology 2019, 109, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Giampetruzzi, A.; Morelli, M.; Saponari, M.; Loconsole, G.; Chiumenti, M.; Boscia, D.; Savino, V.N.; Martelli, G.P.; Saldarelli, P. Transcriptome profiling of two olive cultivars in response to infection by the CoDiRO strain of Xylella fastidiosa subsp. pauca. BMC Genom. 2016, 17, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boscia, D.; Altamura, G.; Ciniero, A.; Di Carolo, M.; Dongiovanni, C.; Fumarola, G.; Giampetruzzi, A.; Greco, P.; La Notte, P.; Loconsole, G. Resistenza a Xylella fastidiosa in diverse cultivar di olivo. L’inform. Agr. 2017, 11, 59–63. [Google Scholar]
- Sabella, E.; Luvisi, A.; Aprile, A.; Negro, C.; Vergine, M.; Nicolì, F.; Miceli, A.; de Bellis, L. Xylella fastidiosa induces differential expression of lignification related-genes and lignin accumulation in tolerant olive trees cv. Leccino. J. Plant Physiol. 2018, 220, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Novelli, S.; Gismondi, A.; Di Marco, G.; Canuti, L.; Nanni, V.; Canini, A. Plant defense factors involved in Olea europaea resistance against Xylella fastidiosa infection. J. Plant Res. 2019, 132, 439–455. [Google Scholar] [CrossRef] [PubMed]
- D’Attoma, G.; Morelli, M.; Saldarelli, P.; Saponari, M.; Giampetruzzi, A.; Boscia, D.; Savino, V.N.; de La Fuente, L.; Cobine, P.A. Ionomic differences between susceptible and resistant olive cultivars infected by Xylella fastidiosa in the outbreak area of salento, italy. Pathogens 2019, 8, 272. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, D.L.; Purcell, A.H. Xylella fastidiosa: Cause of Pierce’s disease of grapevine and other emergent diseases. Plant Dis. 2002, 86, 1056–1066. [Google Scholar] [CrossRef] [Green Version]
- McElrone, A.J.; Sherald, J.L.; Forseth, I.N. Interactive effects of water stress and xylem-limited bacterial infection on the water relations of a host vine. J. Exp. Bot. 2003, 54, 419–430. [Google Scholar] [CrossRef]
- Krivanek, A.; Riaz, S.; Walker, M. Identification and molecular mapping of PdR1, a primary resistance gene to Pierce’s disease in Vitis. Theor. Appl. Genet. 2006, 112, 1125–1131. [Google Scholar] [CrossRef]
- Baccari, C.; Lindow, S.E. Assessment of the process of movement of Xylella fastidiosa within susceptible and resistant grape cultivars. Phytopathology 2011, 101, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, T.R.; James, E.K.; Poole, P.S. The plant microbiome. Genome Biol. 2013, 14, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, G.; Grube, M.; Schloter, M.; Smalla, K. Unraveling the plant microbiome: Looking back and future perspectives. Front. Microbiol. 2014, 5, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlaeppi, K.; Bulgarelli, D. The plant microbiome at work. Mol. Plant Microbe Interact. 2015, 28, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Lucaciu, R.; Pelikan, C.; Gerner, S.; Zioutis, C.; Köstlbacher, S.; Marx, H.; Herbold, C.W.; Schmidt, H.; Rattei, T. A bioinformatics guide to plant microbiome analysis. Front. Plant Sci. 2019, 10, 1313. [Google Scholar] [CrossRef] [Green Version]
- Hillmann, B.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Zhu, Q.; Gohl, D.M.; Beckman, K.B.; Knight, R.; Knights, D. Evaluating the information content of shallow shotgun metagenomics. Msystems 2018, 3, e00069-18. [Google Scholar] [CrossRef] [Green Version]
- Regalado, J.; Lundberg, D.S.; Deusch, O.; Kersten, S.; Karasov, T.L.; Poersch, K.; Shirsekar, G.; Weigel, D. Combining whole genome shotgun sequencing and rDNA amplicon analyses to improve detection of microbe-microbe interaction networks in plant leaves. Nature 2020, 14, 2116–2130. [Google Scholar]
- Dourado, M.N.; Bogas, A.C.; Pomini, A.M.; Andreote, F.D.; Quecine, M.C.; Marsaioli, A.J.; Araújo, W.L. Methylobacterium-plant interaction genes regulated by plant exudate and quorum sensing molecules. Braz. J. Microbiol. 2013, 44, 1331–1339. [Google Scholar] [CrossRef]
- Dourado, M.N.; Santos, D.S.; Nunes, L.R.; Costa de Oliveira, R.L.B.d.; de Oliveira, M.V.; Araujo, W.L. Differential gene expression in Xylella fastidiosa 9a5c during co-cultivation with the endophytic bacterium Methylobacterium mesophilicum SR1. J. Basic Microbiol. 2015, 55, 1357–1366. [Google Scholar] [CrossRef]
- Azevedo, J.L.; Araújo, W.L.; Lacava, P.T. The diversity of citrus endophytic bacteria and their interactions with Xylella fastidiosa and host plants. Genet. Mol. Biol. 2016, 39, 476–491. [Google Scholar] [CrossRef] [Green Version]
- Lacava, P.; Araújo, W.L.; Marcon, J.; Maccheroni, W., Jr.; Azevedo, J.L.D. Interaction between endophytic bacteria from citrus plants and the phytopathogenic bacteria Xylella fastidiosa, causal agent of citrus-variegated chlorosis. Lett. Appl. Microbiol. 2004, 39, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Lacava, P.T.; Li, W.; Araújo, W.L.; Azevedo, J.L.c.; Hartung, J.S. The endophyte Curtobacterium flaccumfaciens reduces symptoms caused by Xylella fastidiosa in Catharanthus roseus. J. Microbiol. 2007, 45, 388. [Google Scholar] [PubMed]
- Deyett, E.; Rolshausen, P.E. Temporal dynamics of the sap microbiome of grapevine under high pierce’s disease pressure. Front. Plant Sci. 2019, 10, 1246. [Google Scholar] [CrossRef]
- Deyett, E.; Roper, M.C.; Ruegger, P.; Yang, J.-I.; Borneman, J.; Rolshausen, P.E. Microbial landscape of the grapevine endosphere in the context of Pierce’s disease. Phytobiomes 2017, 1, 138–149. [Google Scholar] [CrossRef]
- Caliz, J.; Montes-Borrego, M.; Triado-Margarit, X.; Metsis, M.; Landa, B.B.; Casamayor, E.O. Influence of edaphic, climatic, and agronomic factors on the composition and abundance of nitrifying microorganisms in the rhizosphere of commercial olive crops. PLoS ONE 2015, 10, e125787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montes-Borrego, M.; Navas-Cortés, J.A.; Landa, B.B. Linking microbial functional diversity of olive rhizosphere soil to management systems in commercial orchards in southern Spain. Agric. Ecosyst. Environ. 2013, 181, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Fernández-González, A.J.; Cardoni, M.; Cabanás, C.G.-L.; Valverde-Corredor, A.; Villadas, P.J.; Fernández-López, M.; Mercado-Blanco, J. Linking belowground microbial network changes to different tolerance level towards Verticillium wilt of olive. Microbiome 2020, 8, 11. [Google Scholar] [CrossRef]
- Gómez-Lama Cabanás, C.; Legarda, G.; Ruano-Rosa, D.; Pizarro-Tobías, P.; Valverde-Corredor, A.; Niqui, J.L.; Triviño, J.C.; Roca, A.; Mercado-Blanco, J. Indigenous Pseudomonas spp. strains from the olive (Olea europaea L.) rhizosphere as effective biocontrol agents against Verticillium dahliae: From the host roots to the bacterial genomes. Front. Microbiol. 2018, 9, 277. [Google Scholar] [CrossRef] [Green Version]
- Müller, H.; Berg, C.; Landa, B.B.; Auerbach, A.; Moissl-Eichinger, C.; Berg, G. Plant genotype-specific archaeal and bacterial endophytes but similar Bacillus antagonists colonize Mediterranean olive trees. Front. Microbiol. 2015, 6, 138. [Google Scholar] [CrossRef] [Green Version]
- Fausto, C.; Mininni, A.N.; Sofo, A.; Crecchio, C.; Scagliola, M.; Dichio, B.; Xiloyannis, C. Olive orchard microbiome: Characterisation of bacterial communities in soil-plant compartments and their comparison between sustainable and conventional soil management systems. Plant Ecol. Divers. 2018, 11, 597–610. [Google Scholar] [CrossRef]
- Anguita-Maeso, M.; Olivares-García, C.; Haro, C.; Imperial, J.; Navas-Cortés, J.A.; Landa, B.B. Culture-dependent and culture-independent characterization of the olive xylem microbiota: Effect of sap extraction methods. Front. Plant Sci. 2020, 10, 1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, F.; Pereira, J.A.; Bota, P.; Bento, A.; Baptista, P. Fungal endophyte communities in above-and belowground olive tree organs and the effect of season and geographic location on their structures. Fungal Ecol. 2016, 20, 193–201. [Google Scholar] [CrossRef]
- Preto, G.; Martins, F.; Pereira, J.A.; Baptista, P. Fungal community in olive fruits of cultivars with different susceptibilities to anthracnose and selection of isolates to be used as biocontrol agents. Biol. Control 2017, 110, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gomes, T.; Pereira, J.A.; Benhadi, J.; Lino-Neto, T.; Baptista, P. Endophytic and epiphytic phyllosphere fungal communities are shaped by different environmental factors in a Mediterranean ecosystem. Microb. Ecol. 2018, 76, 668–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, T.; Pereira, J.A.; Lino-Neto, T.; Bennett, A.E.; Baptista, P. Bacterial disease induced changes in fungal communities of olive tree twigs depend on host genotype. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Vergine, M.; Meyer, J.B.; Cardinale, M.; Sabella, E.; Hartmann, M.; Cherubini, P.; De Bellis, L.; Luvisi, A. The Xylella fastidiosa-resistant olive cultivar “Leccino” has stable endophytic microbiota during the Olive Quick Decline Syndrome (OQDS). Pathogens 2020, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Tessler, M.; Neumann, J.S.; Afshinnekoo, E.; Pineda, M.; Hersch, R.; Velho, L.F.M.; Segovia, B.T.; Lansac-Toha, F.A.; Lemke, M.; DeSalle, R. Large-scale differences in microbial biodiversity discovery between 16S amplicon and shotgun sequencing. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J. Strategies for taxonomic and functional annotation of metagenomes. In Metagenomics; Elsevier: Amsterdam, The Netherlands, 2018; pp. 55–79. [Google Scholar]
- Breitwieser, F.P.; Lu, J.; Salzberg, S.L. A review of methods and databases for metagenomic classification and assembly. Brief. Bioinform. 2019, 20, 1125–1136. [Google Scholar] [CrossRef]
- Mas-Lloret, J.; Obón-Santacana, M.; Ibáñez-Sanz, G.; Guinó, E.; Pato, M.L.; Rodriguez-Moranta, F.; Mata, A.; García-Rodríguez, A.; Moreno, V.; Pimenoff, V.N. Gut microbiome diversity detected by high-coverage 16S and shotgun sequencing of paired stool and colon sample. Sci. Data 2020, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Cabanillas, D.G.; Zheng, H.; Laliberté, J.-F. Turnip mosaic virus moves systemically through both phloem and xylem as membrane-associated complexes. Plant Physiol. 2015, 167, 1374–1388. [Google Scholar] [CrossRef] [Green Version]
- Richardson, L.G. Viruses on the move in the extracellular space. Plant Physiol. 2019, 180, 1253–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmotte, N.; Knief, C.; Chaffron, S.; Innerebner, G.; Roschitzki, B.; Schlapbach, R.; von Mering, C.; Vorholt, J.A. Community proteogenomics reveals insights into the physiology of phyllosphere bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 16428–16433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelfattah, A.; Nicosia, M.G.L.D.; Cacciola, S.O.; Droby, S.; Schena, L. Metabarcoding analysis of fungal diversity in the phyllosphere and carposphere of olive (Olea europaea). PLoS ONE 2015, 10, e0131069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moissl-Eichinger, C.; Pausan, M.; Taffner, J.; Berg, G.; Bang, C.; Schmitz, R.A. Archaea are interactive components of complex microbiomes. Trends Microbiol. 2018, 26, 70–85. [Google Scholar] [CrossRef]
- Wagner, M.R.; Lundberg, D.S.; Tijana, G.; Tringe, S.G.; Dangl, J.L.; Mitchell-Olds, T. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 2016, 7, 1–15. [Google Scholar] [CrossRef]
- Mina, D.; Pereira, J.A.; Lino-Neto, T.; Baptista, P. Epiphytic and endophytic bacteria on olive tree phyllosphere: Exploring tissue and cultivar effect. Microb. Ecol. 2020, 80, 145–157. [Google Scholar] [CrossRef]
- McNally, L.; Brown, S.P. Building the microbiome in health and disease: Niche construction and social conflict in bacteria. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef]
- Hassani, M.A.; Durán, P.; Hacquard, S. Microbial interactions within the plant holobiont. Microbiome 2018, 6, 58. [Google Scholar] [CrossRef]
- Shen, S.Y.; Fulthorpe, R. Seasonal variation of bacterial endophytes in urban trees. Front. Microbiol. 2015, 6, 427. [Google Scholar] [CrossRef]
- Thongsandee, W.; Matsuda, Y.; Ito, S. Temporal variations in endophytic fungal assemblages of Ginkgo biloba L. J. For. Res. 2012, 17, 213–218. [Google Scholar] [CrossRef]
- Hibbing, M.E.; Fuqua, C.; Parsek, M.R.; Peterson, S.B. Bacterial competition: Surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 2010, 8, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.; Da Costa, M.S. The genus Thermus and related microorganisms. In The Prokaryotes; Springer: New York, NY, USA, 1992; pp. 3745–3753. [Google Scholar]
- Hu, G.; Zhang, L.; Yun, Y.; Peng, Y. Taking insight into the gut microbiota of three spider species: No characteristic symbiont was found corresponding to the special feeding style of spiders. Ecol. Evol. 2019, 9, 8146–8156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lata, R.K.; Divjot, K.; Nath, Y.A. Endophytic microbiomes: Biodiversity, ecological significance and biotechnological applications. Res. J. Biotechnol. 2019, 14, 10. [Google Scholar]
- Sahoo, B.; Ningthoujam, R.; Chaudhuri, S. Isolation and characterization of a lindane degrading bacteria Paracoccus sp. NITDBR1 and evaluation of its plant growth promoting traits. Int. Microbiol. 2019, 22, 155–167. [Google Scholar] [CrossRef]
- Su, J.; Li, L.; Wang, Y.; Ma, X.-X.; Ma, Z.; Peppelenbosch, M.P.; Pan, Q.; Bai, X. Seasonal analysis of the gut microbiota in adult and young captive Asian black bears in northeast China. Anim. Biol. 2020, 70, 109–119. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Weng, C.-C.; Lin, C.-H.; Yang, C.-Y.; Mong, K.-K.T.; Li, Y.-K. Development of a novel engineered antibody targeting Neisseria species. Biotechnol. Lett. 2017, 39, 407–413. [Google Scholar] [CrossRef]
- Belko, J.; Goldmann, D.A.; Macone, A.; ZAIDI, A.K. Clinically significant infections with organisms of the Streptococcus milleri group. Pediatric Infect. Dis. J. 2002, 21, 715–723. [Google Scholar] [CrossRef]
- Prohic, A.; Jovovic Sadikovic, T.; Krupalija-Fazlic, M.; Kuskunovic-Vlahovljak, S. Malassezia species in healthy skin and in dermatological conditions. Int. J. Dermatol. 2016, 55, 494–504. [Google Scholar] [CrossRef]
- Valenzuela-Lopez, N.; Cano-Lira, J.F.; Stchigel, A.M.; Rivero-Menendez, O.; Alastruey-Izquierdo, A.; Guarro, J. Neocucurbitaria keratinophila: An emerging opportunistic fungus causing superficial mycosis in Spain. Med. Mycol. 2019, 57, 733–738. [Google Scholar] [CrossRef]
- Dang, H.X.; Pryor, B.; Peever, T.; Lawrence, C.B. The Alternaria genomes database: A comprehensive resource for a fungal genus comprised of saprophytes, plant pathogens, and allergenic species. BMC Genom. 2015, 16, 239. [Google Scholar] [CrossRef] [Green Version]
- Vaz, A.B.; Mota, R.C.; Bomfim, M.R.Q.; Vieira, M.L.; Zani, C.L.; Rosa, C.A.; Rosa, L.H. Antimicrobial activity of endophytic fungi associated with Orchidaceae in Brazil. Can. J. Microbiol. 2009, 55, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Leal-Dutra, C.A.; Neves, M.A.; Griffith, G.W.; Reck, M.A.; Clasen, L.A.; Dentinger, B.T. Reclassification of Parapterulicium Corner (Pterulaceae, Agaricales), contributions to Lachnocladiaceae and Peniophoraceae (Russulales) and introduction of Baltazaria gen. nov. MycoKeys 2018, 37, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, R.S.; Ariyawansa, H.A.; Singtripop, C.; Li, Y.M.; Yan, J.; Li, X.; Nilthong, S.; Hyde, K.D. A re-assessment of Elsinoaceae (Myriangiales, Dothideomycetes). Phytotaxa 2014, 176, 120–138. [Google Scholar] [CrossRef]
- Zicca, S.; de Bellis, P.; Masiello, M.; Saponari, M.; Saldarelli, P.; Boscia, D.; Sisto, A. Antagonistic activity of olive endophytic bacteria and of Bacillus spp. strains against Xylella fastidiosa. Microbiol. Res. 2020, 236, 126467. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, K.; Makaju, S.; Ibrahim, R.; Missaoui, A. Current progress in nitrogen fixing plants and microbiome research. Plants 2020, 9, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, A.; Cerca, N. Influence of biofilm formation by Gardnerella vaginalis and other anaerobes on bacterial vaginosis. J. Infect. Dis. 2015, 212, 1856–1861. [Google Scholar] [CrossRef] [Green Version]
- Romano-Armada, N.; Yañez-Yazlle, M.F.; Irazusta, V.P.; Rajal, V.B.; Moraga, N.B. Potential of bioremediation and pgp traits in streptomyces as strategies for bio-reclamation of salt-affected soils for agriculture. Pathogens 2020, 9, 117. [Google Scholar] [CrossRef] [Green Version]
- Jeger, M.; Caffier, D.; Candresse, T.; Chatzivassiliou, E.; Dehnen-Schmutz, K.; Gilioli, G.; Grégoire, J.C.; Miret, J.A.J.; MacLeod, A.; Navarro, M.N. Updated pest categorisation of Xylella fastidiosa. EFSA J. 2018, 16. [Google Scholar] [CrossRef] [Green Version]
- European and Mediterranean Plant Protection Organization (EPPO). PM 7/24 (4) Xylella fastidiosa. EPPO Bull. 2019, 49, 175–227. [Google Scholar] [CrossRef] [Green Version]
- Kuklinsky-Sobral, J.; Araújo, W.L.; Mendes, R.; Geraldi, I.O.; Pizzirani-Kleiner, A.A.; Azevedo, J.L. Isolation and characterization of soybean-associated bacteria and their potential for plant growth promotion. Environ. Microbiol. 2004, 6, 1244–1251. [Google Scholar] [CrossRef]
- Loconsole, G.; Potere, O.; Boscia, D.; Altamura, G.; Djelouah, K.; Elbeaino, T.; Frasheri, D.; Lorusso, D.; Palmisano, F.; Pollastro, P. Detection of Xylella fastidiosa in olive trees by molecular and serological methods. J. Plant Pathol. 2014, 96, 7–14. [Google Scholar]
- Harper, S.J.; Ward, L.I.; Clover, G.R.G. Development of LAMP and real-time PCR methods for the rapid detection of Xylella fastidiosa for quarantine and field applications. Phytopathology 2010, 100, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Kraken Database. Available online: https://github.com/mw55309/Kraken_db_install_scripts (accessed on 11 May 2020).
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–812. [Google Scholar] [CrossRef]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome datasets are compositional: And this is not optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [Green Version]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A. Improved bacterial 16S rRNA gene (V4 and V4–5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. Msystems 2016, 1, e00009–e00015. [Google Scholar] [CrossRef] [Green Version]
- Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33) [Software]. Available online: https://github.com/najoshi/sickle (accessed on 11 May 2020).
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ea-Utils: Command-Line Tools for Processing Biological Sequencing Data. Available online: https://expressionanalysis.github.io/ea-utils (accessed on 30 August 2020).
- Albanese, D.; Fontana, P.; De Filippo, C.; Cavalieri, D.; Donati, C. MICCA: A complete and accurate software for taxonomic profiling of metagenomic data. Sci. Rep. 2015, 5, 9743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- SILVA Database. Available online: https://www.arb-silva.de (accessed on 11 May 2020).
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UNITE Database. Available online: https://unite.ut.ee (accessed on 30 August 2020).
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; O’hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package. R Package Version 1.17–4. Available online: http://CRAN.R-project.org/package=vegan (accessed on 1 September 2019).
- The R Project for Statistical Computingfor Statistical Computing. Available online: https://www.r-project.org (accessed on 30 August 2020).
- McCune, B.; Grace, J.B.; Urban, D.L. Analysis of Ecological Communities; MjM Software Design: Gleneden Beach, OR, USA, 2002; Volume 28. [Google Scholar]
- Clarke, K.; Gorley, R. PRIMER v7: User Manual/Tutorial, 3rd ed.; Primer-E Ltd.: Plymouth, UK, 2015. [Google Scholar]
- Dray, S.; Dufour, A.-B. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Season | Cultivar | Sample Name * | Raw Total Reads * | Reads Classified * (%) | Reads Unclassified * (%) | Plants Reads ** (%) | Total Reads Microbes ** | Bacteria *** (%) | Fungi *** (%) | Archaea *** (%) | Viruses *** (%) | Xylella Cq | Xylella CFU/ml | % Xylella/Bacteria Reads | % Average Xylella/ Bacteria |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spring April 2017 | FS17 | FS1-1 | 50,852,732 | 49,963,303 (98.2) | 889,429 (1.75) | 49,808,696 (99.69) | 259,160 | 249,675 (96.34) | 6893 (2.66) | 1735 (0.67) | 857 (0.33) | 28.30 | 34,100 | 0.37 | 2.32 |
| FS1-3 | 46,489,102 | 45,588,956 (98.0) | 900,146 (1.94) | 45,428,720 (99.65) | 261,865 | 251,689 (96.11) | 7435 (2.84) | 1932 (0.74) | 809 (0.31) | 31.20 | 4430 | 0.06 | |||
| FS1-10 | 52,171,471 | 51,356,470 (98.4) | 815,001 (1.56) | 51,202,008 (99.70) | 268,515 | 261,563 (97.41) | 4474 (1.67) | 1623 (0.60) | 855 (0.32) | 27.00 | 84,900 | 0.40 | |||
| FS1-18 | 54,871,547 | 53,962,289 (98.3) | 909,258 (1.66) | 53,845,628 (99.78) | 205,545 | 198,322 (96.49) | 4550 (2.21) | 1801 (0.88) | 872 (0.42) | 30.10 | 9610 | 0.18 | |||
| FS1-43 | 42,746,629 | 42,110,247 (98.5) | 636,382 (1.49) | 42,020,764 (99.79) | 158,999 | 154,117 (96.93) | 2764 (1.74) | 1277 (0.80) | 841 (0.53) | 22.90 | 1,520,000 | 12.87 | |||
| FS1-45 | 39,194,825 | 38,375,168 (97.9) | 819,657 (2.09) | 38,227,260 (99.61) | 240,666 | 230,319 (95.70) | 7825 (3.25 | 1548 (0.64) | 974 (0.40) | 35.10 | 286 | 0.02 | |||
| Kalamata | Kal1-53 | 38,656,227 | 38,101,308 (98.5) | 554,919 (1.44) | 38,028,096 (99.81) | 130,362 | 126,247 (96.84) | 2375 (1.82) | 980 (0.75) | 760 (0.58) | 36.10 | 142 | 0.05 | 8.69 | |
| Kal1-54 | 44,921,146 | 43,839,640 (97.5) | 1,081,506 (2.41) | 43,586,856 (99.42) | 405,349 | 391,036 (96.47) | 11,283 (2.78) | 2135 (0.53) | 895 (0.22) | 28.50 | 29,600 | 0.10 | |||
| Kal1-55 | 39,692,637 | 39,116,264 (98.5) | 576,373 (1.45) | 39,009,116 (99.73) | 178,457 | 172,754 (96.80) | 2438 (1.37) | 1077 (0.60) | 2188 (1.23) | 33.10 | 1170 | 0.02 | |||
| Kal1-57 | 42,445,268 | 41,813,669 (98.5) | 631,599 (1.49) | 41,711,976 (99.76) | 181,331 | 176,926 (97.57) | 2647 (1.46) | 1203 (0.66) | 555 (0.31) | 31.10 | 4760 | 0.22 | |||
| Kal1-65 | 45,136,293 | 44,493,779 (98.5) | 642,514 (1.42) | 44,384,016 (99.75) | 194,201 | 186,304 (95.93) | 2783 (1.43) | 4058 (2.09) | 1056 (0.54) | 34.10 | 577 | 0.04 | |||
| Kal1-89 | 44,631,934 | 43,883,925 (98.3) | 748,009 (1.68) | 43,692,880 (99.56) | 343,522 | 337,244 (98.17) | 3490 (1.02) | 1417 (0.41) | 1371 (0.40) | 20.40 | 8,790,000 | 51.73 | |||
| Autumn November 2018 | FS17 | FS2-1 | 33,462,404 | 32,788,919 (97.9) | 673,485 (2.01) | 32,749,304 (99.88) | 65,201 | 59,793 (91.71) | 3284 (5.04) | 989 (1.52) | 1135 (1.74) | 35.10 | 286 | 0.95 | 31.48 |
| FS2-3 | 32,092,733 | 31,396,019 (97.8) | 696,714 (2.17) | 31,371,784 (99.92) | 38,374 | 34,366 (89.56) | 2222 (5.79) | 770 (2.01) | 1016 (2.65) | 29.96 | 10,600 | 4.89 | |||
| FS2-10 | 33,465,333 | 32,683,674 (97.6) | 781,659 (2.34) | 32,631,210 (99.84) | 90,038 | 84,585 (93.94) | 3636 (4.04) | 934 (1.04) | 883 (0.98) | 25.50 | 244,000 | 46.82 | |||
| FS2-18 | 32,789,969 | 32,080,877 (97.8) | 709,092 (2.16) | 32,029,304 (99.84) | 86,409 | 79,573 (92.09) | 4305 (4.98) | 957 (1.11) | 1574 (1.82) | 31.10 | 4760 | 15.42 | |||
| FS2-43 | 42,453,647 | 41,659,140 (98.1) | 794,507 (1.87) | 41,142,644 (98.76) | 938,782 | 923,823 (98.41) | 10,798 (1.15) | 1082 (0.12) | 3079 (0.33) | 24.57 | 469,000 | 83.11 | |||
| FS2-45 | 31,955,583 | 31,276,525 (97.8) | 679,058 (2.13) | 31,230,648 (99.85) | 77,562 | 72,841 (93.91) | 2862 (3.69) | 857 (1.10) | 1002 (1.29) | 26.60 | 112,000 | 37.68 | |||
| Kalamata | Kal2-53 | 48,745,550 | 47,873,736 (98.2) | 871,814 (1.79) | 47,214,900 (98.62) | 1,198,439 | 1,188,286 (99.15) | 5768 (0.48) | 1299 (0.11) | 3086 (0.26) | 24.20 | 608,000 | 90.05 | 52.67 | |
| Kal2-54 | 33,545,651 | 32,875,120 (98.0) | 670,531 (2.00) | 32,841,592 (99.90) | 56,343 | 51,642 (91.66) | 2265 (4.02) | 738 (1.31) | 1698 (3.01) | 21.50 | 4,060,000 | 34.41 | |||
| Kal2-55 | 38,914,006 | 38,071,869 (97.8) | 842,137 (2.16) | 37,429,772 (98.31) | 1,162,983 | 1,152,955 (99.14) | 6234 (0.54) | 1318 (0.11) | 2476 (0.21) | 22.10 | 2,660,000 | 88.65 | |||
| Kal2-57 | 30,275,514 | 29,663,797 (97.9) | 611,717 (2.02) | 29,635,620 (99.91) | 45,484 | 40,442 (88.91) | 2912 (6.40) | 902 (1.98) | 1228 (2.70) | 21.80 | 3,290,000 | 0.45 | |||
| Kal2-65 | 29,620,002 | 29,066,308 (98.1) | 553,694 (1.87) | 29,033,788 (99.89) | 52,570 | 47,964 (91.24) | 2148 (4.09) | 771 (1.47) | 1687 (3.21) | 22.20 | 2,480,000 | 19.94 | |||
| Kal2-89 | 25,255,482 | 24,756,720 (98.0) | 498,762 (1.97) | 24,597,096 (99.36) | 287,446 | 281,221 (97.83) | 2430 (0.85) | 729 (0.25) | 3066 (1.07) | 22.40 | 2,150,000 | 82.53 |
| Season | Cv. | Sample Name | Total Raw Reads | Sequences Classified (%) | Sequences Unclassified (%) | Plant Sequences (%) | Bacteria (%) | Fungi (%) | % Xylella/ Bacteria | % Average Xylella/Bacteria | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 16S | ITS | 16S | ITS | 16S | ITS | 16S | |||||||
| Spring April 2017 | FS17 | FS1-1 | 13,604 | 110,867 | 13,137 (96.6) | 88,348 (79.7) | 467 (3.4) | 22,519 (20.3) | 12,753 (97.1) | 384 (2.9) | 88,338 (100) | 0.8 | 10.3 |
| FS1-3 | 14,415 | 25,801 | 13,859 (96.1) | 17,759 (68.8) | 556 (3.9) | 8042 (31.2) | 13,386 (96.6) | 473 (3.4) | 17,759 (100) | 0.4 | |||
| FS1-10 | 14,930 | 77,958 | 14,440 (96.7) | 59,012 (75.7) | 490 (3.3) | 18,946 (24.3) | 14,253 (98.7) | 187 (1.3) | 59,012 (100) | 1.6 | |||
| FS1-18 | 12,303 | 62,221 | 11,899 (96.7) | 31,172 (50.1) | 404 (3.3) | 31,049 (49.9) | 11,826 (99.4) | 73 (0.6) | 31,172 (100) | 2.7 | |||
| FS1-43 | 10,505 | 64,245 | 10,259 (97.7) | 34,647 (53.9) | 246 (2.3) | 29,598 (46.1) | 10,125 (98.7) | 134 (1.3) | 34,647 (100) | 56.0 | |||
| FS1-45 | 12,735 | 90,246 | 12,250 (96.2) | 63,942 (70.9) | 485 (3.8) | 26,304 (29.1) | 11,772 (96.1) | 478 (3.9) | 63,942 (100) | 0.0 | |||
| Kalamata | Kal1-53 | 11,780 | 22,772 | 11,335 (96.2) | 15,618 (68.6) | 445 (3.8) | 7154 (31.4) | 11,296 (99.7) | 39 (0.3) | 15,618 (100) | 0.0 | 13.2 | |
| Kal1-54 | 15,279 | 26,9041 | 14,598 (95.5) | 174,764 (65) | 681 (4.5) | 94,277 (35.0) | 13,807 (94.6) | 791 (5.4) | 174,761 (100) | 0.0 | |||
| Kal1-55 | 12,827 | 26,740 | 12,407 (96.7) | 18,577 (69.5) | 420 (3.3) | 8163 (30.5) | 12,156 (98.0) | 251 (2.0) | 18,577 (100) | 0.0 | |||
| Kal1-57 | 9529 | 18,716 | 9259 (97.2) | 12,313 (65.8) | 270 (2.8) | 6403 (34.2) | 9222 (99.6) | 37 (0.4) | 12,313 (100) | 0.0 | |||
| Kal1-65 | 10,275 | 111,806 | 9936 (96.7) | 67,804 (60.6) | 339 (3.3) | 44,002 (39.4) | 9897 (99.6) | 39 (0.4) | 67,804 (100) | 0.0 | |||
| Kal1-89 | 12,140 | 17,612 | 11,612 (95.7) | 15,966 (90.7) | 528 (4.3) | 16,746 (95.1) | 10,808 (93.1) | 804 (6.9) | 15,966 (100) | 79.2 | |||
| Autumn November 2018 | FS17 | FS2-1 | 151,666 | 88,600 | 151,475 (99.9) | 78,642 (88.8) | 191 (0.1) | 9958 (11.2) | 15,1200 (99.8) | 275 (0.2) | 78,642 (100) | 0.0 | 20.5 |
| FS2-3 | 246,844 | 209,701 | 246,317 (99.8) | 188,116 (89.7) | 527 (0.2) | 21,848 (10.4) | 246,201 (99.9) | 116 (0.05) | 187,853 (99.9) | 6.9 | |||
| FS2-10 | 195,548 | 219,077 | 195,319 (99.9) | 203,699 (93.0) | 229 (0.1) | 15,975 (7.3) | 195,168 (99.9) | 150 (0.1) | 203,102 (99.7) | 46.0 | |||
| FS2-18 | 175,303 | 91,227 | 175,011 (99.8) | 73,264 (80.3) | 292 (0.2) | 18,023 (19.8) | 174,853 (99.9) | 158 (0.1) | 73,204 (99.9) | 12.7 | |||
| FS2-43 | 92,265 | 103,006 | 92,257 (100) | 97,801 (94.9) | 8 (0) | 5205 (5.1) | 92,062 (99.8) | 195 (0.2) | 97,801 (100) | 44.1 | |||
| FS2-45 | 131,843 | 65,534 | 131,659 (99.9) | 23,987 (36.6) | 184 (0.1) | 41,709 (63.6) | 131,416 (99.8) | 243 (0.2) | 23,825 (99.3) | 13.2 | |||
| Kalamata | Kal2-53 | 168,034 | 54,316 | 167,912 (99.9) | 23,089 (42.5) | 122 (0.1) | 31,227 (57.5) | 167,418 (99.7) | 494 (0.3) | 23,089 (100) | 77.9 | 45.0 | |
| Kal2-54 | 243,771 | 69,589 | 243,266 (99.8) | 58,880 (84.6) | 505 (0.2) | 10,728 (15.4) | 243,189 (99.9) | 77 (0.03) | 58,861 (100) | 39.0 | |||
| Kal2-55 | 121,959 | 199,625 | 121,878 (99.9) | 143,056 (71.7) | 81 (0.1) | 56,569 (28.3) | 121,179 (99.4) | 699 (0.6) | 143,056 (100) | 81.4 | |||
| Kal2-57 | 162,670 | 47,373 | 162,263 (99.7) | 47,368 (100) | 407 (0.3) | 244 (0.5) | 162,224 (99.9) | 39 (0.02) | 47,129 (99.5) | 0.0 | |||
| Kal2-65 | 69,232 | 78,285 | 69,182 (99.9) | 39,009 (49.8) | 50 (0.1) | 39,276 (50.2) | 69,143 (99.9) | 39 (0.1) | 39,009 (100) | 15.4 | |||
| Kal2-89 | 155,471 | 84,249 | 155,407 (100) | 81,875 (97.2) | 64 (0) | 2374 (2.8) | 155,219 (99.9) | 188 (0.1) | 81,875 (100) | 56.4 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giampetruzzi, A.; Baptista, P.; Morelli, M.; Cameirão, C.; Lino Neto, T.; Costa, D.; D’Attoma, G.; Abou Kubaa, R.; Altamura, G.; Saponari, M.; et al. Differences in the Endophytic Microbiome of Olive Cultivars Infected by Xylella fastidiosa across Seasons. Pathogens 2020, 9, 723. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9090723
Giampetruzzi A, Baptista P, Morelli M, Cameirão C, Lino Neto T, Costa D, D’Attoma G, Abou Kubaa R, Altamura G, Saponari M, et al. Differences in the Endophytic Microbiome of Olive Cultivars Infected by Xylella fastidiosa across Seasons. Pathogens. 2020; 9(9):723. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9090723
Chicago/Turabian StyleGiampetruzzi, Annalisa, Paula Baptista, Massimiliano Morelli, Cristina Cameirão, Teresa Lino Neto, Daniela Costa, Giusy D’Attoma, Raied Abou Kubaa, Giuseppe Altamura, Maria Saponari, and et al. 2020. "Differences in the Endophytic Microbiome of Olive Cultivars Infected by Xylella fastidiosa across Seasons" Pathogens 9, no. 9: 723. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9090723