
Gut Microbiota Associated with Clinical Relapse in Patients with Quiescent Ulcerative Colitis

 , , , , , , , ,
, , , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics
2.2. Patients
2.3. Sample Collection and DNA Extraction
2.4. Sequencing of 16S rRNA Gene
2.5. Microbiome Analysis
2.6. Predictive Functional Profiling of Gut Microbial Communities
2.7. Linear Discriminant Analysis Effect Size (LEfSe) Analysis
3. Results
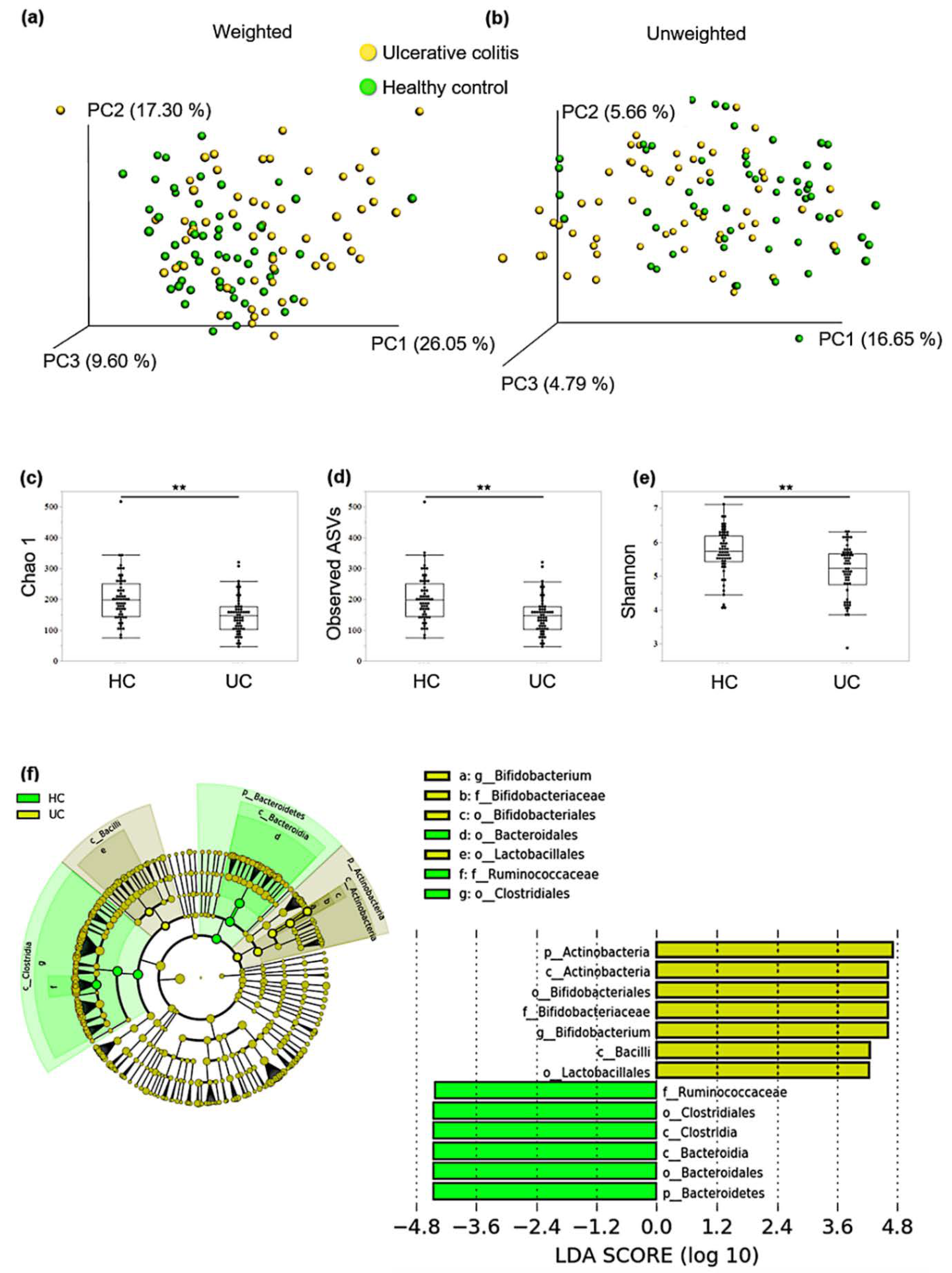
3.1. Comparing the Composition of Gut Microbiota between Patients with qUC and HCs
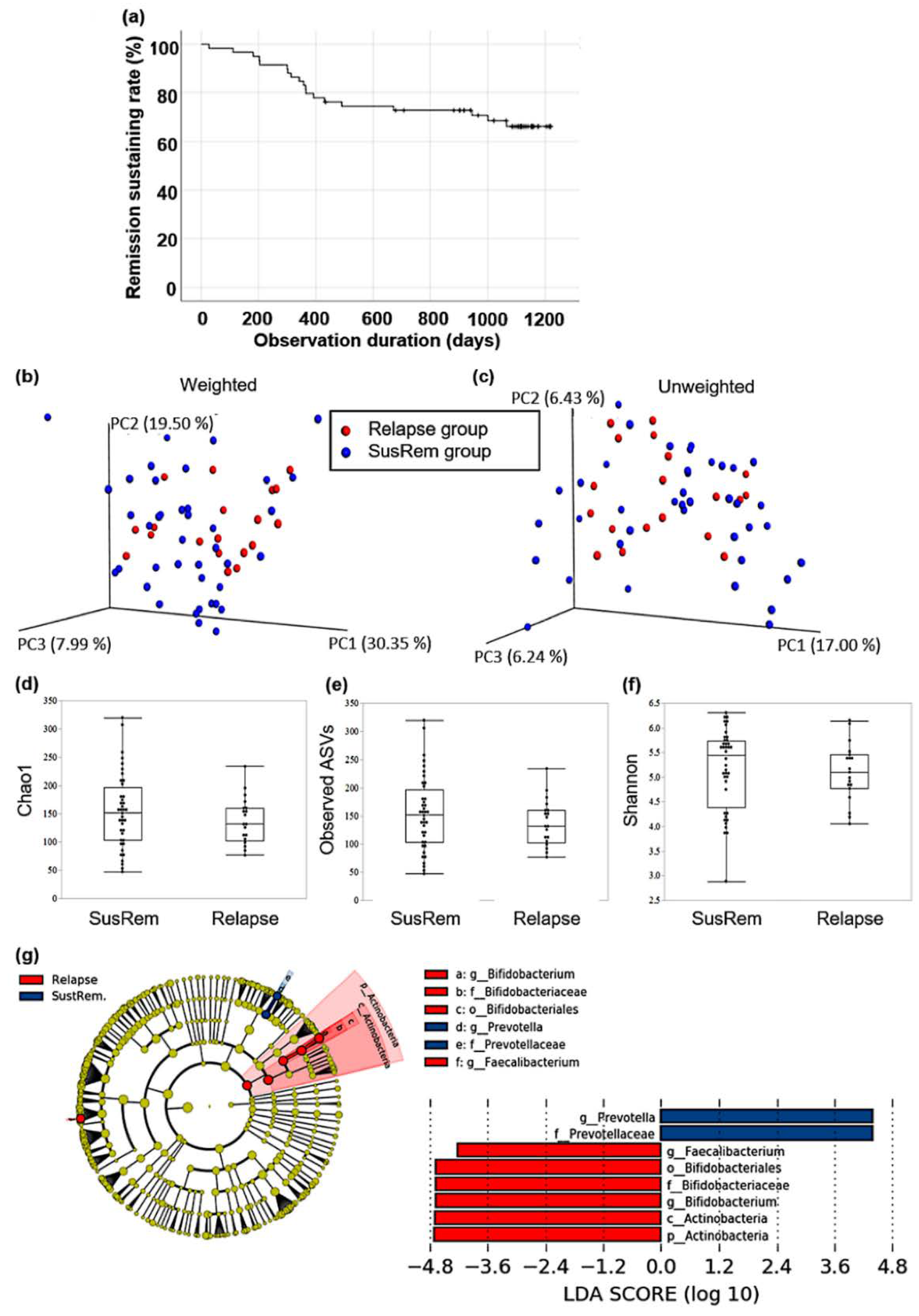
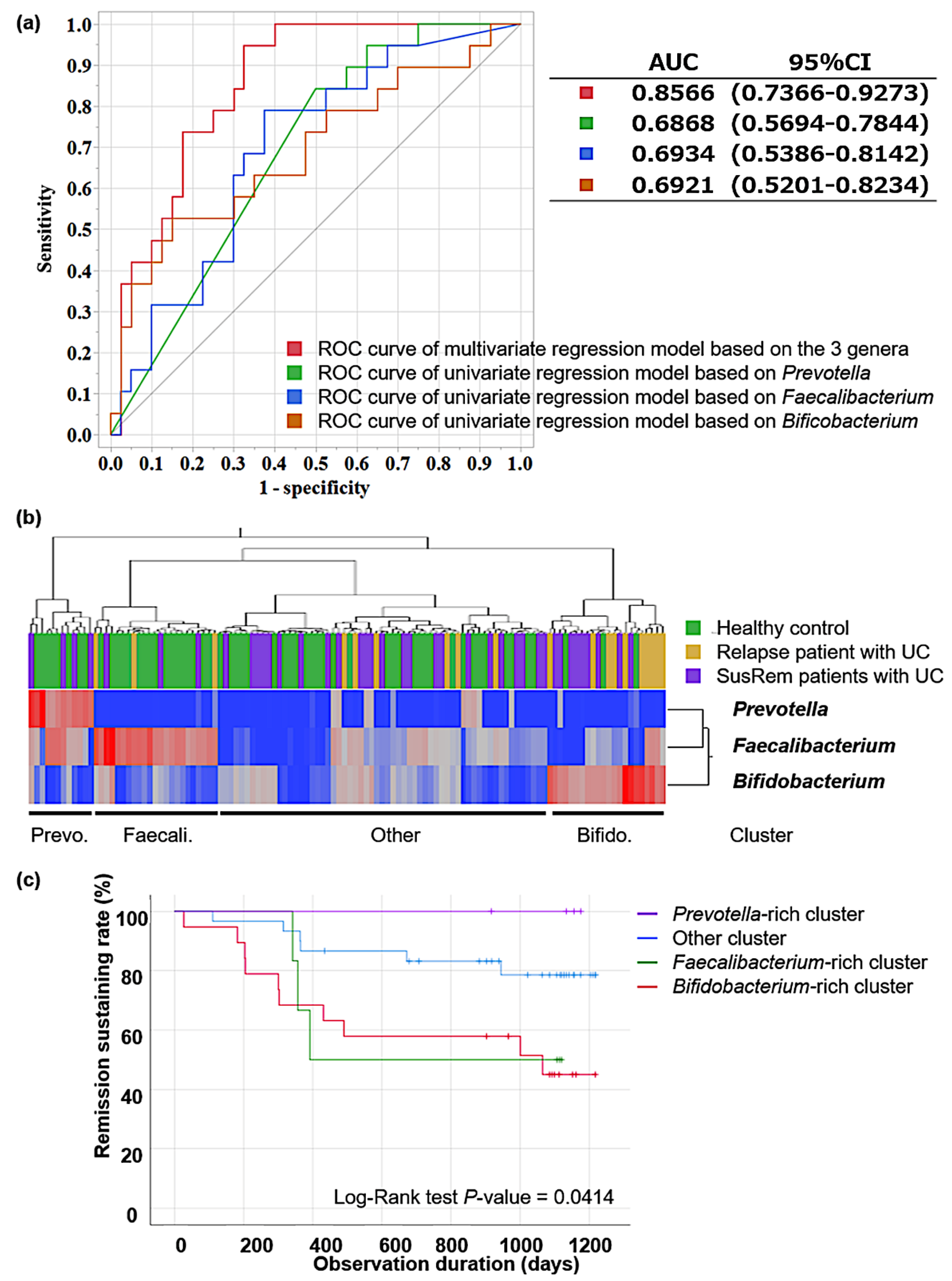
3.2. Fecal Microbiota Associated with Relapse in qUC Patients
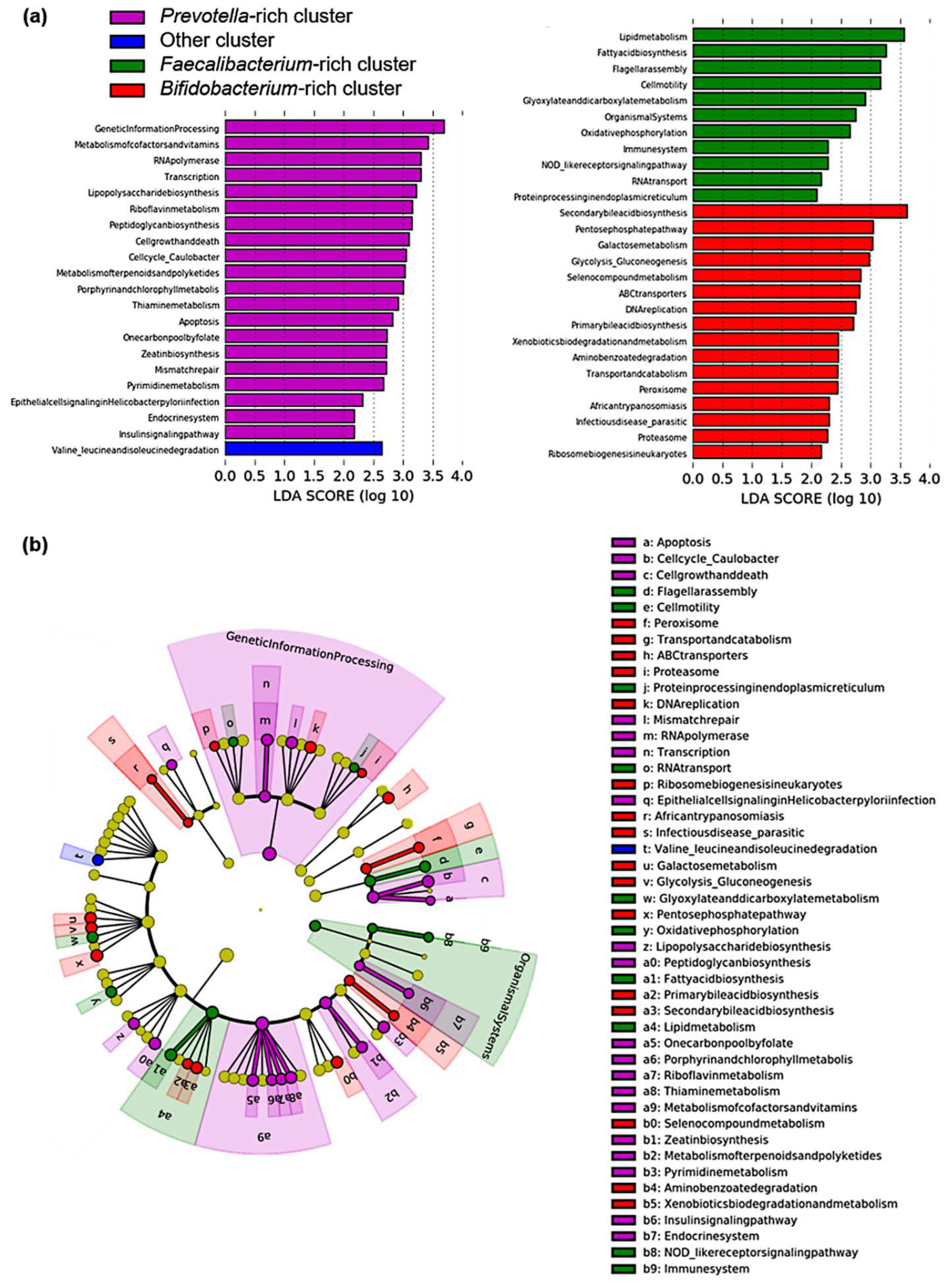
3.3. Pathway Analysis in Identified Clusters
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, T.; Naito, Y.; Inoue, R.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Tsuchiya, S.; Dohi, O.; Yoshida, N.; Kamada, K.; et al. Differences in Gut Microbiota Associated with Age, Sex, and Stool Consistency in Healthy Japanese Subjects. J. Gastroenterol. 2019, 54, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, D.; Moran, C.; Shanahan, F. The Microbiota in Inflammatory Bowel Disease. J. Gastroenterol. 2015, 50, 495–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut Microbiota in the Pathogenesis of Inflammatory Bowel Disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-Phylogenetic Characterization of Microbial Community Imbalances in Human Inflammatory Bowel Diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-Throughput Clone Library Analysis of the Mucosa-Associated Microbiota Reveals Dysbiosis and Differences between Inflamed and Non-Inflamed Regions of the Intestine in Inflammatory Bowel Disease. BMC Microbiol. 2011, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced Diversity of Faecal Microbiota in Crohn’s Disease Revealed by a Metagenomic Approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Michail, S.; Durbin, M.; Turner, D.; Griffiths, A.M.; Mack, D.R.; Hyams, J.; Leleiko, N.; Kenche, H.; Stolfi, A.; Wine, E. Alterations in the Gut Microbiome of Children with Severe Ulcerative Colitis. Inflamm. Bowel Dis. 2012, 18, 1799–1808. [Google Scholar] [CrossRef]
- Imhann, F.; Vich Vila, A.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; van Dullemen, H.M.; et al. Interplay of Host Genetics and Gut Microbiota Underlying the Onset and Clinical Presentation of Inflammatory Bowel Disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef]
- Santoru, M.L.; Piras, C.; Murgia, A.; Palmas, V.; Camboni, T.; Liggi, S.; Ibba, I.; Lai, M.A.; Orrù, S.; Blois, S.; et al. Cross Sectional Evaluation of the Gut-Microbiome Metabolome Axis in an Italian Cohort of IBD Patients. Sci. Rep. 2017, 7, 9523. [Google Scholar] [CrossRef]
- Eun, C.S.; Kwak, M.-J.; Han, D.S.; Lee, A.R.; Park, D.I.; Yang, S.-K.; Kim, Y.S.; Kim, J.F. Does the Intestinal Microbial Community of Korean Crohn’s Disease Patients Differ from That of Western Patients? BMC Gastroenterol. 2016, 16, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liguori, G.; Lamas, B.; Richard, M.L.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Fungal Dysbiosis in Mucosa-Associated Microbiota of Crohn’s Disease Patients. J. Crohn’s Colitis 2016, 10, 296–305. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, X.; Ghozlane, A.; Hu, H.; Li, X.; Xiao, Y.; Li, D.; Yu, G.; Zhang, T. Characteristics of Faecal Microbiota in Paediatric Crohn’s Disease and Their Dynamic Changes During Infliximab Therapy. J. Crohn’s Colitis 2018, 12, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Willing, B.P.; Dicksved, J.; Halfvarson, J.; Andersson, A.F.; Lucio, M.; Zheng, Z.; Järnerot, G.; Tysk, C.; Jansson, J.K.; Engstrand, L. A Pyrosequencing Study in Twins Shows That Gastrointestinal Microbial Profiles Vary With Inflammatory Bowel Disease Phenotypes. Gastroenterology 2010, 139, 1844–1854.e1. [Google Scholar] [CrossRef] [PubMed]
- Pittayanon, R.; Lau, J.T.; Leontiadis, G.I.; Tse, F.; Yuan, Y.; Surette, M.; Moayyedi, P. Differences in Gut Microbiota in Patients With vs. Without Inflammatory Bowel Diseases: A Systematic Review. Gastroenterology 2020, 158, 930–946.e1. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G.; Xia, B.; Forbes, B.A. Increased Proportions of Bifidobacterium and the Lactobacillus Group and Loss of Butyrate-Producing Bacteria in Inflammatory Bowel Disease. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Prosberg, M.; Bendtsen, F.; Vind, I.; Petersen, A.M.; Gluud, L.L. The Association between the Gut Microbiota and the Inflammatory Bowel Disease Activity: A Systematic Review and Meta-Analysis. Scand. J. Gastroenterol. 2016, 51, 1407–1415. [Google Scholar] [CrossRef]
- Sun, Y.; Li, L.; Xia, Y.; Li, W.; Wang, K.; Wang, L.; Miao, Y.; Ma, S. The Gut Microbiota Heterogeneity and Assembly Changes Associated with the IBD. Sci. Rep. 2019, 9, 440. [Google Scholar] [CrossRef]
- Lichtiger, S.; Present, D.H.; Kornbluth, A.; Gelernt, I.; Bauer, J.; Galler, G.; Michelassi, F.; Hanauer, S. Cyclosporine in Severe Ulcerative Colitis Refractory to Steroid Therapy. N. Engl. J. Med. 1994, 330, 1841–1845. [Google Scholar] [CrossRef]
- Inoue, R.; Ohue-Kitano, R.; Tsukahara, T.; Tanaka, M.; Masuda, S.; Inoue, T.; Yamakage, H.; Kusakabe, T.; Hasegawa, K.; Shimatsu, A.; et al. Prediction of Functional Profiles of Gut Microbiota from 16S RRNA Metagenomic Data Provides a More Robust Evaluation of Gut Dysbiosis Occurring in Japanese Type 2 Diabetic Patients. J. Clin. Biochem. Nutr. 2017, 61, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Nishino, K.; Nishida, A.; Inoue, R.; Kawada, Y.; Ohno, M.; Sakai, S.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Kawahara, M.; et al. Analysis of Endoscopic Brush Samples Identified Mucosa-Associated Dysbiosis in Inflammatory Bowel Disease. J. Gastroenterol. 2018, 53, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, T.; Naito, Y.; Inoue, R.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Tsuchiya, S.; Okayama, T.; Dohi, O.; Yoshida, N.; et al. The Influence of Long-Term Use of Proton Pump Inhibitors on the Gut Microbiota: An Age-Sex-Matched Case-Control Study. J. Clin. Biochem. Nutr. 2018, 62, 100–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive Functional Profiling of Microbial Communities Using 16S RRNA Marker Gene Sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Murray, A.; Nguyen, T.M.; Parker, C.E.; Feagan, B.G.; MacDonald, J.K. Oral 5-Aminosalicylic Acid for Maintenance of Remission in Ulcerative Colitis. Cochrane Database Syst. Rev. 2020, 2020, CD000544. [Google Scholar] [CrossRef]
- Yamamoto, T.; Shiraki, M.; Bamba, T.; Umegae, S.; Matsumoto, K. Fecal Calprotectin and Lactoferrin as Predictors of Relapse in Patients with Quiescent Ulcerative Colitis during Maintenance Therapy. Int. J. Colorectal Dis. 2014, 29, 485–491. [Google Scholar] [CrossRef]
- Nishihara, Y.; Ogino, H.; Tanaka, M.; Ihara, E.; Fukaura, K.; Nishioka, K.; Chinen, T.; Tanaka, Y.; Nakayama, J.; Kang, D.; et al. Mucosa-Associated Gut Microbiota Reflects Clinical Course of Ulcerative Colitis. Sci. Rep. 2021, 11, 13743. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of Diet in Shaping Gut Microbiota Revealed by a Comparative Study in Children from Europe and Rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- von Martels, J.Z.H.; Bourgonje, A.R.; Klaassen, M.A.Y.; Alkhalifah, H.A.A.; Sadaghian Sadabad, M.; Vich Vila, A.; Gacesa, R.; Gabriëls, R.Y.; Steinert, R.E.; Jansen, B.H.; et al. Riboflavin Supplementation in Patients with Crohn’s Disease [the RISE-UP Study]. J. Crohn’s Colitis 2020, 14, 595–607. [Google Scholar] [CrossRef]
- Toffano, R.; Hillesheim, E.; Mathias, M.; Coelho-Landell, C.; Salomão, R.; Almada, M.; Camarneiro, J.; Barros, T.; Camelo-Junior, J.; Rezzi, S.; et al. Validation of the Brazilian Healthy Eating Index-Revised Using Biomarkers in Children and Adolescents. Nutrients 2018, 10, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, J.P.; Riis, L.B.; Malham, M.; Høgdall, E.; Langholz, E.; Nielsen, B.S. MicroRNA Biomarkers in IBD-Differential Diagnosis and Prediction of Colitis-Associated Cancer. Int. J. Mol. Sci. 2020, 21, 7893. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-K.; Kim, H.-N.; Choi, C.H.; Im, J.P.; Cha, J.M.; Eun, C.S.; Kim, T.-O.; Kang, S.-B.; Bang, K.B.; Kim, H.G.; et al. Differentially Abundant Bacterial Taxa Associated with Prognostic Variables of Crohn’s Disease: Results from the IMPACT Study. J. Clin. Med. 2020, 9, 1748. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Xu, H.; Chen, S.; He, J.; Zhou, Y.; Nie, Y. Systematic Review and Meta-Analysis of the Role of Faecalibacterium Prausnitzii Alteration in Inflammatory Bowel Disease. J. Gastroenterol. Hepatol. 2021, 36, 320–328. [Google Scholar] [CrossRef]
- Duncan, S.H.; Hold, G.L.; Harmsen, H.J.M.; Stewart, C.S.; Flint, H.J. Growth Requirements and Fermentation Products of Fusobacterium Prausnitzii, and a Proposal to Reclassify It as Faecalibacterium Prausnitzii Gen. Nov., Comb. Nov. Int. J. Syst. Evol. Microbiol. 2002, 52, 2141–2146. [Google Scholar] [CrossRef] [Green Version]
- Gill, P.A.; van Zelm, M.C.; Muir, J.G.; Gibson, P.R. Review Article: Short Chain Fatty Acids as Potential Therapeutic Agents in Human Gastrointestinal and Inflammatory Disorders. Aliment. Pharmacol. Ther. 2018, 48, 15–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcenilla, A.; Pryde, S.E.; Martin, J.C.; Duncan, S.H.; Stewart, C.S.; Henderson, C.; Flint, H.J. Phylogenetic Relationships of Butyrate-Producing Bacteria from the Human Gut. Appl. Environ. Microbiol. 2000, 66, 1654–1661. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Huo, D.; Peng, Q.; Pan, Y.; Jiang, S.; Liu, B.; Zhang, J. Lactobacillus Plantarum HNU082-Derived Improvements in the Intestinal Microbiome Prevent the Development of Hyperlipidaemia. Food Funct. 2017, 8, 4508–4516. [Google Scholar] [CrossRef]
- Song, H.; Yoo, Y.; Hwang, J.; Na, Y.-C.; Kim, H.S. Faecalibacterium Prausnitzii Subspecies–Level Dysbiosis in the Human Gut Microbiome Underlying Atopic Dermatitis. J. Allergy Clin. Immunol. 2016, 137, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Climent, E.; Martinez-Blanch, J.F.; Llobregat, L.; Ruzafa-Costas, B.; Carrión-Gutiérrez, M.Á.; Ramírez-Boscá, A.; Prieto-Merino, D.; Genovés, S.; Codoñer, F.M.; Ramón, D.; et al. Changes in Gut Microbiota Correlates with Response to Treatment with Probiotics in Patients with Atopic Dermatitis. A Post Hoc Analysis of a Clinical Trial. Microorganisms 2021, 9, 854. [Google Scholar] [CrossRef]
- Cox, S.R.; Lindsay, J.O.; Fromentin, S.; Stagg, A.J.; McCarthy, N.E.; Galleron, N.; Ibraim, S.B.; Roume, H.; Levenez, F.; Pons, N.; et al. Effects of Low FODMAP Diet on Symptoms, Fecal Microbiome, and Markers of Inflammation in Patients with Quiescent Inflammatory Bowel Disease in a Randomized Trial. Gastroenterology 2020, 158, 176–188.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, M.; Izquierdo, E.; Ennahar, S.; Sanz, Y. Differential Immunomodulatory Properties of Bifidobacterium Logum Strains: Relevance to Probiotic Selection and Clinical Applications. Clin. Exp. Immunol. 2007, 150, 531–538. [Google Scholar] [CrossRef]
- Jakubczyk, D.; Leszczyńska, K.; Górska, S. The Effectiveness of Probiotics in the Treatment of Inflammatory Bowel Disease (IBD)—A Critical Review. Nutrients 2020, 12, 1973. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile Salt Biotransformations by Human Intestinal Bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizuka, S. Variation of Molecular Species of Bile Acids: Structure, Metabolism, and Physiological Influence. Kagaku Seibutsu 2014, 52, 301–306. [Google Scholar] [CrossRef]
- Kurdi, P.; Tanaka, H.; van Veen, H.W.; Asano, K.; Tomita, F.; Yokota, A. Cholic Acid Accumulation and Its Diminution by Short-Chain Fatty Acids in Bifidobacteria. Microbiology 2003, 149, 2031–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, B. Bile Acid-Microbiota Crosstalk in Gastrointestinal Inflammation and Carcinogenesis: A Role for Bifidobacteria and Lactobacilli? Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 205. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Siles, M.; Duncan, S.H.; Garcia-Gil, L.J.; Martinez-Medina, M. Faecalibacterium Prausnitzii: From Microbiology to Diagnostics and Prognostics. ISME J. 2017, 11, 841–852. [Google Scholar] [CrossRef]
- Nishijima, S.; Suda, W.; Oshima, K.; Kim, S.-W.; Hirose, Y.; Morita, H.; Hattori, M. The Gut Microbiome of Healthy Japanese and Its Microbial and Functional Uniqueness. DNA Res. 2016, 23, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Loftus, E.V. Clinical Epidemiology of Inflammatory Bowel Disease: Incidence, Prevalence, and Environmental Influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut Microbiota and IBD: Causation or Correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwagi, S.; Naito, Y.; Inoue, R.; Takagi, T.; Nakano, T.; Inada, Y.; Fukui, A.; Katada, K.; Mizushima, K.; Kamada, K.; et al. Mucosa-Associated Microbiota in the Gastrointestinal Tract of Healthy Japanese Subjects. Digestion 2020, 101, 107–120. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HC (n = 59) | qUC (n = 59) | p-Value | |
|---|---|---|---|
| Sex, n (%) | 1 | ||
| Male | 32 (54.2) | 32 (54.2) | |
| Female | 27 (42.8) | 27 (42.8) | |
| Age (years) | 1 | ||
| Mean ± SD | 50.5 ± 15.1 | 50.4 ± 15.5 | |
| Median (min, max) | 51.0 (24, 82) | 51 (21, 84) |
| Healthy Controls | Patients with Quiescent Ulcerative Colitis | |||
|---|---|---|---|---|
| Genus | Mean | Genus | Mean | |
| 1 | Bacteroides | 17.77% | Bacteroides | 15.71% |
| 2 | Faecalibacterium | 8.73% | Bifidobacterium | 14.82% |
| 3 | Bifidobacterium | 6.55% | Faecalibacterium | 6.58% |
| 4 | Blautia | 5.87% | Blautia | 6.08% |
| 5 | Lachnospiraceae; Other | 5.15% | Lachnospiraceae; Other | 4.32% |
| 6 | Enterobacteriaceae; Other | 3.86% | Roseburia | 3.67% |
| 7 | Prevotella | 3.48% | Enterobacteriaceae; Other | 2.98% |
| 8 | Ruminococcaceae; Ruminococcus | 3.44% | Collinsella | 2.91% |
| 9 | Coprococcus | 2.80% | Streptococcus | 2.91% |
| 10 | Roseburia | 2.49% | Ruminococcus | 2.72% |
| SusRem | Relapse | p-Value | |
|---|---|---|---|
| (n = 40) | (n = 19) | ||
| Sex, n (%) | 0.69 | ||
| Male | 21 (52.5) | 11 (57.9) | |
| Female | 19 (47.5) | 8 (42.1) | |
| Age (years) | 0.99 | ||
| Mean ± SD | 50.4 ± 16.2 | 50.3 ± 14.3 | |
| Median (min, max) | 50.5 (21, 84) | 51 (32, 77) | |
| Body mass index | 0.77 | ||
| Mean ± SD | 21.8 ± 3.4 | 22.1 ± 4.0 | |
| Median (IQR) | 21.43 (14.8, 29.6) | 22.2 (16.1, 29.7) | |
| UC disease duration (months) | 0.12 | ||
| Mean ± SD | 209.6 ± 21.8 | 156.4 ± 31.6 | |
| Median (min, max) | 178 (18, 31) | 126 (31, 349) | |
| Observation period until relapse (days) | - | ||
| Mean ± SD | 1058.9 ± 164.1 | 424.8 ± 49.0 | |
| Median (min, max) | 1115 (434, 1218) | 357 (27, 1065) | |
| Extent of disease, n (%) | 0.02 | ||
| Ulcerative proctitis | 10 (25.0) | 0 (0.0) | |
| Left-sided colitis | 7 (17.5) | 8 (42.1) | |
| Panulcerative colitis | 23 (57.5) | 11 (57.9) | |
| Medication, n (%) | |||
| 5-Aminosalicylicacid compounds | 33 (82.5) | 18 (94.7) | 0.19 |
| Sulfasalazine | 9 (27.3) | 0 (0.0) | |
| Pentasa® | 12 (36.4) | 10 (55.6) | |
| Asacol® | 11 (33.3) | 6 (33.3) | |
| Rialda® | 1 (3.0) | 2 (11.1) | |
| Azathioprine | 6 (15.0) | 5 (26.3) | 0.3 |
| Infliximab | 2 (5.0) | 2 (10.5) | 0.43 |
| Adalimumab | 1 (2.5) | 0 (0.0) | 0.38 |
| Tacrolimus | 1 (2.5) | 1 (5.2) | 0.6 |
| Systemic steroid | 0 (0.0) | 0 (0.0) | - |
| Local steroid | 1 (2.5) | 1 (5.2) | 0.6 |
| Probiotics, n (%) | |||
| Intake of probiotics | 19 (47.5) | 11 (57.9) | 0.46 |
| Biofermin | 4 (21.1) | 3 (27.3) | |
| Lac-B | 6 (31.6) | 2 (18.2) | |
| Bio-three | 0 (0.0) | 1 (9.1) | |
| Miya-BM | 9 (47.4) | 7 (63.6) |
| Prevotella-Rich Cluster | Faecalibacterium-Rich Cluster | Bifidobacterium-Rich Cluster | Other Cluster | |||||
|---|---|---|---|---|---|---|---|---|
| Genus | Mean | Genus | Mean | Genus | Mean | Genus | Mean | |
| 1 | Prevotella | 22.02% | Bacteroides | 21.56% | Bifidobacterium | 29.62% | Bacteroides | 19.27% |
| 2 | Faecalibacterium | 10.59% | Faecalibacterium | 16.73% | Bacteroides | 9.69% | Blautia | 6.94% |
| 3 | Bacteroides | 7.55% | Bifidobacterium | 6.13% | Lachnospiraceae; Other | 4.76% | Bifidobacterium | 6.85% |
| 4 | Blautia | 5.59% | Blautia | 5.44% | Faecalibacterium | 4.34% | Faecalibacterium | 4.85% |
| 5 | Lachnospiraceae; Other | 4.38% | Lachnospiraceae; Other | 4.82% | Blautia | 4.09% | Lachnospiraceae; Other | 4.77% |
| 6 | Bifidobacterium | 4.18% | Roseburia | 3.66% | Streptococcus | 4.01% | Enterobacteriaceae; Other | 3.67% |
| 7 | Enterobacteriaceae; Other | 2.88% | Enterobacteriaceae; Other | 3.29% | Collinsella | 3.55% | Ruminococcaceae; Ruminococcus | 3.58% |
| 8 | Coprococcus | 2.72% | Coprococcus | 3.25% | Ruminococcus | 3.17% | Roseburia | 3.47% |
| 9 | Ruminococcaceae; Ruminococcus | 2.68% | Lachnospira | 2.88% | Enterobacteriaceae; Other | 3.15% | Megamonas | 2.96% |
| 10 | Gemmiger | 2.66% | Ruminococcaceae; Ruminococcus | 2.24% | Coprococcus | 2.49% | Ruminococcus | 2.77% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitae, H.; Takagi, T.; Naito, Y.; Inoue, R.; Azuma, Y.; Torii, T.; Mizushima, K.; Doi, T.; Inoue, K.; Dohi, O.; et al. Gut Microbiota Associated with Clinical Relapse in Patients with Quiescent Ulcerative Colitis. Microorganisms 2022, 10, 1044. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10051044
Kitae H, Takagi T, Naito Y, Inoue R, Azuma Y, Torii T, Mizushima K, Doi T, Inoue K, Dohi O, et al. Gut Microbiota Associated with Clinical Relapse in Patients with Quiescent Ulcerative Colitis. Microorganisms. 2022; 10(5):1044. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10051044
Chicago/Turabian StyleKitae, Hiroaki, Tomohisa Takagi, Yuji Naito, Ryo Inoue, Yuka Azuma, Takashi Torii, Katsura Mizushima, Toshifumi Doi, Ken Inoue, Osamu Dohi, and et al. 2022. "Gut Microbiota Associated with Clinical Relapse in Patients with Quiescent Ulcerative Colitis" Microorganisms 10, no. 5: 1044. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10051044