Gut Bacterial Communities in Geographically Distant Populations of Farmed Sea Bream (Sparus aurata) and Sea Bass (Dicentrarchus labrax)

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
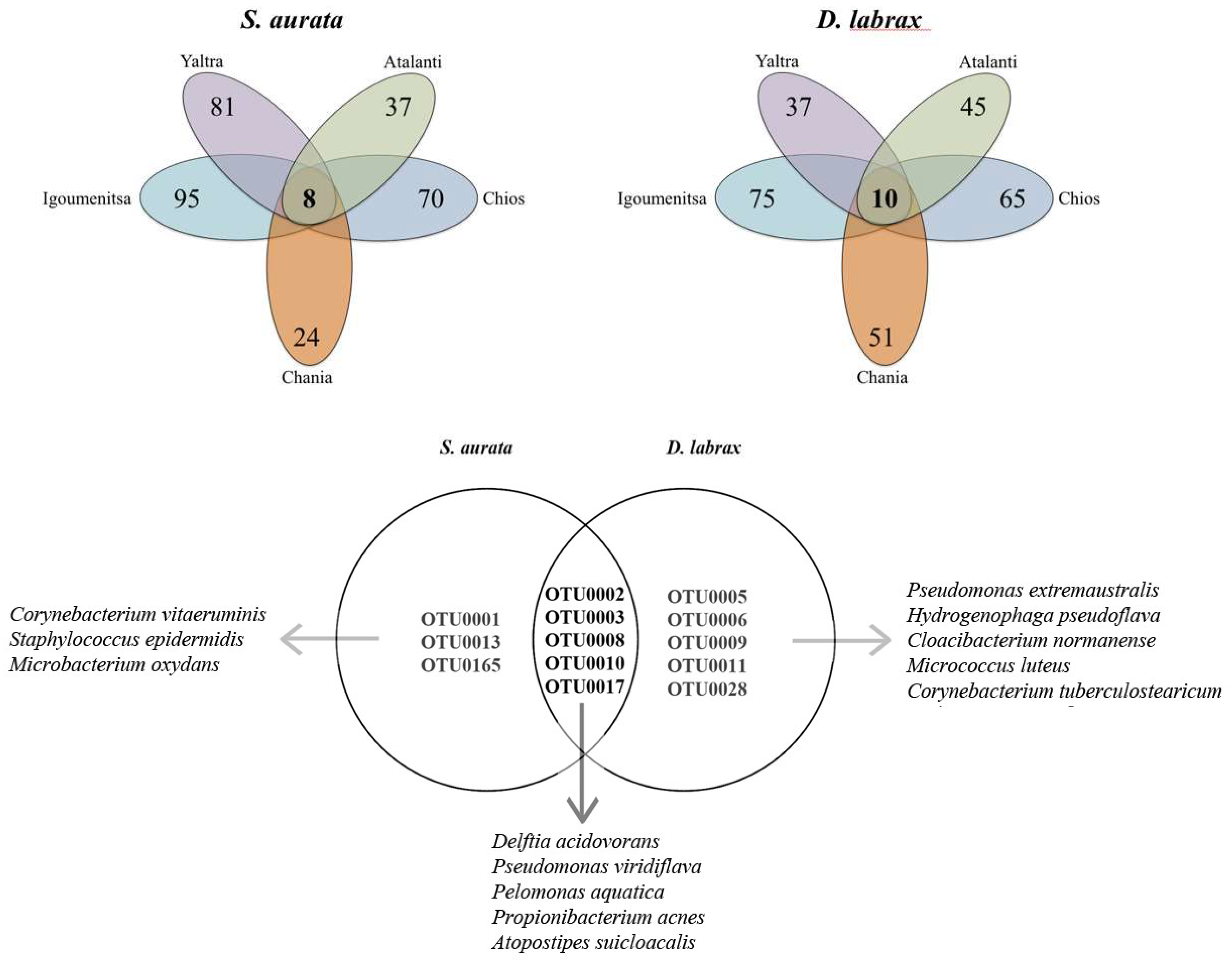
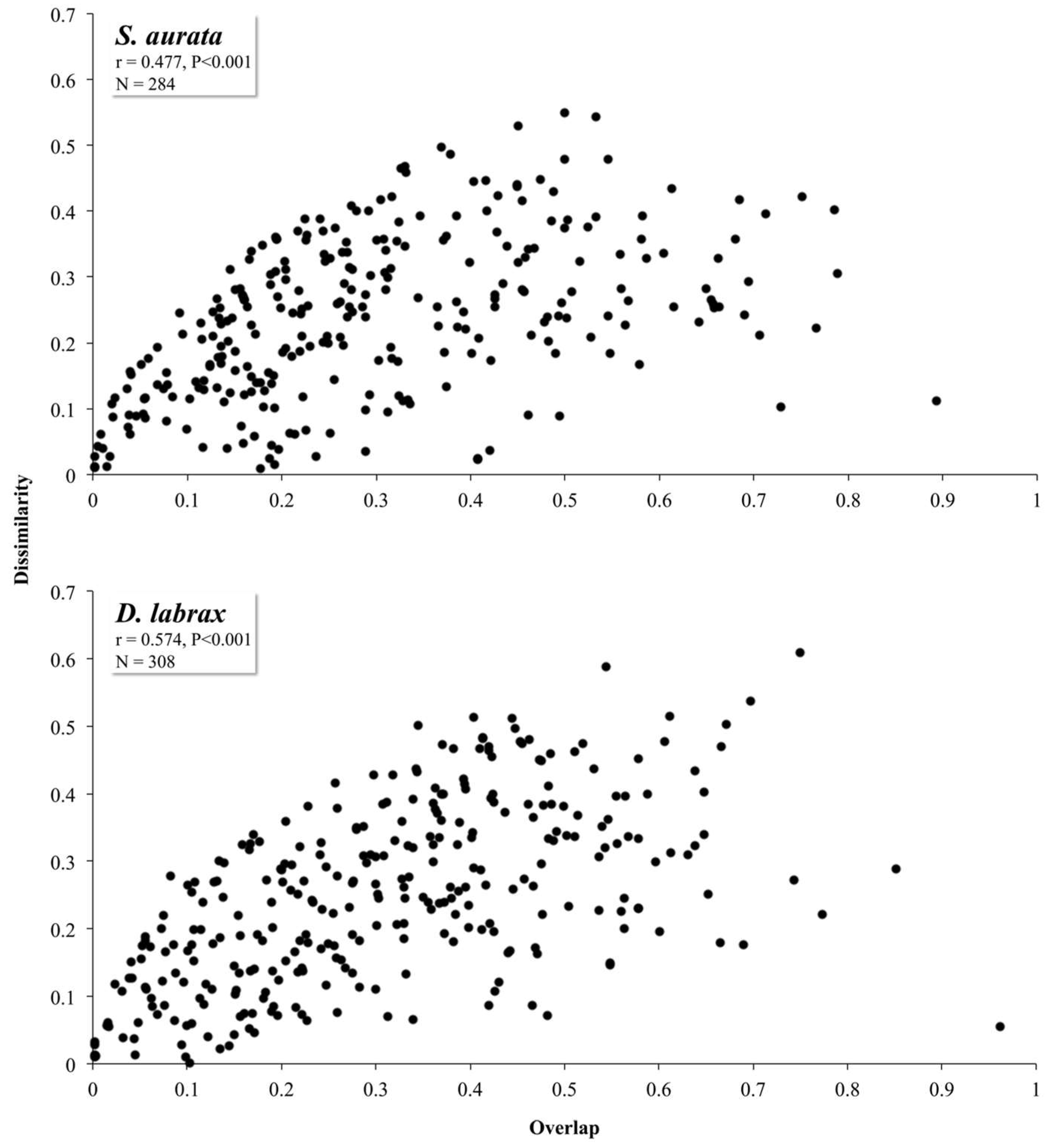
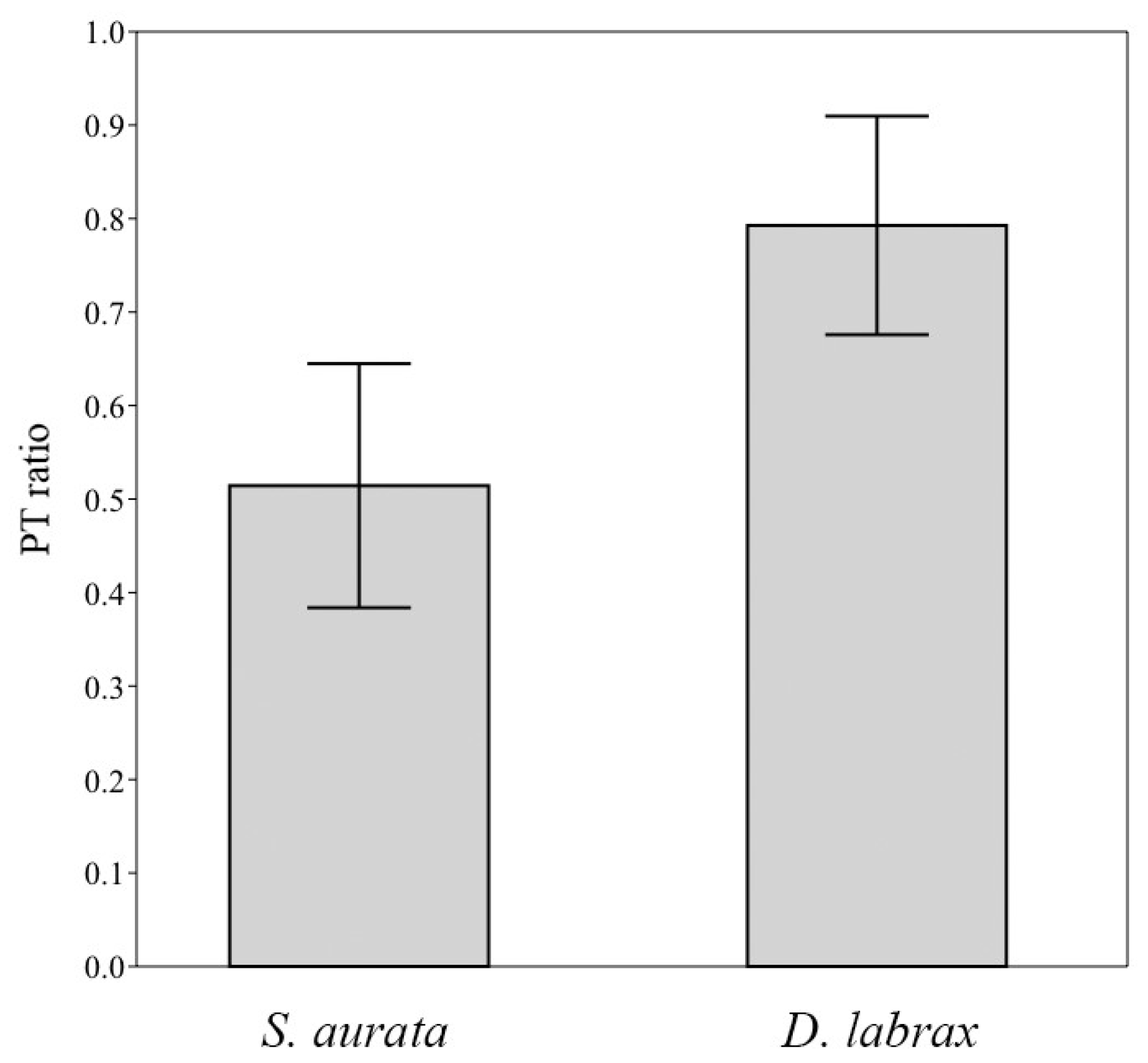
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Hai, N.V. The use of probiotics in aquaculture. J. Appl. Microbiol. 2015, 119, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Sullam, K.E.; Essinger, S.D.; Lozupone, C.A.; O’Connor, M.P.; Rosen, G.L.; Knight, R.O.B.; Kilham, S.S.; Russell, J.A. Environmental and ecological factors that shape the gut bacterial communities of fish: A meta-analysis. Mol. Ecol. 2012, 21, 3363–3378. [Google Scholar] [CrossRef] [PubMed]
- Hamady, M.; Knight, R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Res. 2009, 19, 1141–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llewellyn, M.S.; Boutin, S.; Hoseinifar, S.H.; Derome, N. Teleost microbiomes: The state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front. Microbiol. 2014, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Shade, A.; Handelsman, J. Beyond the venn diagram: The hunt for a core microbiome. Environ. Microbiol. 2012, 14, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Zelezniak, A.; Andrejev, S.; Ponomarova, O.; Mende, D.R.; Bork, P.; Patil, K.R. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc. Natl. Acad. Sci. USA 2015, 112, 6449–6454. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.A.; Worobey, M. Geographical variation of human gut microbial composition. Biol. Lett. 2014, 10, 20131037. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Duncan, S.H.; Louis, P. The impact of nutrition on intestinal bacterial communities. Curr. Opin. Microbiol. 2017, 38, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Rawls, J.F. Intestinal microbiota composition in fishes is influenced by host ecology and environment. Mol. Ecol. 2012, 21, 3100–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.R.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16s ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2012, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16s rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. Sina: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Hammer, Ø.; Harper, D.; Ryan, P. Past: Paleontological statistics software package for education and data analysis. Palaeontol Electr. 2001, 4, 1–9. [Google Scholar]
- Team, R. Rstudio. Integrated development for R. Rstudio, Inc., Boston, MA. Available online: http://www.rstudio.com/ (accessed on 2 July 2018).
- Lennon, J.T.; Locey, K.J. Macroecology for microbiology. Environ. Microbiol. Rep. 2017, 9, 38–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashan, A.; Gibson, T.E.; Friedman, J.; Carey, V.J.; Weiss, S.T.; Hohmann, E.L.; Liu, Y.-Y. Universality of human microbial dynamics. Nature 2016, 534, 259–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morueta-Holme, N.; Blonder, B.; Sandel, B.; McGill, B.J.; Peet, R.K.; Ott, J.E.; Violle, C.; Enquist, B.J.; Jørgensen, P.M.; Svenning, J.-C. A network approach for inferring species associations from co-occurrence data. Ecography 2016, 39, 1139–1150. [Google Scholar] [CrossRef]
- Meziti, A.; Mente, E.; Kormas, K.A. Gut bacteria associated with different diets in reared Nephrops norvegicus. Syst. Appl. Microbiol. 2012, 35, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Estruch, G.; Collado, M.C.; Peñaranda, D.S.; Tomás Vidal, A.; Jover Cerdá, M.; Pérez Martínez, G.; Martinez-Llorens, S. Impact of fishmeal replacement in diets for gilthead sea bream Sparus aurata on the gastrointestinal microbiota determined by pyrosequencing the 16s rRNA gene. PLoS ONE 2015, 10, e0136389. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, V.T.; Smith, K.F.; Melvin, D.W.; Amaral-Zettler, L.A. Community assembly of a euryhaline fish microbiome during salinity acclimation. Mol. Ecol. 2015, 24, 2537–2550. [Google Scholar] [CrossRef] [PubMed]
- Borsodi, A.K.; Szabó, A.; Krett, G.; Felföldi, T.; Specziár, A.; Boros, G. Gut content microbiota of introduced bigheaded carps (Hypophthalmichthys spp.) inhabiting the largest shallow lake in central Europe. Microbiol. Res. 2017, 195, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.A.; Young, C.R.; Gifford, S.M.; Eppley, J.M.; Marin, R.; Schuster, S.C.; Scholin, C.A.; DeLong, E.F. Multispecies diel transcriptional oscillations in open ocean heterotrophic bacterial assemblages. Science 2014, 345, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aylward, F.O.; Eppley, J.M.; Smith, J.M.; Chavez, F.P.; Scholin, C.A.; DeLong, E.F. Microbial community transcriptional networks are conserved in three domains at ocean basin scales. Proc. Natl. Acad. Sci. USA 2015, 112, 5443–5448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrman, J.; Steele, J.A. Community structure of marine bacterioplankton: Patterns, networks, and relationships to function. Aquat. Microb. Ecol. 2008, 53, 69–81. [Google Scholar] [CrossRef]
- Meziti, A.; Kormas, K.A.; Moustaka-Gouni, M.; Karayanni, H. Spatially uniform but temporally variable bacterioplankton in a semi-enclosed coastal area. Syst. Appl. Microbiol. 2015, 38, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Bunse, C.; Pinhassi, J. Marine bacterioplankton seasonal succession dynamics. Trends Microbiol. 2017, 25, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Troussellier, M.; Escalas, A.; Bouvier, T.; Mouillot, D. Sustaining rare marine microorganisms: Macroorganisms as repositories and dispersal agents of microbial diversity. Front. Microbiol. 2017, 8, 947. [Google Scholar] [CrossRef] [PubMed]
- Floris, R.; Manca, S.; Fois, N. Microbial ecology of intestinal tract of gilthead sea bream (Sparus aurata Linnaeus, 1758) from two coastal lagoons of Sardinia (Italy). Transit. Waters Bull. 2013, 7, 4–12. [Google Scholar] [CrossRef]
- Silvi, S.; Nardi, M.; Sulpizio, R.; Orpianesi, C.; Caggiano, M.; Carnevali, O.; Cresci, A. Effect of the addition of Lactobacillus delbrueckii subsp. delbrueckii on the gut microbiota composition and contribution to the well-being of European sea bass (Dicentrarchus labrax, L.). Microbial Ecology in Health and Disease 2008, 20, 53–59. [Google Scholar] [CrossRef]
- Bourouni, O.C.; El Bour, M.; Calo-Mata, P.; Mraouna, R.; Abedellatif, B.; Barros-Velàzquez, J. Phylogenetic analysis of antimicrobial lactic acid bacteria from farmed seabass Dicentrarchus labrax. Can. J. Microbiol. 2012, 58, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Lamari, F.; Mahdhi, A.; Chakroun, I.; Esteban, M.A.; Mazurais, D.; Amina, B.; Gatesoupe, F.J. Interactions between candidate probiotics and the immune and antioxidative responses of European sea bass (Dicentrarchus labrax) larvae. J. Fish Dis. 2016, 39, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Mladineo, I.; Bušelić, I.; Hrabar, J.; Radonić, I.; Vrbatović, A.; Jozić, S.; Trumbić, Ž. Autochthonous bacterial isolates successfully stimulate in vitro peripheral blood leukocytes of the European sea bass (Dicentrarchus labrax). Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Torrecillas, S.; Mompel, D.; Caballero, M.J.; Montero, D.; Merrifield, D.; Rodiles, A.; Robaina, L.; Zamorano, M.J.; Karalazos, V.; Kaushik, S.; et al. Effect of fishmeal and fish oil replacement by vegetable meals and oils on gut health of European sea bass (Dicentrarchus labrax). Aquaculture 2017, 468, 386–398. [Google Scholar] [CrossRef]
- Givens, C.; Ransom, B.; Bano, N.; Hollibaugh, J. Comparison of the gut microbiomes of 12 bony fish and 3 shark species. Mar. Ecol. Prog. Ser. 2015, 518, 209–223. [Google Scholar] [CrossRef]
- Tarnecki, A.M.; Burgos, F.A.; Ray, C.L.; Arias, C.R. Fish intestinal microbiome: Diversity and symbiosis unravelled by metagenomics. J. Appl. Microbiol. 2017, 123, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Roller, B.R.K.; Stoddard, S.F.; Schmidt, T.M. Exploiting rRNA operon copy number to investigate bacterial reproductive strategies. Nat. Microbiol. 2016, 1, 16160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Yang, H.; Ling, Z.; Chang, J.; Ye, J.D. Gut microbiota of fast and slow growing grouper Epinephelus coioides. Afr. J. Microbiol. Res. 2009, 3, 713–720. [Google Scholar] [CrossRef]
- Navarrete, P.; Magne, F.; Araneda, C.; Fuentes, P.; Barros, L.; Opazo, R.; Espejo, R.; Romero, J. PCR-TTGE analysis of 16s rRNA from rainbow trout (Oncorhynchus mykiss) gut microbiota reveals host-specific communities of active bacteria. PLoS ONE 2012, 7, e31335. [Google Scholar] [CrossRef] [PubMed]
- Gajardo, K.; Rodiles, A.; Kortner, T.M.; Krogdahl, Å.; Bakke, A.M.; Merrifield, D.L.; Sørum, H. A high-resolution map of the gut microbiota in Atlantic salmon (Salmo salar): A basis for comparative gut microbial research. Sci. Rep. 2016, 6, 30893. [Google Scholar] [CrossRef] [PubMed]
- Wen, A.; Fegan, M.; Hayward, C.; Chakraborty, S.; Sly, L.I. Phylogenetic relationships among members of the Comamonadaceae, and description of Delftia acidovorans (Den Dooren De Jong 1926 and Tamaoka et al. 1987) gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 1999, 49, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Rhman, A.M.; Khattab, Y.A.E.; Shalaby, A.M.E. Micrococcus luteus and Pseudomonas species as probiotics for promoting the growth performance and health of Nile tilapia, Oreochromis niloticus. Fish Shellfish Immunol. 2009, 27, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Korkea-aho, T.L.; Heikkinen, J.; Thompson, K.D.; von Wright, A.; Austin, B. Pseudomonas sp. M174 inhibits the fish pathogen Flavobacterium psychrophilum. J. Appl. Microbiol. 2011, 111, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Lazado, C.C.; Caipang, C.M.A.; Kiron, V. Enzymes from the gut bacteria of Atlantic cod, Gadus morhua and their influence on intestinal enzyme activity. Aquacult. Nutr. 2012, 18, 423–431. [Google Scholar] [CrossRef]
- Rasheeda, M.K.; Rangamaran, V.R.; Srinivasan, S.; Ramaiah, S.K.; Gunasekaran, R.; Jaypal, S.; Gopal, D.; Ramalingam, K. Comparative profiling of microbial community of three economically important fishes reared in sea cages under tropical offshore environment. Mar. Genomics 2017, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Carda-Diéguez, M.; Mira, A.; Fouz, B. Pyrosequencing survey of intestinal microbiota diversity in cultured sea bass (Dicentrarchus labrax) fed functional diets. FEMS Microbiol. Ecol. 2014, 87, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Kormas, K.A.; Meziti, A.; Mente, E.; Frentzos, A. Dietary differences are reflected on the gut prokaryotic community structure of wild and commercially reared sea bream (Sparus aurata). MicrobiologyOpen 2014, 3, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Godoy, F.A.; Miranda, C.D.; Wittwer, G.D.; Aranda, C.P.; Calderón, R. High variability of levels of Aliivibrio and lactic acid bacteria in the intestinal microbiota of farmed Atlantic salmon Salmo salar L. Ann. Microbiol. 2015, 65, 2343–2353. [Google Scholar] [CrossRef]
- Rurangwa, E.; Sipkema, D.; Kals, J.; ter Veld, M.; Forlenza, M.; Bacanu, G.M.; Smidt, H.; Palstra, A.P. Impact of a novel protein meal on the gastrointestinal microbiota and the host transcriptome of larval zebrafish Danio rerio. Front. Physiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Koleva, Z.; Dedov, I.; Kizheva, J.; Lipovanska, R.; Moncheva, P.; Hristova, P. Lactic acid microflora of the gut of snail Cornu aspersum. Biotechnol. Biotechnol. Equip. 2014, 28, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Krutmann, J. Pre- and probiotics for human skin. J Dermatol. Sci. 2009, 54, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.I.; Kim, G.; Hwang, C.Y.; Cho, B.C. Prokaryotic community composition in alkaline-fermented skate (Raja pulchra). Food Microbiol. 2017, 61, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Montes, C.; Altimira, F.; Canchignia, H.; Castro, Á.; Sánchez, E.; Miccono, M.; Tapia, E.; Sequeida, Á.; Valdés, J.; Tapia, P.; et al. A draft genome sequence of Pseudomonas veronii R4: A grapevine (Vitis vinifera L.) root-associated strain with high biocontrol potential. Stand. Genomic Sci. 2016, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- De Schryver, P.; Dierckens, K.; Bahn Thi, Q.Q.; Amalia, R.; Marzorati, M.; Bossier, P.; Boon, N.; Verstraete, W. Convergent dynamics of the juvenile European sea bass gut microbiota induced by poly-β-hydroxybutyrate. Environ. Microbiol. 2011, 13, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.C.D.P.; Nicoli, J.R.; Zambonino-Infante, J.L.; Kaushik, S.; Gatesoupe, F.-J. Influence of the diet on the microbial diversity of faecal and gastrointestinal contents in gilthead sea bream (Sparus aurata) and intestinal contents in goldfish (Carassius auratus). FEMS Microbiol. Ecol. 2011, 78, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial community variation in human body habitats across space and time. Science 2009, 326. [Google Scholar] [CrossRef] [PubMed]
- Gatesoupe, F.-J.; Huelvan, C.; Le Bayon, N.; Le Delliou, H.; Madec, L.; Mouchel, O.; Quazuguel, P.; Mazurais, D.; Zambonino-Infante, J.-L. The highly variable microbiota associated to intestinal mucosa correlates with growth and hypoxia resistance of sea bass, Dicentrarchus labrax, submitted to different nutritional histories. BMC Microbiol. 2016, 16, 266. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. Host-microbe interaction: Rules of the game for microbiota. Nature 2016, 534, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Hovda, M.B.; Lunestad, B.T.; Fontanillas, R.; Rosnes, J.T. Molecular characterisation of the intestinal microbiota of farmed Atlantic salmon (Salmo salar L.). Aquaculture 2007, 272, 581–588. [Google Scholar] [CrossRef]
- Zarkasi, K.Z.; Taylor, R.S.; Glencross, B.D.; Abell, G.C.J.; Tamplin, M.L.; Bowman, J.P. In vitro characteristics of an Atlantic salmon (Salmo salar L.) hind gut microbial community in relation to different dietary treatments. Res. Microbiol. 2017, 168, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, B.; Delgado, S.; Blanco-Míguez, A.; Lourenço, A.; Gueimonde, M.; Margolles, A. Probiotics, gut microbiota, and their influence on host health and disease. Mol. Nutr. Food Res. 2017, 61, 1600240. [Google Scholar] [CrossRef] [PubMed]
- Coyte, K.Z.; Schluter, J.; Foster, K.R. The ecology of the microbiome: Networks, competition, and stability. Science 2015, 350, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Widder, S.; Allen, R.J.; Pfeiffer, T.; Curtis, T.P.; Wiuf, C.; Sloan, W.T.; Cordero, O.X.; Brown, S.P.; Momeni, B.; Shou, W.; et al. Challenges in microbial ecology: Building predictive understanding of community function and dynamics. ISME J. 2016, 10, 2557–2568. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Site | Reads | OTUs | No. of the Most Dominant OTUs (Cumulative Relative Dominance ≥ 80%) | Most Abundant OTU, Dominance (%) and Closest Relative (≥97%) | ||||
|---|---|---|---|---|---|---|---|---|
| S. aurata | D. labrax | S. aurata | D. labrax | S. aurata | D. labrax | S. aurata | D. labrax | |
| Chania | 827 ± 512.4 N = 4 | 2395 ± 725.4 N = 5 | 11 ± 2.2 | 16 ± 8.7 | 10 (80.0) | 17 (79.9) | OTU0011 (22.7) Micrococcus luteus | OTU0014 (17.2) Paracocccus denitrificans |
| Igoumenitsa | 2360 ± 1972.7 N = 5 | 1809 ± 571.3 N = 4 | 25 ± 28.3 | 27 ± 20.3 | 13 (81.0) | 14 (80.0) | OTU0004 (27.8) Bacillus hisashii | OTU0001 (19.9) Corynebacterium vitaeruminis |
| Chios | 2407 ± 1771.0 N = 6 | 2148 ± 1785.2 N = 6 | 18 ± 11.4 | 17 ± 9.8 | 13 (79.5) | 13 (80.9) | OTU0004 (22.0) Bacillus hisashii | OTU0001 (24.7) Corynebacterium vitaeruminis |
| Yaltra | 2656 ± 1529.0 N = 6 | 697 ± 367.3 N = 6 | 19 ± 12.2 | 11 ± 2.4 | 21 (80.0) | 10 (79.9) | OTU0002 (16.9) Delftia acidovorans | OTU0025 (21.9) Acinetobacter lwoffii |
| Atalanti | 1574 ± 1005.9 N = 4 | 2533 ± 1052.7 N = 5 | 13 ± 6.1 | 14 ± 8.1 | 12 (80.1) | 11 (80.0) | OTU0005 (14.7) Pseudomonas extremaustralis | OTU0002 (17.7) Delftia acidovorans |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikouli, E.; Meziti, A.; Antonopoulou, E.; Mente, E.; Kormas, K.A. Gut Bacterial Communities in Geographically Distant Populations of Farmed Sea Bream (Sparus aurata) and Sea Bass (Dicentrarchus labrax). Microorganisms 2018, 6, 92. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms6030092
Nikouli E, Meziti A, Antonopoulou E, Mente E, Kormas KA. Gut Bacterial Communities in Geographically Distant Populations of Farmed Sea Bream (Sparus aurata) and Sea Bass (Dicentrarchus labrax). Microorganisms. 2018; 6(3):92. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms6030092
Chicago/Turabian StyleNikouli, Eleni, Alexandra Meziti, Efthimia Antonopoulou, Eleni Mente, and Konstantinos A. Kormas. 2018. "Gut Bacterial Communities in Geographically Distant Populations of Farmed Sea Bream (Sparus aurata) and Sea Bass (Dicentrarchus labrax)" Microorganisms 6, no. 3: 92. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms6030092