A Metabolomic-Based Evaluation of the Role of Commensal Microbiota throughout the Gastrointestinal Tract in Mice

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Sample Collection and Preparation for Metabolome Analysis
2.3. LC-MS/MS Conditions
2.4. CE-TOFMS Conditions
2.5. Processing of Metabolome Data
2.6. Metabolome Data Analysis
2.7. DNA Extraction, PCR Amplification, and Pyrosequencing of Gastrointestinal Luminal Contents
2.8. Analysis of Microbial 16S rRNA Gene Sequences
2.9. Statistical Analysis
3. Results
3.1. Characteristics of Gastrointestinal Luminal Metabolites in SPF and GF Mice
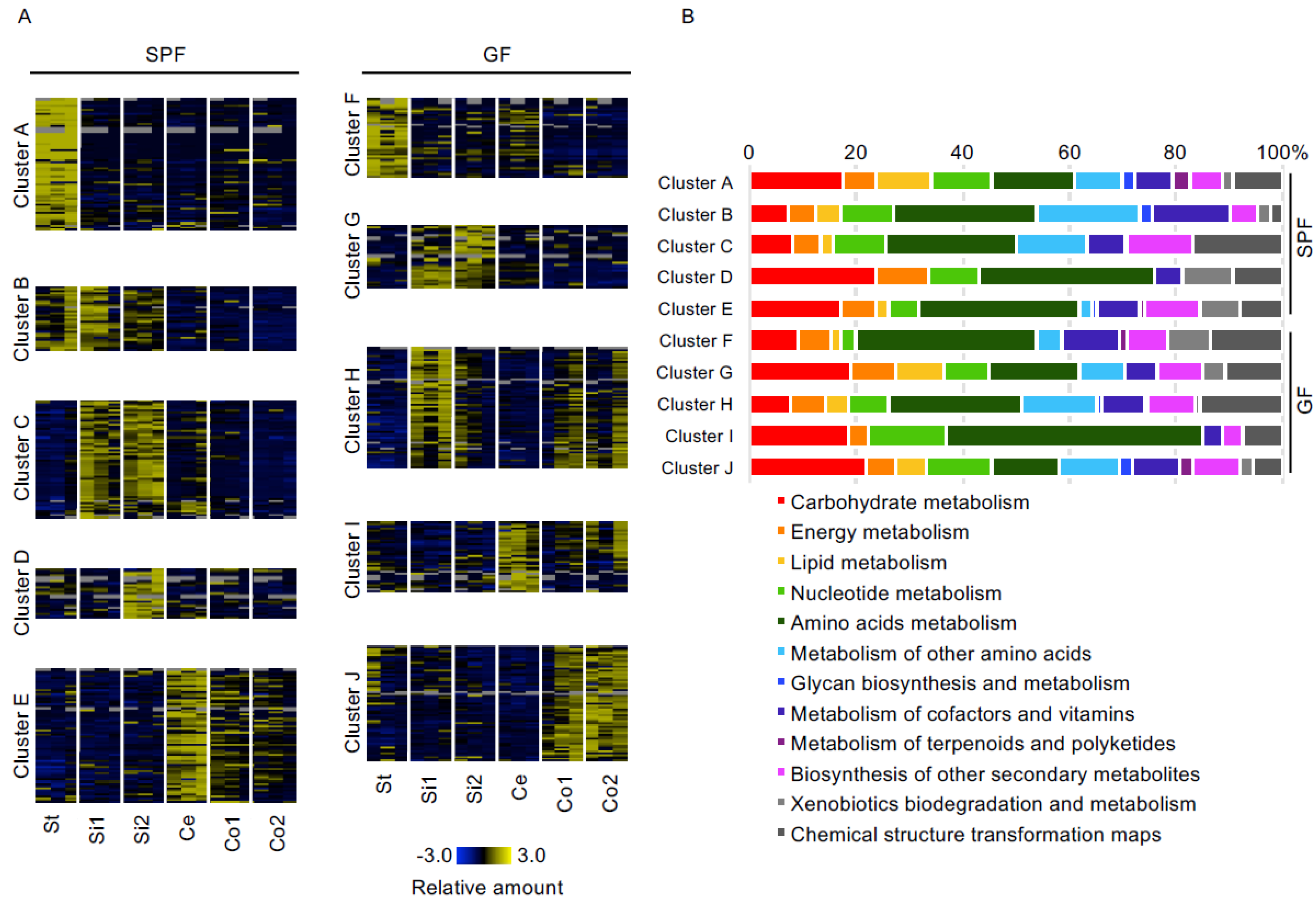
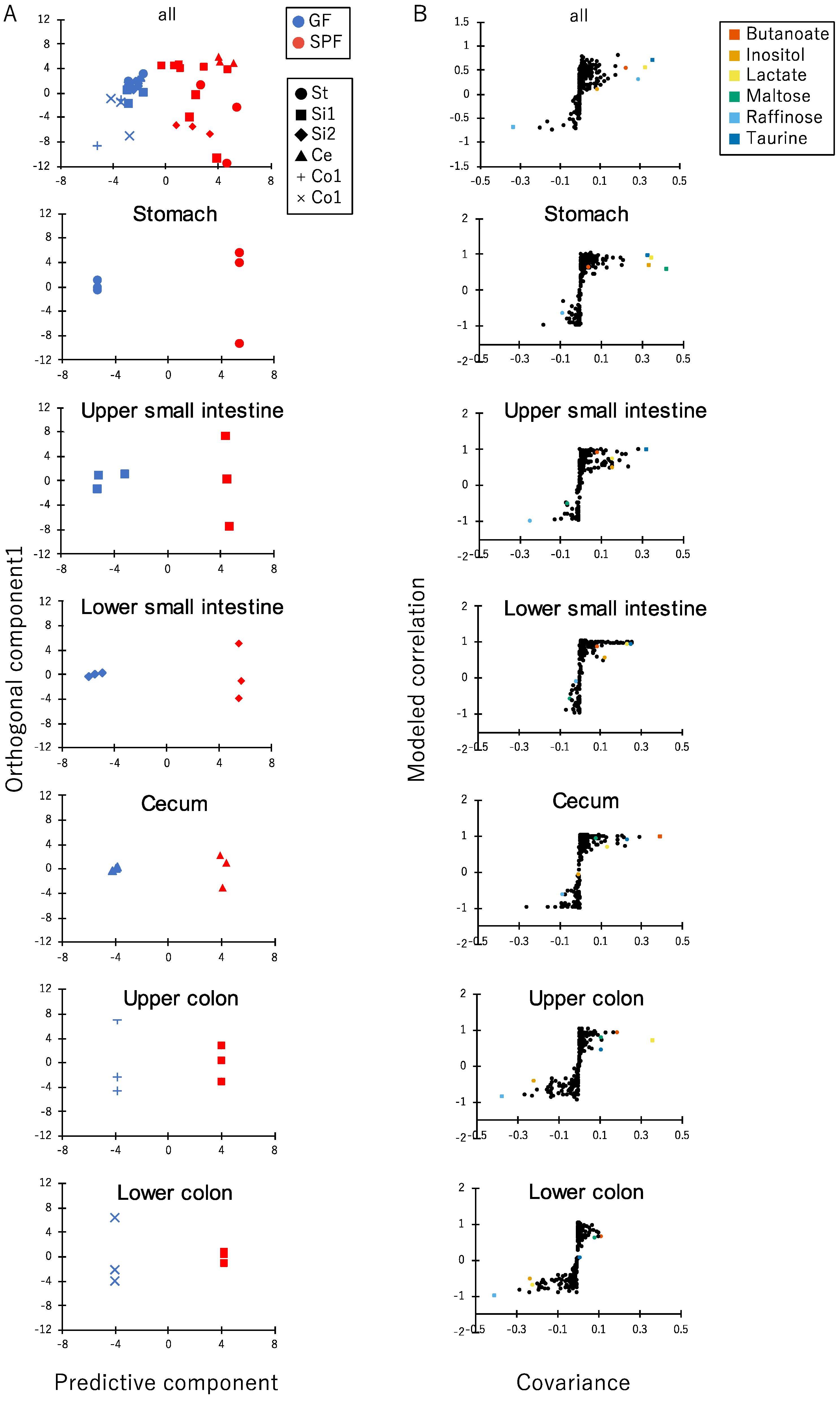
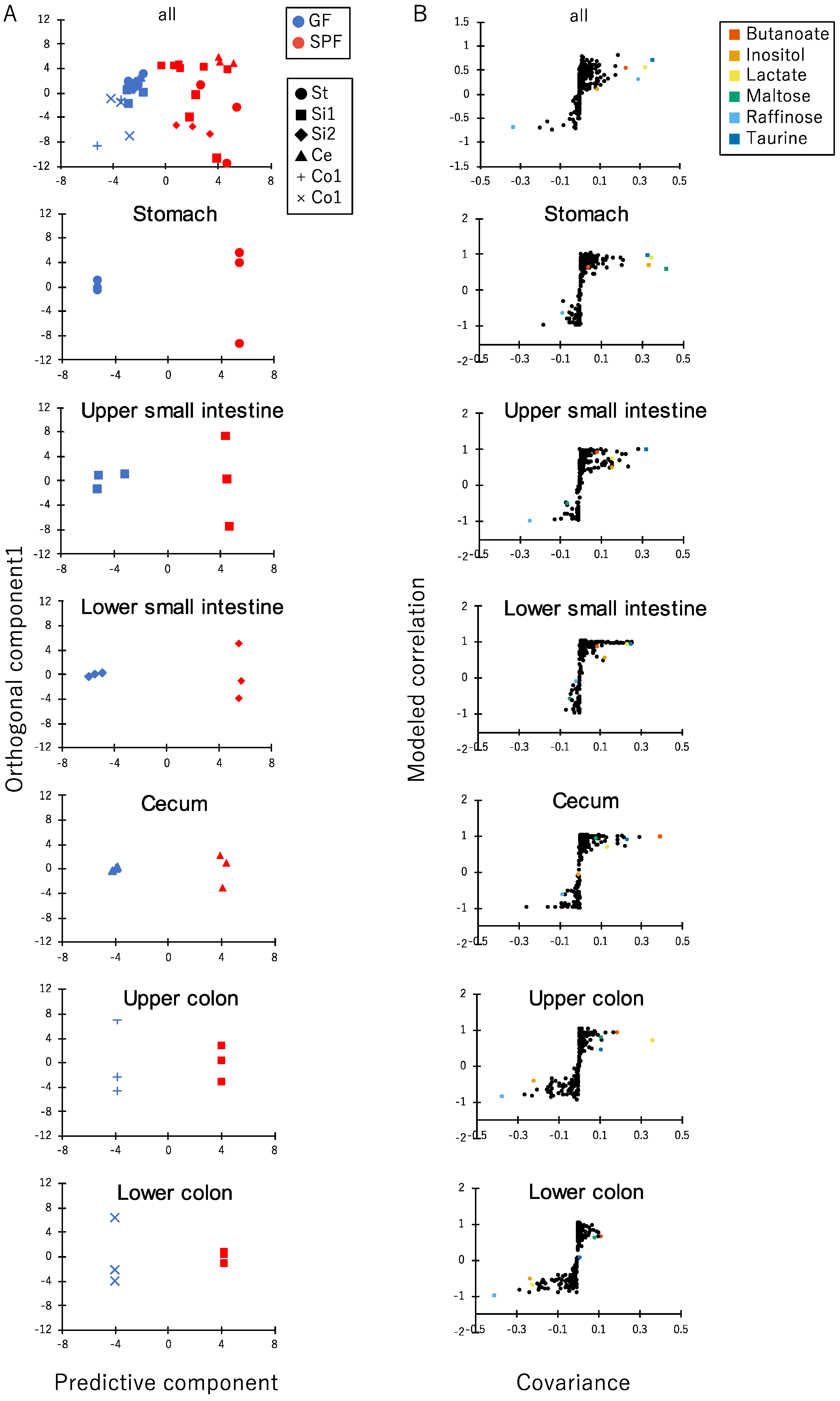
3.2. Comparison of Luminal Metabolome Profiles among Each Part of the Gastrointestinal Tracts in SPF and GF Mice
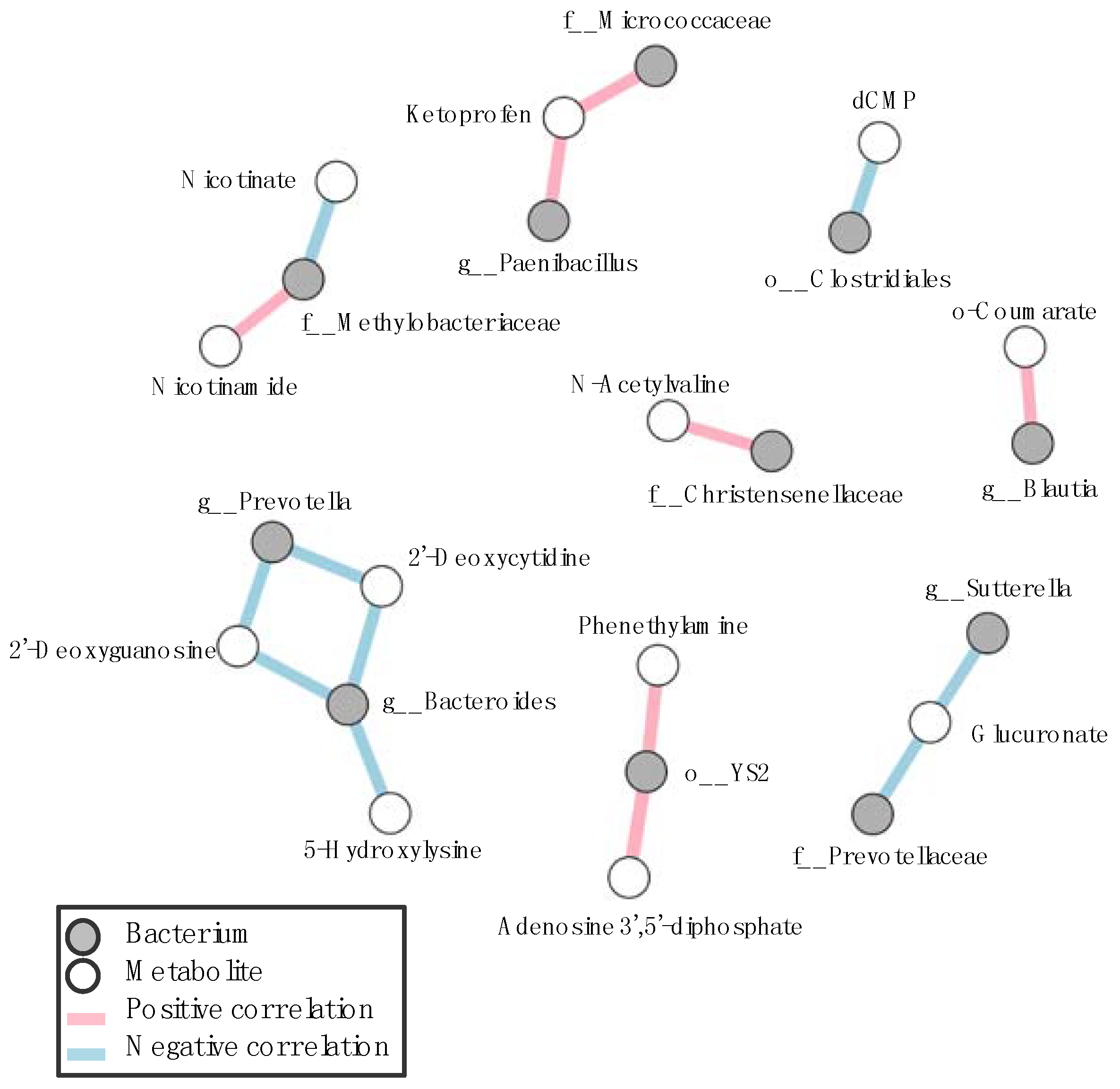
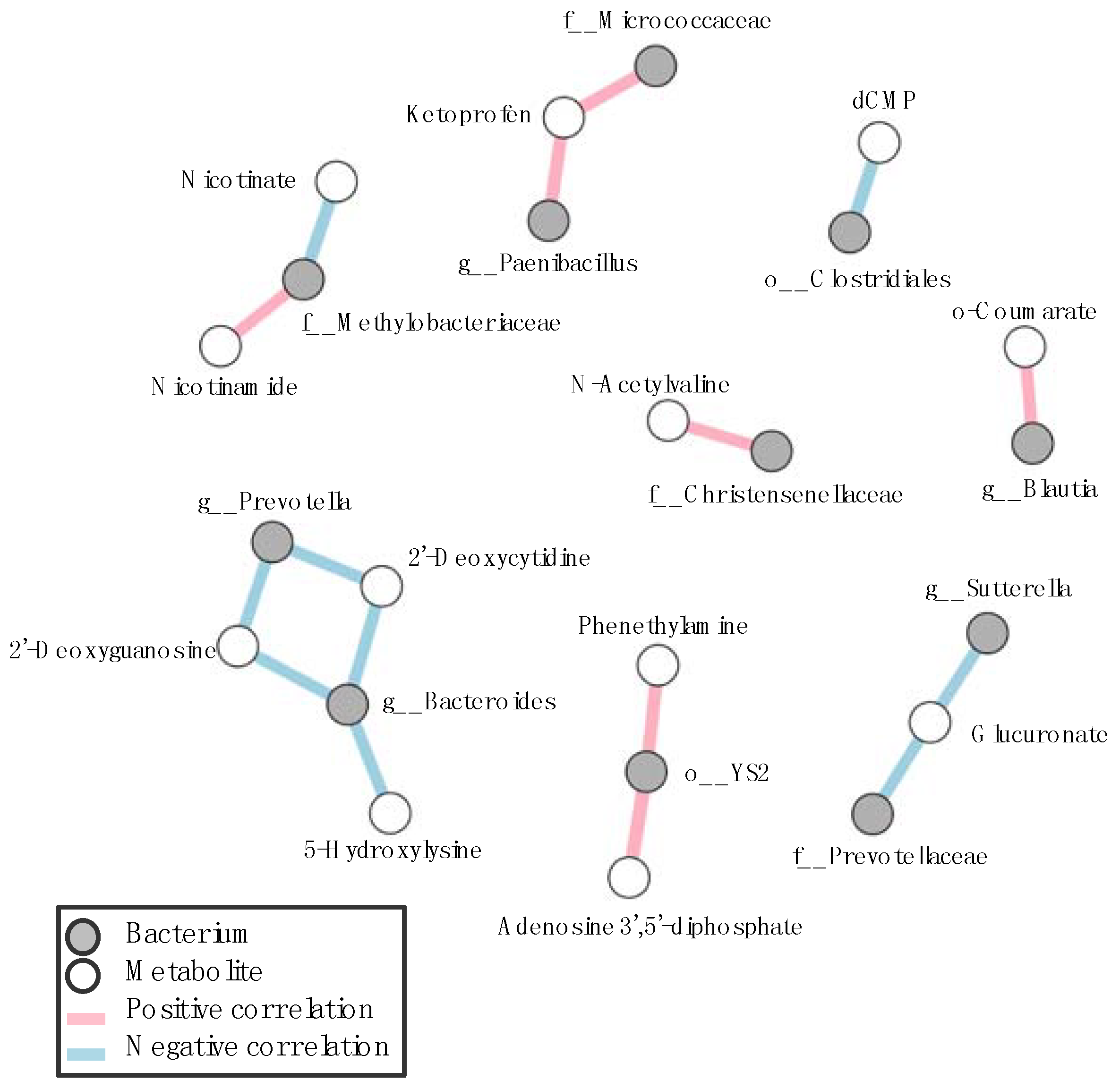
3.3. Correlation between Gastrointestinal Microbes and Metabolites in SPF Mice
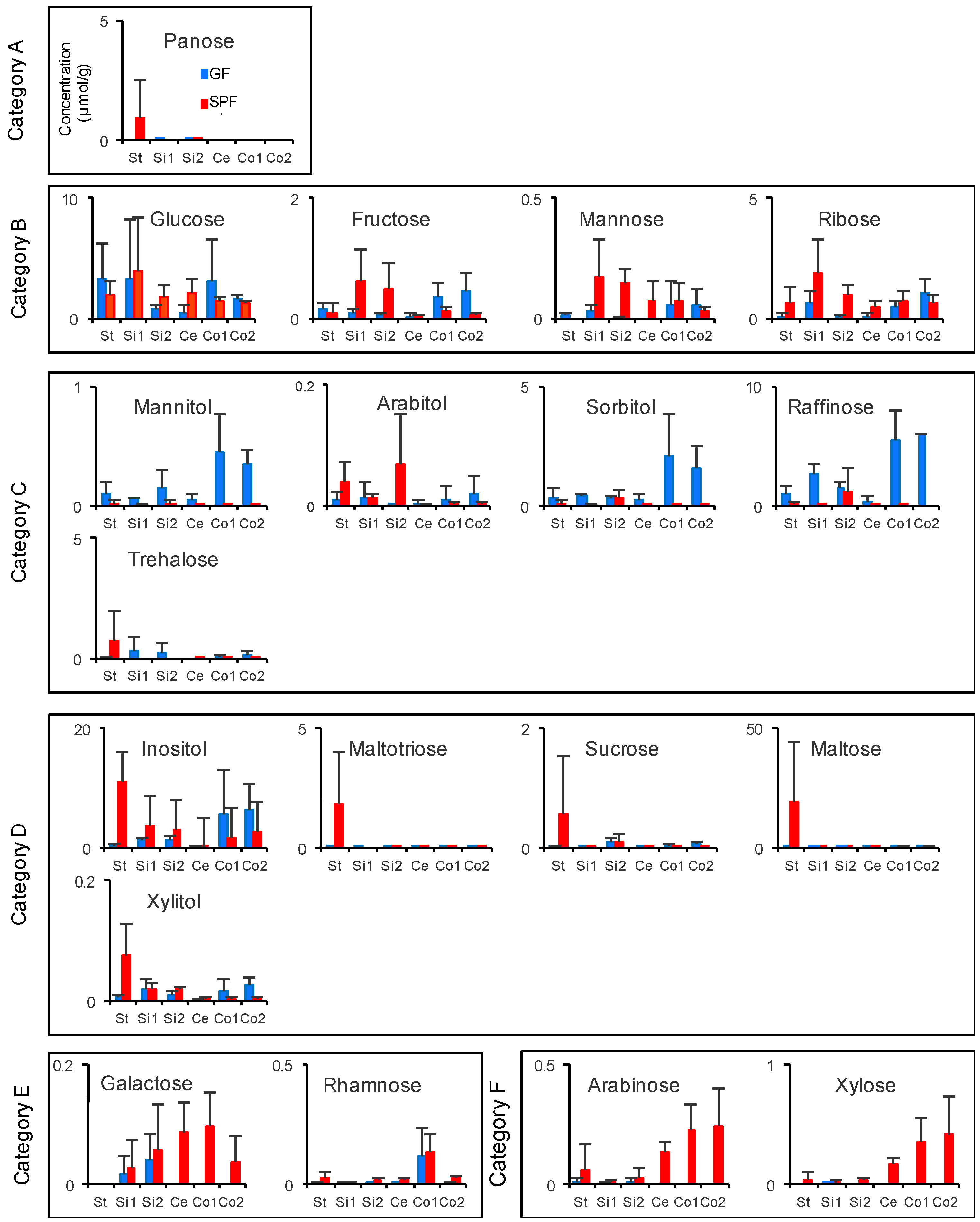
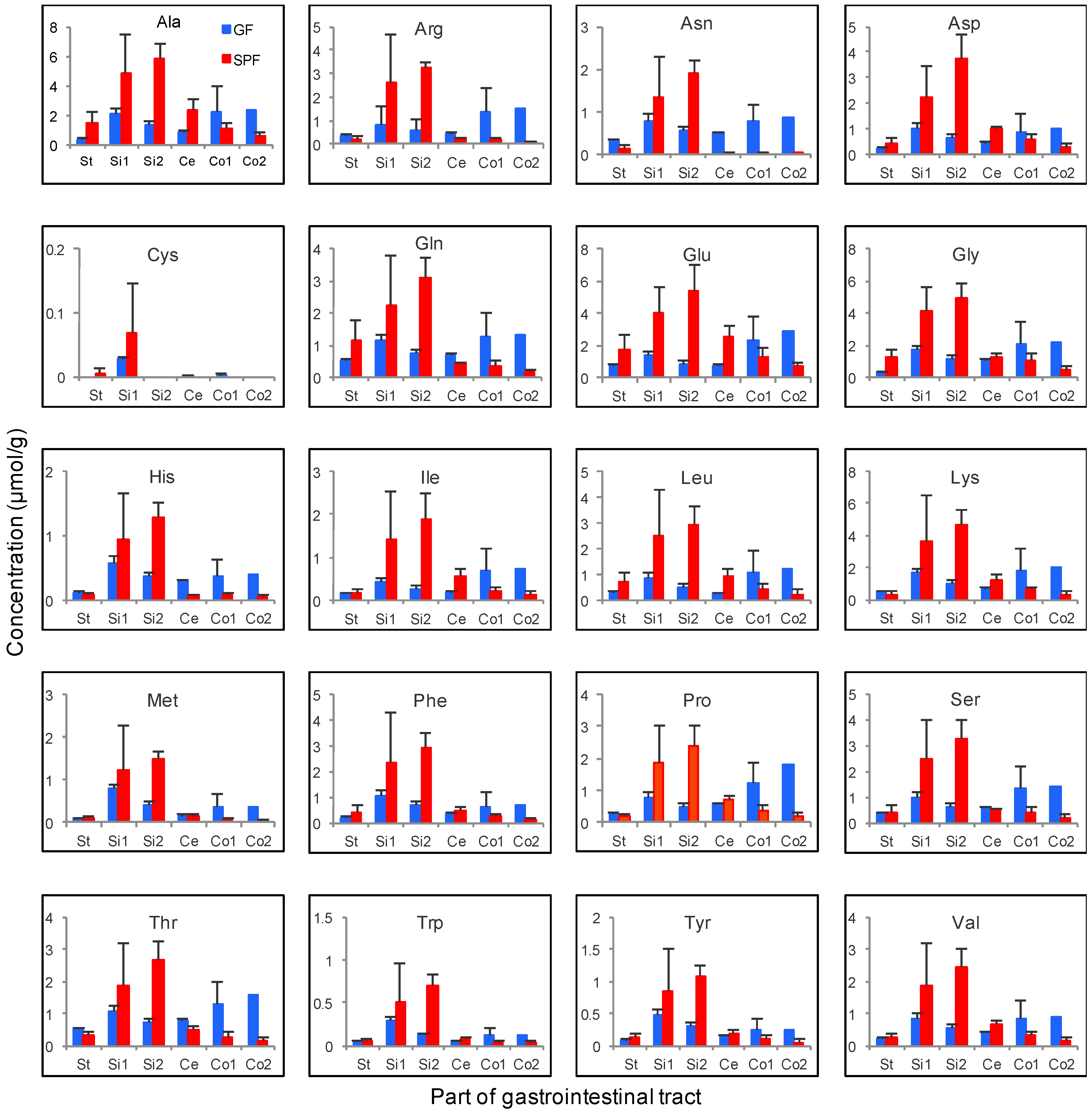
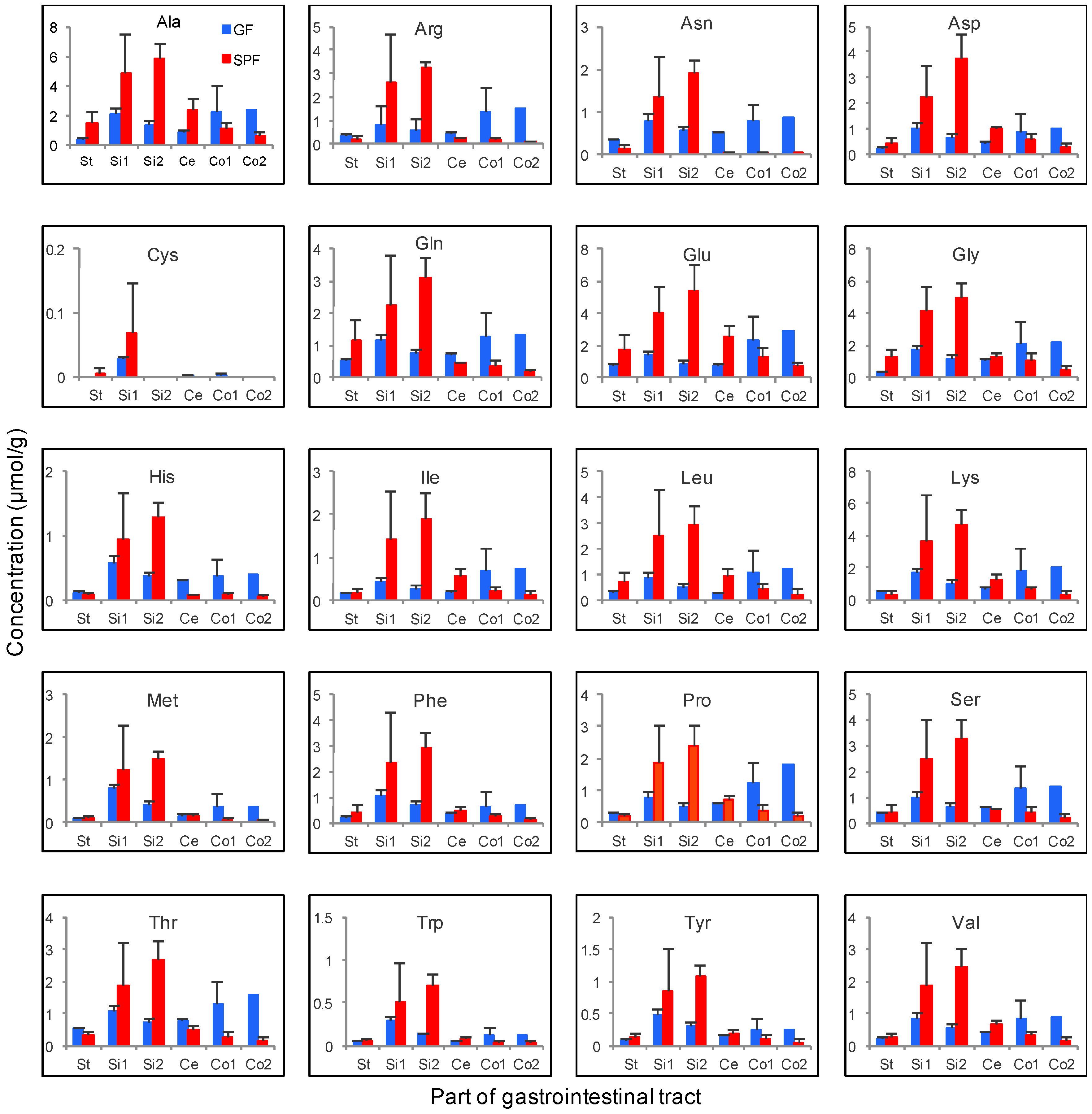
3.4. Gastrointestinal Luminal Features of Sugars and Amino Acids in SPF and GF Mice
4. Discussion
Supplementary Materials
Author contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holmes, E.; Li, J.V.; Marchesi, J.R.; Nicholson, J.K. Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab. 2012, 16, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Bjorksten, B.; Naaber, P.; Sepp, E.; Mikelsaar, M. The intestinal microflora in allergic Estonian and Swedish 2-year-old children. Clin. Exp. Allergy 1999, 29, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Bjorksten, B.; Sepp, E.; Julge, K.; Voor, T.; Mikelsaar, M. Allergy development and the intestinal microflora during the first year of life. J. Allergy Clin. Immunol. 2001, 108, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Kalliomaki, M.; Kirjavainen, P.; Eerola, E.; Kero, P.; Salminen, S.; Isolauri, E. Distinct patterns of neonatal gut microflora in infants in whom atopy was and was not developing. J. Allergy Clin. Immunol. 2001, 107, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Cahenzli, J.; Koller, Y.; Wyss, M.; Geuking, M.B.; McCoy, K.D. Intestinal microbial diversity during early-life colonization shapes long-term IgE levels. Cell Host Microbe 2013, 14, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A.N.; McKenzie, C.I.; Shen, S.; Stanley, D.; Macia, L.; Mason, L.J.; Roberts, L.K.; Wong, C.H.; Shim, R.; Robert, R.; et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat. Commun. 2015, 6, 7320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadowaki, A.; Miyake, S.; Saga, R.; Chiba, A.; Mochizuki, H.; Yamamura, T. Gut environment-induced intraepithelial autoreactive CD4(+) T cells suppress central nervous system autoimmunity via LAG-3. Nat. Commun. 2016, 7, 11639. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Buffington, S.A.; Di Prisco, G.V.; Auchtung, T.A.; Ajami, N.J.; Petrosino, J.F.; Costa-Mattioli, M. Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 2016, 165, 1762–1775. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 173, 1728–1741. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Chen, D.; Zhang, J.N.; Lv, X.; Wang, K.; Duan, L.P.; Nie, Y.; Wu, X.L. Bacterial community mapping of the mouse gastrointestinal tract. PLoS ONE 2013, 8, e74957. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Kibe, R.; Ooga, T.; Aiba, Y.; Kurihara, S.; Sawaki, E.; Koga, Y.; Benno, Y. Impact of intestinal microbiota on intestinal luminal metabolome. Sci. Rep. 2012, 2, 233. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhang, L.; Wang, Y.; Tang, H. Age-related topographical metabolic signatures for the rat gastrointestinal contents. J. Proteome Res. 2012, 11, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Soga, T.; Ohashi, Y.; Ueno, Y.; Naraoka, H.; Tomita, M.; Nishioka, T. Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J. Proteome Res. 2003, 2, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Soga, T.; Baran, R.; Suematsu, M.; Ueno, Y.; Ikeda, S.; Sakurakawa, T.; Kakazu, Y.; Ishikawa, T.; Robert, M.; Nishioka, T.; et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006, 281, 16768–16776. [Google Scholar] [CrossRef] [PubMed]
- Nagao-Kitamoto, H.; Shreiner, A.B.; Gillilland, M.G., III; Kitamoto, S.; Ishii, C.; Hirayama, A.; Kuffa, P.; El-Zaatari, M.; Grasberger, H.; Seekatz, A.M.; et al. Functional Characterization of Inflammatory Bowel Disease-Associated Gut Dysbiosis in Gnotobiotic Mice. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Kaneko, M.; Onuma, H.; Sakaguchi, Y.; Mori, M.; Abe, S.; Soga, T.; Tomita, M. Changes in the charged metabolite and sugar profiles of pasteurized and unpasteurized Japanese sake with storage. J. Agric. Food Chem. 2012, 60, 2586–2593. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Wong, D.T.; Hirayama, A.; Soga, T.; Tomita, M. Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics 2010, 6, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, K.; Edgett, B.A.; Burrows, H.W.; Castro, C.; Griffin, J.L.; Schwertani, A.G.; Gurd, B.J.; Funk, C.D. Whole blood transcriptomics and urinary metabolomics to define adaptive biochemical pathways of high-intensity exercise in 50–60 year old masters athletes. PLoS ONE 2014, 9, e92031. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Fukuda, S.; Fujiwara, A.; Suda, W.; Hattori, M.; Kikuchi, J.; Ohno, H. Multiple omics uncovers host-gut microbial mutualism during prebiotic fructooligosaccharide supplementation. DNA Res. 2014, 21, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Suda, W.; Kim, S.; Oshima, K.; Fukuda, S.; Ohno, H.; Morita, H.; Hattori, M. Robustness of gut microbiota of healthy adults in response to probiotic intervention revealed by high-throughput pyrosequencing. DNA Res. 2013, 20, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Goto, Y.; Ito, K.; Hayasaka, S.; Kurihara, S.; Soga, T.; Tomita, M.; Fukuda, S. The Consumption of Bicarbonate-Rich Mineral Water Improves Glycemic Control. Evid. Based Complement. Altern. Med. 2015, 2015, 824395. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Fukuda, S.; Hase, K.; Nishiumi, S.; Izumi, Y.; Yoshida, M.; Hagiwara, T.; Kawashima, R.; Yamazaki, M.; Oshio, T.; et al. Microbiota-derived lactate accelerates colon epithelial cell turnover in starvation-refed mice. Nat. Commun. 2013, 4, 1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Sayin, S.I.; Wahlstrom, A.; Felin, J.; Jantti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyotylainen, T.; Oresic, M.; Backhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Parseus, A.; Sommer, N.; Sommer, F.; Caesar, R.; Molinaro, A.; Stahlman, M.; Greiner, T.U.; Perkins, R.; Backhed, F. Microbiota-induced obesity requires farnesoid X receptor. Gut 2017, 66, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Gazzaniga, F.; Stebbins, R.; Chang, S.Z.; McPeek, M.A.; Brenner, C. Microbial NAD metabolism: Lessons from comparative genomics. Microbiol. Mol. Biol. Rev. 2009, 73, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, M.; Kurihara, S.; Kibe, R.; Ashida, H.; Benno, Y. Longevity in mice is promoted by probiotic-induced suppression of colonic senescence dependent on upregulation of gut bacterial polyamine production. PLoS ONE 2011, 6, e23652. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Sakamoto, K.; Seo, S.U.; Pickard, J.M.; Gillilland, M.G., 3rd; Pudlo, N.A.; Hoostal, M.; Li, X.; Wang, T.D.; Feehley, T.; et al. Neonatal acquisition of Clostridia species protects against colonization by bacterial pathogens. Science 2017, 356, 315–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugahara, H.; Odamaki, T.; Fukuda, S.; Kato, T.; Xiao, J.Z.; Abe, F.; Kikuchi, J.; Ohno, H. Probiotic Bifidobacterium longum alters gut luminal metabolism through modification of the gut microbial community. Sci. Rep. 2015, 5, 13548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthulakshmi, S.; Saravanan, R. Efficacy of azelaic acid on hepatic key enzymes of carbohydrate metabolism in high fat diet induced type 2 diabetic mice. Biochimie 2013, 95, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Nichols, B.L.; Quezada-Calvillo, R.; Robayo-Torres, C.C.; Ao, Z.; Hamaker, B.R.; Butte, N.F.; Marini, J.; Jahoor, F.; Sterchi, E.E. Mucosal maltase-glucoamylase plays a crucial role in starch digestion and prandial glucose homeostasis of mice. J. Nutr. 2009, 139, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Quezada-Calvillo, R.; Robayo-Torres, C.C.; Opekun, A.R.; Sen, P.; Ao, Z.; Hamaker, B.R.; Quaroni, A.; Brayer, G.D.; Wattler, S.; Nehls, M.C.; et al. Contribution of mucosal maltase-glucoamylase activities to mouse small intestinal starch alpha-glucogenesis. J. Nutr. 2007, 137, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.E.; Leese, H.J. Sucrose absorption by the rat small intestine in vivo and in vitro. J. Physiol. 1977, 267, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, Y.; Nakanishi, Y.; Murakami, S.; Aw, W.; Tsukimi, T.; Nozu, R.; Ueno, M.; Hioki, K.; Nakahigashi, K.; Hirayama, A.; et al. A Metabolomic-Based Evaluation of the Role of Commensal Microbiota throughout the Gastrointestinal Tract in Mice. Microorganisms 2018, 6, 101. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms6040101
Yamamoto Y, Nakanishi Y, Murakami S, Aw W, Tsukimi T, Nozu R, Ueno M, Hioki K, Nakahigashi K, Hirayama A, et al. A Metabolomic-Based Evaluation of the Role of Commensal Microbiota throughout the Gastrointestinal Tract in Mice. Microorganisms. 2018; 6(4):101. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms6040101
Chicago/Turabian StyleYamamoto, Yuri, Yumiko Nakanishi, Shinnosuke Murakami, Wanping Aw, Tomoya Tsukimi, Ryoko Nozu, Masami Ueno, Kyoji Hioki, Kenji Nakahigashi, Akiyoshi Hirayama, and et al. 2018. "A Metabolomic-Based Evaluation of the Role of Commensal Microbiota throughout the Gastrointestinal Tract in Mice" Microorganisms 6, no. 4: 101. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms6040101