An Alternative Platform for Protein Expression Using an Innate Whole Expression Module from Metagenomic DNA

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strain and Plasmid DNA
2.2. Deletion and Sequence Analysis of an Expression Module
2.3. Construction of Expression Vectors and Analysis of Vector Stability
2.4. Cloning and Expression of Foreign Genes in Novel Expression Vectors
2.5. Prediction of Translation Initiation Rate of Foreign ORFs in the pBEM Series Vector
3. Results and Discussion
3.1. Selection of a Clone Having an Innate Module with a Hyper-Expressed ORF from a Metagenomic Library
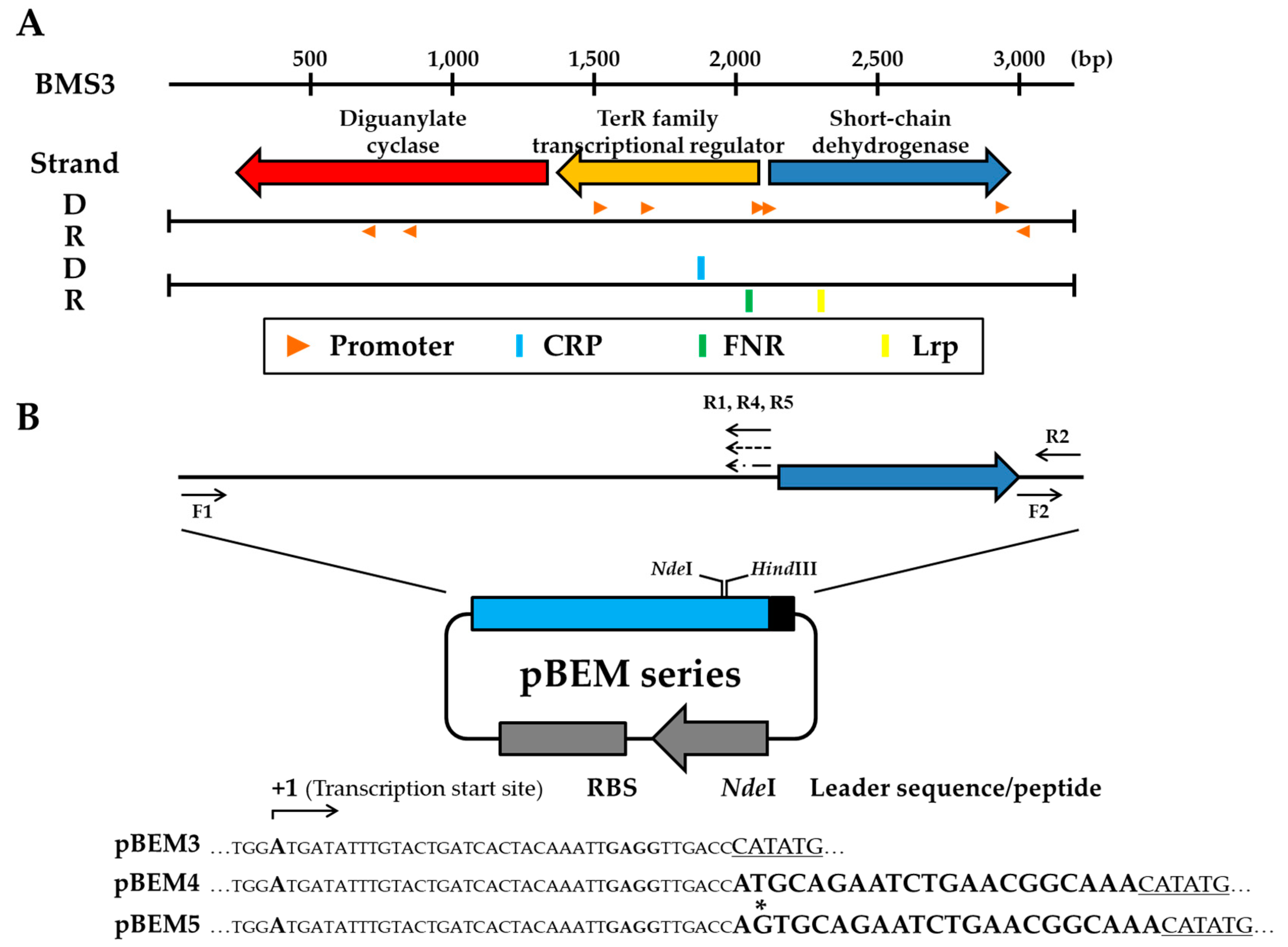
3.2. Sequence Analyses of the Minimized BMS3 DNA for Further Use as an Expression Module
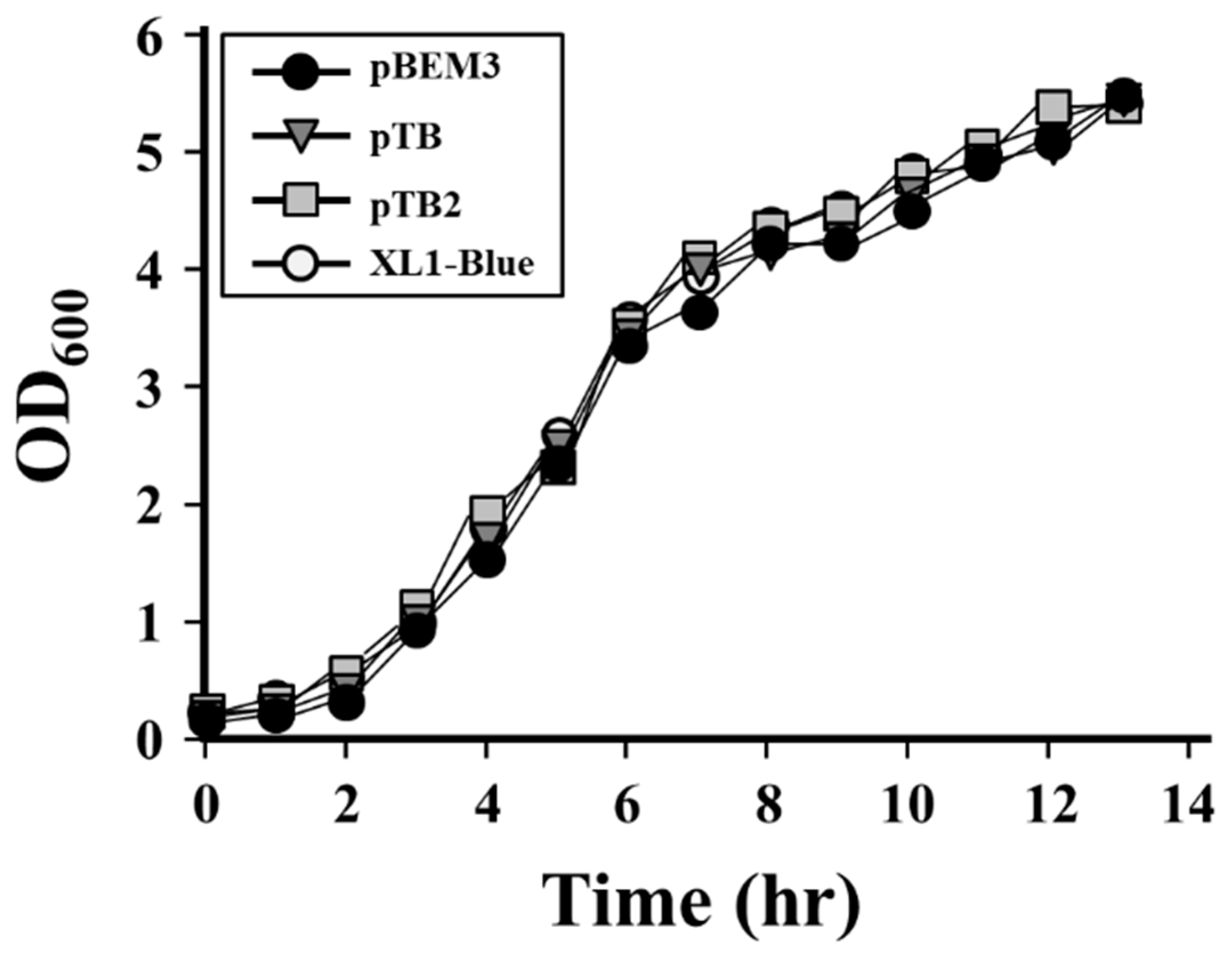
3.3. Construction and Stability Analysis of the Expression Vector Using the BMS3 Module
3.4. Analysis of Recombinant Protein Expression Using the pBEM3 Vector in E. coli
3.5. Analyses of Protein Expression in Recombinant Cells with Expression Vectors pBEM4 and pBEM5
Author Contributions
Funding
Conflicts of Interest
References
- Jana, S.; Deb, J.K. Strategies for efficient production of heterologous proteins in Escherichia coli. Appl. Microbiol. Biot. 2005, 67, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Jonasson, P.; Liljeqvist, S.; Nygren, P.A.; Stahl, S. Genetic design for facilitated production and recovery of recombinant proteins in Escherichia coli. Biotechnol. Appl. Biochem. 2002, 35, 91–105. [Google Scholar] [CrossRef]
- Roodveldt, C.; Aharoni, A.; Tawfik, D.S. Directed evolution of proteins for heterologous expression and stability. Curr. Opin. Struct. Biol. 2005, 15, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Makrides, S.C. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol. Rev. 1996, 60, 512–538. [Google Scholar] [PubMed]
- Swami, M. Gene expression: Directly linking transcription and translation. Nat. Rev. Genet. 2010, 11, 388–389. [Google Scholar] [CrossRef]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-sequence determinants of gene expression in Escherichia coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Alper, H.; Fischer, C.; Nevoigt, E.; Stephanopoulos, G. Tuning genetic control through promoter engineering. Proc. Natl. Acad. Sci. USA 2005, 102, 12678–12683. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Morel, L.; Le Francois, T.; Bourque, D.; Bourget, L.; Groleau, D.; Massie, B.; Miguez, C.B. Novel, Versatile, and Tightly Regulated Expression System for Escherichia coli Strains. Appl. Environ. Microb. 2010, 76, 5058–5066. [Google Scholar] [CrossRef]
- Salis, H.M.; Mirsky, E.A.; Voigt, C.A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009, 27, 946–950. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.H.; Kang, T.J.; Kim, D.M. Tuning the expression level of recombinant proteins by modulating mRNA stability in a cell-free protein synthesis system. Biotechnol. Bioeng. 2008, 101, 422–427. [Google Scholar] [CrossRef]
- Bandmann, N.; Nygren, P.A. Combinatorial expression vector engineering for tuning of recombinant protein production in Escherichia coli. Nucleic Acids Res. 2007, 35, e32. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, B.F.; Pitera, D.J.; Smolke, C.D.; Keasling, J.D. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat. Biotechnol. 2006, 24, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Lee, J.Y.; Kim, W.H.; Shin, H.J.; Kim, G.J. Screening of Promoters from Metagenomic DNA and Their Use for the Construction of Expression Vectors. J. Microbiol. Biotechn. 2008, 18, 1634–1640. [Google Scholar]
- Park, W.J.; You, S.H.; Choi, H.A.; Chu, Y.J.; Kim, G.J. Over-expression of recombinant proteins with N-terminal His-tag via subcellular uneven distribution in Escherichia coli. Acta Biochim. Biophys. Sin. 2015, 47, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Cheong, D.E.; Chang, W.S.; Kim, G.J. A cloning vector employing a versatile beta-glucosidase as an indicator for recombinant clones. Anal. Biochem. 2012, 425, 166–168. [Google Scholar] [CrossRef] [PubMed]
- Salgado, H.; Gama-Castro, S.; Peralta-Gil, M.; Diaz-Peredo, E.; Sanchez-Solano, F.; Santos-Zavaleta, A.; Martinez-Flores, I.; Jimenez-Jacinto, V.; Bonavides-Martinez, C.; Segura-Salazar, J.; et al. RegulonDB (version 5.0): Escherichia coli K-12 transcriptional regulatory network, operon organization, and growth conditions. Nucleic Acids Res. 2006, 34, D394–D397. [Google Scholar] [CrossRef]
- Salis, H.M. The ribosome binding site calculator. Methods Enzymol. 2011, 498, 19–42. [Google Scholar]
- Uchiyama, T.; Abe, T.; Ikemura, T.; Watanabe, K. Substrate-induced gene-expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat. Biotechnol. 2004, 23, 88–93. [Google Scholar] [CrossRef]
- Hwang, C.S.; Choi, E.S.; Han, S.S.; Kim, G.J. Screening of a highly soluble and oxygen-independent blue fluorescent protein from metagenome. Biochem. Bioph. Res. Co. 2012, 419, 676–681. [Google Scholar] [CrossRef]
- Martinez-Antonio, A.; Collado-Vides, J. Identifying global regulators in transcriptional regulatory networks in bacteria. Curr. Opin. Microbiol. 2003, 6, 482–489. [Google Scholar] [CrossRef]
- Ishihama, A. Prokaryotic genome regulation: multifactor promoters, multitarget regulators and hierarchic networks. FEMS Microbiol. Rev. 2010, 34, 628–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Lee, J.Y.; Shin, Y.S.; Kim, G.J. Characterization of an indican-hydrolyzing enzyme from Sinorhizobium meliloti. Process Biochem. 2010, 45, 892–896. [Google Scholar] [CrossRef]
- Goodman, D.B.; Church, G.M.; Kosuri, S. Causes and effects of N-terminal codon bias in bacterial genes. Science 2013, 342, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Cheong, D.E.; Ko, K.C.; Han, Y.; Jeon, H.G.; Sung, B.H.; Kim, G.J.; Choi, J.H.; Song, J.J. Enhancing functional expression of heterologous proteins through random substitution of genetic codes in the 5′ coding region. Biotechnol. Bioeng. 2015, 112, 822–826. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′→3′) | REase |
|---|---|---|
| BMS3 DNA F1 | TTTGAATTCCTCGCCGCC 1 | EcoRI |
| BMS3 DNA R1 | TTTGATATCCATATGGGTCAACCTCAAT | EcoRV, NdeI |
| BMS3 DNA F2 | TTTAAGCTTGCGTGCAGGGC | HindIII |
| BMS3 DNA R2 | TTTGAATTCTGGTGGGCTGTGAG | EcoRI |
| pTB F | ATATCTAGAGGCTGTTTTGGCGGA | XbaI |
| pTB R | TATCAGCTGCGGTGTGAAATACC | PvuII |
| BMS promoter F | TTTGTGTTGATCGATAAGAAAATC | |
| BEM4 common R | ATACATATGTTTGCCGTTCAGATTCTGCAT | |
| BEM5 common R | ATACATATGTTTGCCGTTCAGATTCTGCAC 2T | SpeI |
| mBFP F | TATCATATGCAGAATCTGAACGGCAAAGTGGCTT | NdeI |
| mBFP F | TATAAGCTTTCAAGCGGCGAAGCC | HindIII |
| MBP F | CGCCATATGAAAATCGAAGAAGGTAAAC | NdeI |
| MBP R | AAACATATGTCATCCGCCAAAA | NdeI |
| GFPuv F | TTTAAGCTTAAAGGAGAAGAACTTTTCACTG | HindIII |
| GFPuv R | TTTAAGCTTTTATTTGTAGAGCTCATCC | HindIII |
| SmGlu F | CCCATATGATGATCGAAGCCAAG | NdeI |
| SmGlu R | ATAAGCTTTCATCCCGGCTTGT | HindIII |
| 1767 F | ATACATATGGTGCAGATTCAGGGT | NdeI |
| 1767 R | TATAAGCTTTTACAGACAACCGGC | HindIII |
| Vector | RBS Sequence | ORF | Translation Initiation Rate |
|---|---|---|---|
| pBEM3 | GAGGTTGACCCAT | Mbfp | 5796.89 |
| GFPuv | 724.78 | ||
| SmGlu | 7358.43 | ||
| Esterase 1767 | 420.45 | ||
| pBEM4 | GAGGTTGACC | GFPuv | 5633.32 |
| SmGlu | 2472.22 | ||
| Esterase 1767 | 1104.66 | ||
| pBEM5 | GAGGTTGACCAGTGCAGAATCTGAACGGCAAACAT | GFPuv | 20.9 |
| SmGlu | 6.66 | ||
| Esterase 1767 | 6.66 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheong, D.-E.; Park, S.-Y.; Lim, H.-D.; Kim, G.-J. An Alternative Platform for Protein Expression Using an Innate Whole Expression Module from Metagenomic DNA. Microorganisms 2019, 7, 9. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7010009
Cheong D-E, Park S-Y, Lim H-D, Kim G-J. An Alternative Platform for Protein Expression Using an Innate Whole Expression Module from Metagenomic DNA. Microorganisms. 2019; 7(1):9. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7010009
Chicago/Turabian StyleCheong, Dae-Eun, So-Youn Park, Ho-Dong Lim, and Geun-Joong Kim. 2019. "An Alternative Platform for Protein Expression Using an Innate Whole Expression Module from Metagenomic DNA" Microorganisms 7, no. 1: 9. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7010009