Genetic Diversity and Demographic History of Ganoderma boninense in Oil Palm Plantations of Sarawak, Malaysia Inferred from ITS Regions

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Sequence Variations
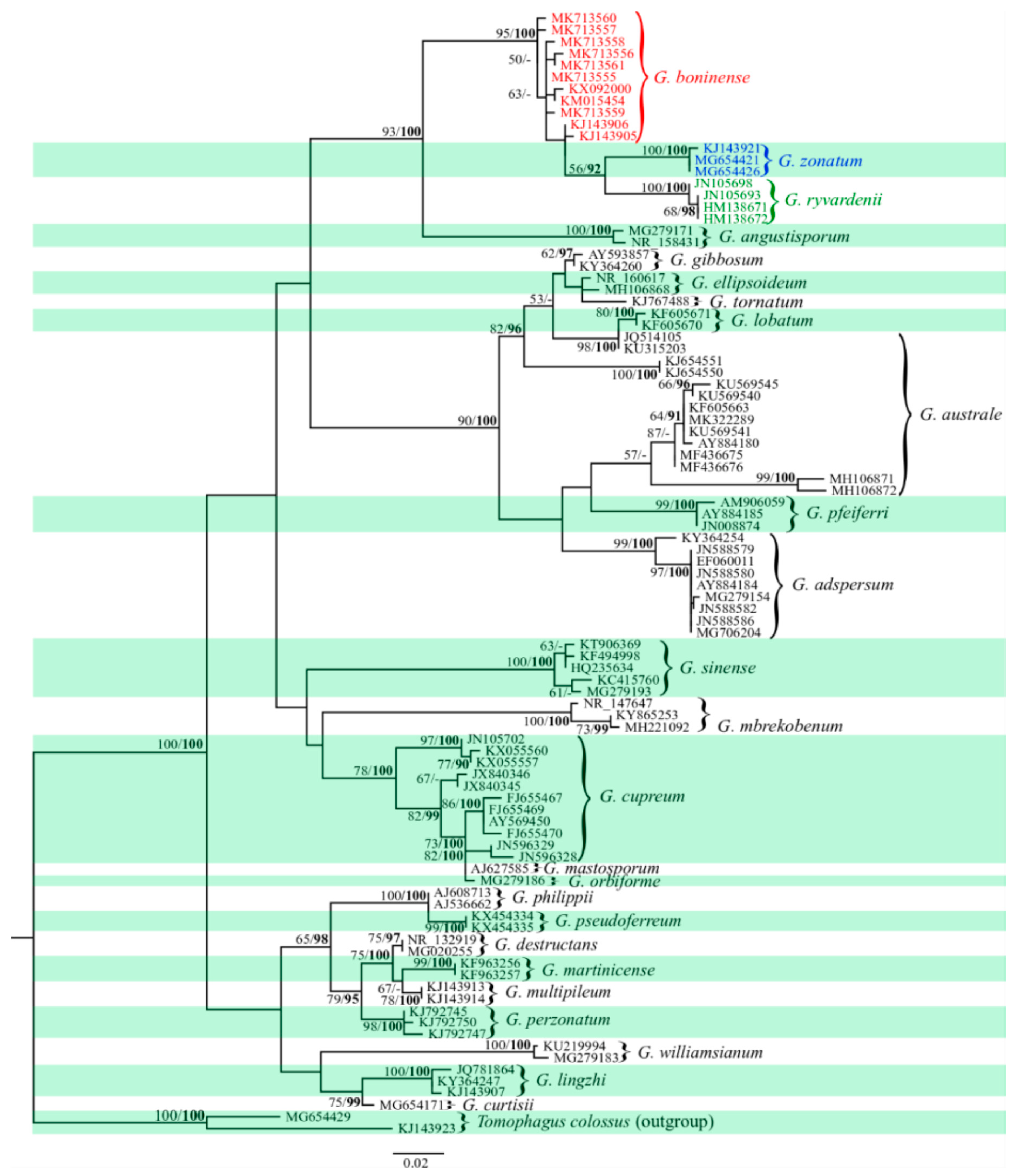
2.2. Phylogenetic Relationships among Haplotypes
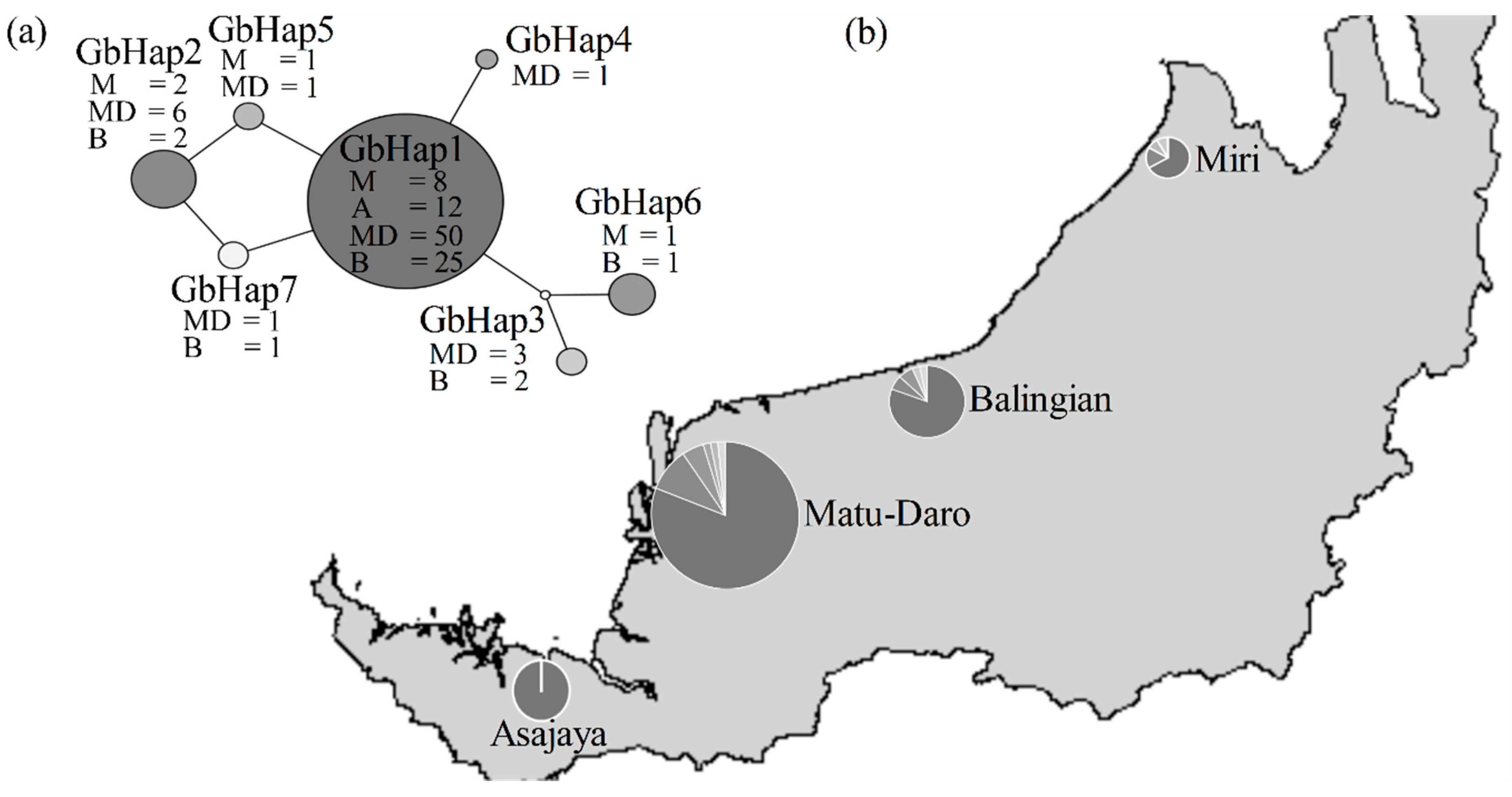
2.3. Population Structure Analyses
2.4. Demographic History of G. boninense Populations
3. Discussion
3.1. High Morphological Plasticity in Ganoderma spp. Leads to Misidentification
3.2. Ganoderma boninense SNPs-ITS Variation Analyses Showed a Single Population Throughout Sarawak
3.3. Haplotype GbHap1 as a Dominant Ancestral Isolate in Recent Population Expansion
3.4. Challenges and Recommendations
4. Materials and Methods
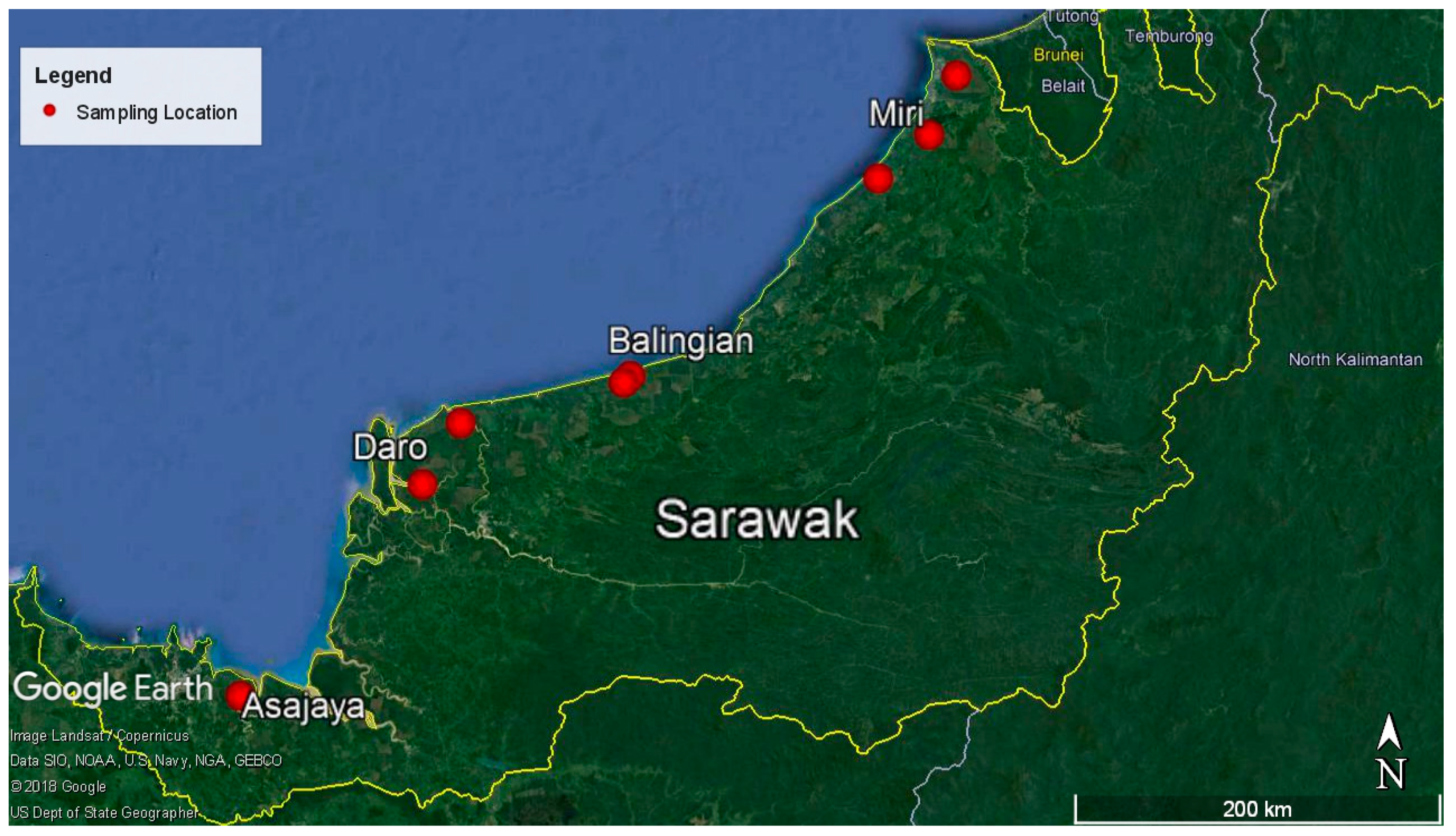
4.1. Sampling Location and Sample Collection
4.2. Ganoderma Culture
4.3. DNA Extraction, PCR Amplification and Sequencing
4.4. Data Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- USDAFAS. World Agricultural Production. United States Department of Agriculture Foreign Agricultural Service; 2018. Available online: https://www.fas.usda.gov/data (accessed on 31 January 2019).
- MPOB. Oil Palm Planted Area, Dec. Malaysian Palm Oil Board. 2018. Available online: http://bepi.mpob.gov.my/index.php/en/statistics/area.html (accessed on 31 January 2019).
- Hasan, Y.; Foster, H.L.; Flood, J. Investigations on the causes of upper stem rot (USR) on standing mature oil palms. Mycopathologia 2005, 159, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Pilotti, C.A. Stem rots of oil palm caused by Ganoderma boninense: Pathogen biology and epidemiology. Mycopathologia 2005, 159, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Roslan, A.; Idris, A.S. Economic impact of Ganoderma incidence on Malaysian Oil Palm Plantation—A case study in Johor. Oil Palm Ind. Econ. J. 2012, 12, 24–30. Available online: http://palmoilis.mpob.gov.my/publications/OPIEJ/opiejv12n1-Roslan.pdf (accessed on 7 June 2019).
- Abdullah, F.; Liew, S.B.; Malik, N. Upper stem rot of oil palms (E. guineensis) in Langkon, Sabah. In Sustainable Crop Protection Practices in the Next Millennium, Proceeding of MCB-MAPPS Plant Protection Conference, Kota Kinabalu, Sabah, Malaysia, 2–3 November 1999; Sidek, Z., Bong, S.L., Vijaya, S.K., Ong, C.A., Husan, A.K., Eds.; Malaysia Plant Protection Society: Kuala Lumpur, Malaysia, 1999; pp. 101–103. [Google Scholar]
- Utomo, C.; Werner, S.; Niepold, F.; Deising, H.B. Identification of Ganoderma, the causal agent of basal stem rot disease in oil palm using a molecular method. Mycopathologia 2005, 159, 159–170. [Google Scholar] [CrossRef]
- Rakib, M.R.M.; Bong, C.F.J.; Khairulmazmi, A.; Idris, A.S. Genetic and morphological diversity of Ganoderma species isolated from infected oil palms (Elaeis guineensis). Int. J. Agric. Biol. 2014, 16, 691–699. [Google Scholar]
- Ariffin, D.; Singh, G.; Lim, T.K. Ganoderma in Malaysia- Current Status and Research Strategy. In Module II: Agriculture, Proceedings of the PORIM International Palm Oil Development Conference, Bangi, Selangor, Malaysia, 5–9 September 1989; Palm Oil Research Institute of Malaysia: Bangi, Selangor, Malaysia, 1989; pp. 249–297. [Google Scholar]
- Idris, A.S.; Ariffin, D.; Swinburne, T.R.; Watt, T.A. The identity of Ganoderma species responsible for basal stem rot (BSR) disease of oil palm in Malaysia—Pathogenicity test. MPOB Inf. Ser. 2000, 103. Available online: http://www.mpob.gov.my/tot/tt77b.pdf (accessed on 4 May 2017).
- Hushiarian, R.; Yusof, N.A.; Dutse, S.W. Detection and control of Ganoderma boninense: Strategies and perspectives. SpringerPlus 2013, 2, 555. [Google Scholar] [CrossRef]
- Chong, K.P.; Dayou, J.; Alexander, A. Detection and control of Ganoderma boninense in oil palm crop; Springer Brief in Agriculture. Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Pilotti, C.A.; Gorea, A.E.; Bonneau, L. Basidiospores as sources of inoculum in the spread of Ganoderma boninense Pat. in oil palm plantations in Papua New Guinea. Plant Pathol. 2018, 67, 1841–1849. [Google Scholar] [CrossRef]
- Parthiban, K.; Vanitah, R.; Jusoff, K.; Nordiana, A.A.; Anuar, A.R.; Wahid, O.; Hamdan, A.B. GIS mapping of basal stem rot disease in relation to soil series among oil palm smallholders. Am. J. Agric. Biol. Sci. 2016, 11, 2–12. [Google Scholar] [CrossRef]
- Turner, P.D. Oil Palm Diseases and Disorders; Oxford University Press: Oxford, UK, 1981. [Google Scholar]
- Azahar, T.M.; Mustapha, J.C.; Mazliham, S.; Boursier, P. Temporal analysis of basal stem rot disease in oil palm plantations: An analysis on peat soil. Int. J. Eng. Technnol. 2011, 11, 96–101. [Google Scholar]
- Hashim, Z.; Subramaniam, V.; Harun, M.H. Carbon footprint of oil palm planted on peat in Malaysia. Int. J. Life Cycle Assess. 2018, 23, 1201–1217. [Google Scholar] [CrossRef]
- Cooper, R.M.; Flood, J.; Rees, R.W. Ganoderma boninense in oil palm plantations: Current thinking on epidemiology, resistance and pathology. Planter 2011, 87, 515–526. [Google Scholar]
- Miller, R.N.G.; Holderness, M.; Bridge, P.D.; Chung, G.F.; Zakaria, M.H. Genetic diversity of Ganoderma in oil palm plantings. Plant Pathol. 1999, 48, 595–603. [Google Scholar] [CrossRef]
- Pilotti, C.A.; Sanderson, F.R.; Aitken, E.A.B. Genetic structure of a population of Ganoderma boninense on oil palm. Plant Pathol. 2003, 52, 455–463. [Google Scholar] [CrossRef]
- Moncalvo, J.M. Systematics of Ganoderma. In Ganoderma Diseases of Perennial Crops; Flood, J., Bridge, P.D., Holderness, M., Eds.; CABI Publishing: Oxford, UK, 2000; pp. 23–45. [Google Scholar]
- Hong, K.K.; Geon, S.S.; Hong, G.K. Comparison of characteristics of Ganoderma lucidum according to geographical origins: Consideration of morphological characteristics. Microbiology 2001, 29, 80–84. [Google Scholar]
- Wang, D.M.; Wu, S.H.; Yao, Y.J. Clarification of the concept of Ganoderma orbiforme with high morphological plasticity. PLoS ONE 2014, 9, e98733. [Google Scholar] [CrossRef]
- Idris, A.S.; Rajinder, S.; Madihah, A.Z.; Wahid, M.B. Multiplex PCR-DNA kit for early detection and identification of Ganoderma species in oil palm. MPOB Inf. Ser. 2010, 531. Available online: http://palmoilis.mpob.gov.my/publications/TOT/TS-73.pdf (accessed on 4 May 2017).
- Wong, L.C.; Bong, C.F.J.; Idris, A.S. Ganoderma species associated with basal stem rot disease of oil palm. Am. J. Appl. Sci. 2012, 9, 879–885. [Google Scholar] [CrossRef]
- Nicolotti, G.; Gonthier, P.; Guglielmo, F. Advances in detection and identification of wood rooting fungi in timber and standing trees. In Molecular Fungal Identification; Gherbawy, Y., Voigt, K., Eds.; Springer: Berlin, Germany, 2010; pp. 251–276. [Google Scholar]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W. Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef]
- Utomo, C.; Niepold, F. Development of diagnostic methods for detecting Ganoderma infected oil palms. J. Phytopathol. 2000, 148, 507–514. [Google Scholar] [CrossRef]
- Zakaria, L.; Kulaverasingham, H.; Tan, S.G.; Abdullah, F.; Ho, Y.W. Random amplified polymorphic DNA (RAPD) and random amplified microsatellite (RAMS) of Ganoderma from infected oil palm and coconut stumps in Malaysia. Asia Pac. J. Mol. Biol. Biotechnol. 2005, 13, 23–34. [Google Scholar]
- Bridge, P.D.; O’Grady, E.B.; Pilotti, C.A.; Sanderson, F.R. Development of molecular diagnostics for the detection of Ganoderma isolates pathogenic to oil palm. In Ganoderma Diseases in Perennial Crops; Flood, J., Bridge, P.D., Holderness, M., Eds.; CABI Publishing: Wallingford, UK, 2000; pp. 225–234. [Google Scholar]
- Loyd, A.L.; Barnes, C.W.; Held, B.W.; Schink, M.J.; Smith, M.E.; Smith, J.A.; Blanchette, R.A. Elucidating “lucidum”: Distinguishing the diverse laccate Ganoderma species of the United States. PLoS ONE 2018, 13, e0199738. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [PubMed]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [PubMed]
- Zhou, L.W.; Cao, Y.; Wu, S.H.; Li, D.W.; Li, M.J.; Dai, Y.C. Global diversity of the Ganoderma lucidum complex (Ganodermataceae, Polyporales) inferred from morphology and multilocous phylogeny. Phytochemistry 2015, 114, 7–15. [Google Scholar] [CrossRef]
- Farr, D.F.; Rossman, A.Y. Fungal Databases. U.S. National Fungus Collection, ARS, USDA, 2019. Available online: https://nt.ars-grin.gov/fungaldatabases/ (accessed on 11 July 2019).
- Ahmad, M.F.; Wan Azhar, W.M.A.; Syazwan, S.A.; Mohamed, R. First record of basal stem rot of foxtail palm Wodyetia bifurcate caused by Ganoderma boninense in Malaysia. Plant Dis. 2018, 102, 1461. [Google Scholar]
- Elliott, M.L.; Des Jardin, E.A.; Ortiz, J.V.; Macias, T. Genetic variability of Ganoderma zonatum infecting palms in Florida. Mycologia 2018, 110, 339–346. [Google Scholar] [CrossRef]
- Kinge, T.R.; Mih, A.M. Ganoderma ryvardense sp. nov. associated with basal stem rot (BSR) disease of oil palm in Cameroon. Mycosphere 2011, 2, 179–188. [Google Scholar]
- Xing, J.; Sun, Y.; Han, Y.; Cui, B.; Dai, Y. Morphological and molecular identification of two new Ganoderma species on Casuarina equisetifolia from China. MycoKeys 2018, 34, 93–108. [Google Scholar] [CrossRef]
- Jargalmaa, S.; Eimes, J.A.; Park, M.S.; Park, J.Y.; Oh, S.Y.; Lim, Y.W. Taxonomic evaluation of selected Ganoderma species and database sequence validation. PeerJ 2017, 5, e3596. [Google Scholar] [CrossRef]
- Hong, S.G.; Jung, H.S. Phylogenetic analysis of Ganoderma based on nearly complete mitochondrial small-subunit ribosomal DNA sequences. Mycologia 2004, 96, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, D.; Kumar, M.K.P.; Chandrakanth, R.; Devaki, N.S. Molecular diversity of internal transcribed spacer among the monoconidial isolates of Magnaporthe oryzae isolated from rice in Southern Karnataka, India. J. Genet. Eng. Biotechnol. 2018, 16, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Ramdial, H.; Latchoo, R.K.; Hosein, F.N.; Rampersad, S.N. Phylogeny and haplotype analysis of fungi within the Fusarium incarnatum-equiseti species complex. Phytopathology 2017, 107, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.M.; Yao, Y.J. Intra-strain internal transcribed spacer heterogeneity in Ganoderma species. Can. J Microbiol. 2005, 51, 113–121. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, Z.; Pei, H.; Chen, Z.; Tan, X.; Hu, J.; Yang, B.; Sun, J. Intraspecific variation and phylogenetic relationships are revealed by ITS1 secondary structure analysis and single-nucleotide polymorphism in Ganoderma lucidum. PLoS ONE 2017, 12, e0169042. [Google Scholar] [CrossRef]
- Moncalvo, J.M.; Buchanan, P.K. Molecular evidence for long distance dispersal across the Southern Hemisphere in the Ganoderma applanatum-australe species complex (Basidiomycota). Mycol. Res. 2008, 112, 425–436. [Google Scholar] [CrossRef]
- Li, M.; Liang, J.; Li, Y.; Feng, B.; Yang, Z.; James, T.Y.; Xu, J. Genetic diversity of Dahonjun, the commercially important “Big Red Mushroom” from Southern China. PLoS ONE 2010, 5, e10684. [Google Scholar]
- Brewer, M.T.; Milgroom, M.G. Phylogeography and population structure of the grape powdery mildew fungus, Erysiphe necator, from diverse Vitis species. BMC Evolut. Biol. 2010, 10, 268. [Google Scholar] [CrossRef]
- Merciere, M.; Boulord, R.; Carasco-Lacombe, C.; Klopp, C.; Lee, Y.P.; Tan, J.S.; Syed Alwee, S.S.R.; Zaremski, A.; De Franqueville, H.; Breton, F.; et al. About Ganoderma boninense in oil palm plantations of Sumatra and Peninsular Malaysia: Ancient population expansion, extensive gene flow and large-scale dispersion ability. Fungal Biol. 2017, 121, 529–540. [Google Scholar] [CrossRef]
- Moller, M.; Stukenbrock, E.H. Evolution and genome architecture in fungal plant pathogens. Nat. Rev. Microbiol. 2017, 15, 756–771. [Google Scholar] [CrossRef]
- Linde, C.C.; Liles, J.A.; Thrall, P.H. Expansion of genetic diversity in randomly mating founder populations of Altenaria brassicicola infecting Cakile maritima in Australia. Appl. Environ. Microbiol. 2010, 76, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Soriano, A.; Ramos-Onsins, S.E.; Rozas, J.; Calafell, F.; Navarro, A. Statistical power analysis of neutrality tests under demographic expansions, contractions and bottlenecks with recombination. Genetics 2008, 179, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Rees, R.W.; Flood, J.; Hasan, Y.; Wills, M.A.; Cooper, R.M. Ganoderma boninense basidiospores in oil palm plantations: Evaluation of their possible role in stem rots of Elaeis guineensis. Plant Pathol. 2012, 61, 567–578. [Google Scholar] [CrossRef]
- Ferreri, M.; Qu, W.; Han, B. Phylogenetic networks: A tool to display character conflict and demographic history. Afr. J. Biotechnol. 2011, 10, 12799–12803. [Google Scholar] [Green Version]
- Edwards, S.V.; Potter, S.; Schmitt, C.J.; Bragg, J.G.; Moritz, C. Reticulation, divergence, and the phylogeography-phylogenetics continuum. Procs. Natl. Acad. Sci. USA 2016, 113, 8025–8032. [Google Scholar] [CrossRef] [PubMed]
- Othman, H.; Darus, F.M.; Mohammed, A.T. Experiences in peat development for oil palm planting in the MPOB Research Station at Sessang, Sarawak. Oil Palm Bulletin 2009, 58, 1–13. Available online: http://palmoilis.mpob.gov.my/publications/OPB/opb58-hasnol.pdf (accessed on 7 June 2019).
- McDonald, B.A.; Linde, C. Pathogen population genetics, evolutionary potential and durable resistance. Annu. Rev. Phytopathol. 2002, 40, 349–379. [Google Scholar] [CrossRef]
- Rosenberg, N.N.; Nordborg, M. Genealogical tress, coalescent theory and the analysis of genetic polymorphism. Nat. Rev. Genet. 2002, 3, 380–390. [Google Scholar] [CrossRef]
- Grant, W.S. Problems and cautions with sequence mismatch analysis and Bayesian Skyline plots to infer history demography. J. Hered. 2015, 106, 333–346. [Google Scholar] [CrossRef]
- Heled, J.; Drummond, A.J. Bayesian inference of population size history from multiple loci. BMC Evolut. Biol. 2008, 8, 289. [Google Scholar] [CrossRef]
- Google Earth Pro v7.3. Map of Sarawak 2°55’16.83” N 112°39’26.39” E. 2019. Available online: https://www.google.com/earth/ (accessed on 2 April 2019).
- Ariffin, D.; Idris, A.S. The Ganoderma selective medium (GSM). PORIM Information Series, 8, November 1992. Available online: http://palmoilis.mpob.gov.my/images/PORIM%20IS/0008/PORIM%20IS%200008.pdf (accessed on 4 May 2017).
- Tung, H.J.; Ong, C.E.; Goh, Y.K.; Goh, Y.K.; Goh, K.J. Survival and pathogenicity of monokaryotic and dikaryotic Ganoderma boninense following three different preservation methods. Trans. Sci. Technol. 2018, 5, 46–52. Available online: http://tost.unise.org/pdfs/vol5/no1/5x1x46x52.pdf (accessed on 1 February 2019).
- Zhang, Y.J.; Zhang, S.; Liu, X.Z.; Wen, H.A.; Wang, M. A simple method of genomic DNA extraction suitable for analysis of bulk fungal strains. Lett. Appl. Microbiol. 2010, 51, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes-application to identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Villesen, P. FaBox: An Online Fasta Sequence Toolbox. 2007. Available online: http://www.birc.au.dk/software/fabox (accessed on 14 March 2019).
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evolut. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guiro-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evolut. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Kimura, M. Genetic variability maintained in a finite population due to mutational production of neutral and nearly neutral isoalleles. Genet. Res. 1968, 11, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linus and Windows. Mol. Ecol. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate Maximum-Likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayre, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Effecienty Bayesian Phylogenetic Inference and model choice across a large model space. Syst. Biol. 2010, 61, 539–542. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Rambaut A. FigTree 1.4.4. Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, 2018. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 7 April 2019).
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.M.; Cabezas, M.P.; Tavares, A.I.; Xavier, R.; Branco, M. tcsBU: A tool to extend TCS network layout and visualization. Bioinformatics 2016, 32, 627–628. [Google Scholar] [CrossRef] [PubMed]
- Inkscape’s Contributors. Inkscape 0.92.4. The Inkscape Project: Oregon, USA, 2019. Available online: http://inkscape.org/en/ (accessed on 8 April 2019).




{kind=link}
{kind=link}
{kind=link}
| Variables | 17 | 64 | 84 | 130 | 152 | 503 | |
|---|---|---|---|---|---|---|---|
| Haplotype | |||||||
| GBHap1 | T | T | T | G | T | C | |
| GbHap2 | ∙ | ∙ | ∙ | T | C | ∙ | |
| GbHap3 | C | ∙ | C | ∙ | ∙ | ∙ | |
| GbHap4 | ∙ | C | ∙ | ∙ | ∙ | ∙ | |
| GbHap5 | ∙ | ∙ | ∙ | ∙ | C | ∙ | |
| GbHap6 | ∙ | ∙ | C | ∙ | ∙ | T | |
| GbHap7 | ∙ | ∙ | ∙ | T | ∙ | ∙ | |
| Haplotypes | Populations | |||
|---|---|---|---|---|
| Miri (n = 12) | Asajaya (n = 12) | Matu-Daro (n = 62) | Balingian (n = 31) | |
| GbHap1 | 0.667 | 1.000 | 0.820 | 0.806 |
| GbHap2 | 0.167 | 0 | 0.098 | 0.065 |
| GbHap3 | 0 | 0 | 0.049 | 0.065 |
| GbHap4 | 0 | 0 | 0.016 | 0 |
| GbHap5 | 0.083 | 0 | 0.016 | 0 |
| GbHap6 | 0.083 | 0 | 0 | 0.032 |
| GbHap7 | 0 | 0 | 0.016 | 0.032 |
| Sequence Variation | ||||
| Total no. of polymorphic loci (S) | 4 | 0 | 5 | 5 |
| Total no. mutation (Eta) | 4 | 0 | 5 | 5 |
| Ave. no. nucleotide differences (k) | 1.045 | 0 | 0.627 | 0.675 |
| Molecular Diversity Indices | ||||
| Nucleotide diversity (PiJC) | 0.002 | 0 | 0.001 | 0.001 |
| Theta S (per sequence) | 1.325 | 0 | 1.065 | 1.252 |
| Number of haplotypes (h) | 4 | 1 | 6 | 5 |
| Haplotype diversity (Hd) | 0.561 | 0 | 0.343 | 0.351 |
| Tajima’s D | −0.741 * | N/A | −0.955 * | −1.246 * |
| Fu’s FS | −0.524 * | N/A | −2.272 * | −1.634 * |
| Population | Miri | Asajaya | Matu-Daro | Balingian |
|---|---|---|---|---|
| Miri | N/A | |||
| Asajaya | 0.104 * | N/A | ||
| Matu-Daro | −0.003 * | 0.008 * | N/A | |
| Balingian | −0.014 * | 0.048 * | −0.014 * | N/A |
| Source of Variations | Sum of Squares | Variance Components | Percentage of Variations | p-Value |
|---|---|---|---|---|
| Among populations | 0.871 | −0.001 | −0.254 | 0.503 * |
| Within populations | 34.992 | 0.310 | 100.254 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Midot, F.; Lau, S.Y.L.; Wong, W.C.; Tung, H.J.; Yap, M.L.; Lo, M.L.; Jee, M.S.; Dom, S.P.; Melling, L. Genetic Diversity and Demographic History of Ganoderma boninense in Oil Palm Plantations of Sarawak, Malaysia Inferred from ITS Regions. Microorganisms 2019, 7, 464. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7100464
Midot F, Lau SYL, Wong WC, Tung HJ, Yap ML, Lo ML, Jee MS, Dom SP, Melling L. Genetic Diversity and Demographic History of Ganoderma boninense in Oil Palm Plantations of Sarawak, Malaysia Inferred from ITS Regions. Microorganisms. 2019; 7(10):464. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7100464
Chicago/Turabian StyleMidot, Frazer, Sharon Yu Ling Lau, Wei Chee Wong, Hun Jiat Tung, Mui Lan Yap, Mei Lieng Lo, Mui Sie Jee, Simon Peter Dom, and Lulie Melling. 2019. "Genetic Diversity and Demographic History of Ganoderma boninense in Oil Palm Plantations of Sarawak, Malaysia Inferred from ITS Regions" Microorganisms 7, no. 10: 464. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7100464